
Current Pharmacotherapy and Multi-Target Approaches for Alzheimer’s Disease

Abstract
:
1. Introduction
2. Pathogenesis of Alzheimer’s Disease
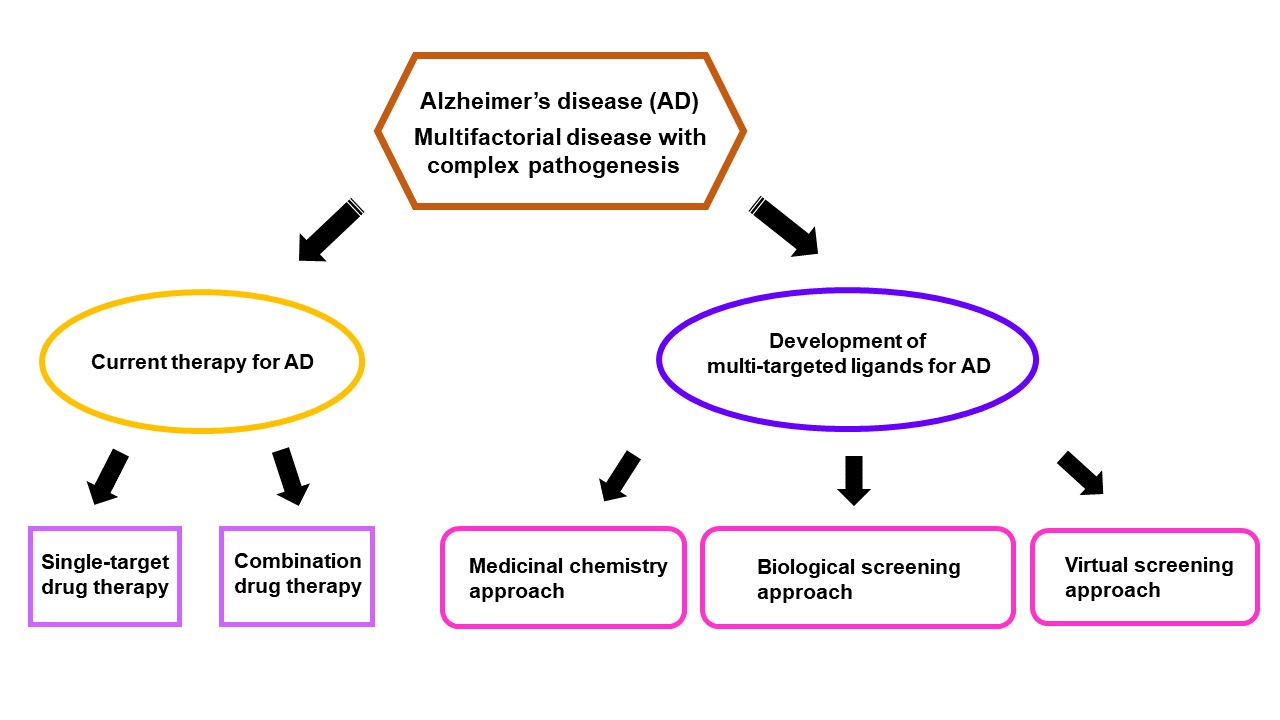
3. Current Drug Therapies for Alzheimer’s Disease
4. Multi-Target Approaches in the Discovery of Polypharmacological Ligands for Alzheimer’s Disease
4.1. Medicinal Chemistry-Based Approaches
4.1.1. Conjugate Hybrids
4.1.2. Merged Hybrids
4.1.3. Fused Hybrids
4.2. Biological Screening-Based Approaches
4.3. Virtual Screening-Based Approaches
4.4. Other Related Targets for Multi-Target Drug Discovery of Alzheimer’s Disease
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Association. 2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022, 18, 700–789. [Google Scholar] [CrossRef] [PubMed]
- National Institute on Aging USA. What Is Alzheimer’s Disease? Available online: https://www.nia.nih.gov/health/what-alzheimers-disease (accessed on 18 August 2022).
- National Institute on Aging USA. What Are the Signs of Alzheimer’s Disease? Available online: https://www.nia.nih.gov/health/what-are-signs-alzheimers-disease (accessed on 22 August 2022).
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments in Alzheimer disease: An update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef]
- McDade, E.; Bateman, R.J. Stop Alzheimer’s before it starts. Nature 2017, 547, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Tariq, S.; Barber, P.A. Dementia risk and prevention by targeting modifiable vascular risk factors. J. Neurochem. 2018, 144, 565–581. [Google Scholar] [CrossRef] [Green Version]
- Cuyvers, E.; Sleegers, K. Genetic variations underlying Alzheimer’s disease: Evidence from genome-wide association studies and beyond. Lancet Neurol. 2016, 15, 857–868. [Google Scholar] [CrossRef]
- Van der Schyf, C.J.; Youdim, M.B. Multifunctional drugs as neurotherapeutics. Neurotherapeutics 2009, 6, 1–3. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Pasqualetti, G.; Tognini, S.; Calsolaro, V.; Polini, A.; Monzani, F. Potential drug-drug interactions in Alzheimer patients with behavioral symptoms. Clin. Interv. Aging 2015, 10, 1457–1466. [Google Scholar]
- Polaka, S.; Koppisetti, H.P.; Tekade, M.; Sharma, M.C.; Sengupta, P.; Tekade, R.K. Drug–Drug Interactions and Their Implications on the Pharmacokinetics of the Drugs. In Pharmacokinetics and Toxicokinetic Considerations; Academic Press: Cambridge, MA, USA, 2022; pp. 291–322. [Google Scholar]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Pathogenesis of Alzheimer’s disease. Clin. Interv. Aging. 2007, 2, 347–359. [Google Scholar]
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain. 2011, 4, 3. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New insights into the pathogenesis of Alzheimer’s disease. Front. Neurol. 2020, 10, 1312. [Google Scholar] [CrossRef]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.; Klein, P.S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Wang, Z.; Hu, H.; Zhao, M.; Sun, L. Microglia in Alzheimer’s disease: A target for therapeutic intervention. Front. Cell. Neurosci. 2021, 15, 749587. [Google Scholar] [CrossRef]
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and inflammation in Alzheimers disease. CNS Neurol. Disord. Drug Targets 2010, 9, 156–167. [Google Scholar] [CrossRef]
- Kocahan, S.; Doğan, Z. Mechanisms of Alzheimer’s disease pathogenesis and prevention: The brain, neural pathology, N-methyl-D-Aspartate receptors, tau protein and other risk factors. Clin. Psychopharmacol. Neurosci. 2017, 15, 1–8. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, P.; Feng, J.; Wu, M. Dysfunction of NMDA receptors in Alzheimer’s disease. Neurol. Sci. 2016, 37, 1039–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, P. Challenging the cholinergic hypothesis in Alzheimer disease. JAMA 1999, 281, 1433–1434. [Google Scholar] [CrossRef] [PubMed]
- Craig, L.A.; Hong, N.S.; McDonald, R.J. Revisiting the cholinergic hypothesis in the development of Alzheimer’s disease. Neurosci. Biobehav. Rev. 2011, 35, 1397–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terry, A.V., Jr.; Buccafusco, J.J. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: Recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.; Martinez, A. Targeting beta-amyloid pathogenesis through acetylcholinesterase inhibitors. Curr. Pharm. Des. 2006, 12, 4377–4387. [Google Scholar] [CrossRef]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Chu, L.W. Alzheimer’s disease: Early diagnosis and treatment. Hong Kong Med. J. 2012, 18, 228–237. [Google Scholar]
- Zhang, N.; Gordon, M.L. Clinical efficacy and safety of donepezil in the treatment of Alzheimer’s disease in Chinese patients. Clin. Interv. Aging 2018, 13, 1963–1970. [Google Scholar] [CrossRef] [Green Version]
- Cacabelos, R. Donepezil in Alzheimer’s disease: From conventional trials to pharmacogenetics. Neuropsychiatr. Dis. Treat. 2007, 3, 303–333. [Google Scholar]
- Black, S.; Román, G.C.; Geldmacher, D.S.; Salloway, S.; Hecker, J.; Burns, A.; Perdomo, C.; Kumar, D.; Pratt, R.; Donepezil 307 Vascular Dementia Study Group. Efficacy and tolerability of donepezil in vascular dementia: Positive results of a 24-week, multicenter, international, randomized, placebo-controlled clinical trial. Stroke 2003, 34, 2323–2330. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, D.G.; Passmore, A.P.; Bullock, R.; Hopker, S.W.; Smith, R.; Potocnik, F.C.; Maud, C.M.; Engelbrecht, I.; Hock, C.; Ieni, J.R.; et al. A multinational, randomised, 12-week, comparative study of donepezil and rivastigmine in patients with mild to moderate Alzheimer’s disease. Int. J. Clin. Pract. 2002, 56, 441–446. [Google Scholar]
- Farlow, M.R.; Salloway, S.; Tariot, P.N.; Yardley, J.; Moline, M.L.; Wang, Q.; Brand-Schieber, E.; Zou, H.; Hsu, T.; Satlin, A. Effectiveness and tolerability of high-dose (23 mg/d) versus standard-dose (10 mg/d) donepezil in moderate to severe Alzheimer’s disease: A 24-week, randomized, double-blind study. Clin. Ther. 2010, 32, 1234–1251. [Google Scholar] [CrossRef] [Green Version]
- Thavichachart, N.; Phanthumchinda, K.; Chankrachang, S.; Praditsuwan, R.; Nidhinandana, S.; Senanarong, V.; Poungvarin, N. Efficacy study of galantamine in possible Alzheimer’s disease with or without cerebrovascular disease and vascular dementia in Thai patients: A slow-titration regimen. Int. J. Clin. Pract. 2006, 60, 533–540. [Google Scholar] [CrossRef] [Green Version]
- Wallin, A.K.; Wattmo, C.; Minthon, L. Galantamine treatment in Alzheimer’s disease: Response and long-term outcome in a routine clinical setting. Neuropsychiatr. Dis. Treat. 2011, 7, 565–576. [Google Scholar] [CrossRef]
- Kandiah, N.; Pai, M.C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging 2017, 12, 697–707. [Google Scholar] [CrossRef] [Green Version]
- Onor, M.L.; Trevisiol, M.; Aguglia, E. Rivastigmine in the treatment of Alzheimer’s disease: An update. Clin. Interv. Aging 2007, 2, 17–32. [Google Scholar] [CrossRef]
- Khoury, R.; Rajamanickam, J.; Grossberg, G.T. An update on the safety of current therapies for Alzheimer’s disease: Focus on rivastigmine. Ther. Adv. Drug Saf. 2018, 9, 171–178. [Google Scholar] [CrossRef]
- Rösler, M.; Anand, R.; Cicin-Sain, A.; Gauthier, S.; Agid, Y.; Dal-Bianco, P.; Stähelin, H.B.; Hartman, R.; Gharabawi, M. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: International randomised controlled trial. BMJ 1999, 318, 633–638. [Google Scholar] [CrossRef] [Green Version]
- Karaman, Y.; Erdoğan, F.; Köseoğlu, E.; Turan, T.; Ersoy, A.O. A 12-month study of the efficacy of rivastigmine in patients with advanced moderate Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2005, 19, 51–56. [Google Scholar] [CrossRef]
- Folch, J.; Busquets, O.; Ettcheto, M.; Sánchez-López, E.; Castro-Torres, R.D.; Verdaguer, E.; Garcia, M.L.; Olloquequi, J.; Casadesús, G.; Beas-Zarate, C.; et al. Memantine for the treatment of dementia: A review on its current and future applications. J. Alzheimer’s Dis. 2018, 62, 1223–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendiola-Precoma, J.; Berumen, L.C.; Padilla, K.; Garcia-Alcocer, G. Therapies for prevention and treatment of Alzheimer’s disease. BioMed Res. Int. 2016, 2016, 2589276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.M.; Keating, G.M. Memantine: A review of its use in Alzheimer’s disease. Drugs 2006, 66, 1515–1534. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Léveillé, F.; El Gaamouch, F.; Gouix, E.; Lecocq, M.; Lobner, D.; Nicole, O.; Buisson, A. Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB J. 2008, 22, 4258–4271. [Google Scholar] [CrossRef]
- Bordji, K.; Becerril-Ortega, J.; Buisson, A. Synapses, NMDA receptor activity and neuronal Aβ production in Alzheimer’s disease. Rev. Neurosci. 2011, 22, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Schulz, J.B.; Rainer, M.; Klünemann, H.H.; Kurz, A.; Wolf, S.; Sternberg, K.; Tennigkeit, F. Sustained effects of once-daily memantine treatment on cognition and functional communication skills in patients with moderate to severe Alzheimer’s disease: Results of a 16-week open-label trial. J. Alzheimer’s Dis. 2011, 25, 463–475. [Google Scholar] [CrossRef] [Green Version]
- Responder analyses on memantine in Alzheimer’s disease: Executive summary of rapid report A10-06, Version 1.0. In Institute for Quality and Efficiency in Health Care: Executive Summaries; IQWiG (Institute for Quality and Efficiency in Health Care): Cologne, Germany, 2005.
- Deardorff, W.J.; Grossberg, G.T. A fixed-dose combination of memantine extended-release and donepezil in the treatment of moderate-to-severe Alzheimer’s disease. Drug Des. Dev. Ther. 2016, 10, 3267–3279. [Google Scholar] [CrossRef] [Green Version]
- Tariot, P.N.; Farlow, M.R.; Grossberg, G.T.; Graham, S.M.; McDonald, S.; Gergel, I.; Memantine Study Group. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: A randomized controlled trial. JAMA 2004, 291, 317–324. [Google Scholar] [CrossRef]
- Atri, A.; Hendrix, S.B.; Pejović, V.; Hofbauer, R.K.; Edwards, J.; Molinuevo, J.L.; Graham, S.M. Cumulative, additive benefits of memantine-donepezil combination over component monotherapies in moderate to severe Alzheimer’s dementia: A pooled area under the curve analysis. Alzheimer’s Res. Ther. 2015, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Boinpally, R.; Chen, L.; Zukin, S.R.; McClure, N.; Hofbauer, R.K.; Periclou, A. A novel once-daily fixed-dose combination of memantine extended release and donepezil for the treatment of moderate to severe alzheimer’s disease: Two phase I studies in healthy volunteers. Clin. Drug Investig. 2015, 35, 427–435. [Google Scholar] [CrossRef] [Green Version]
- Zerkak, D.; Dougados, M. Benefit/risk of combination therapies. Clin. Exp. Rheumatol. 2004, 22, S71–S76. [Google Scholar]
- Van der Schyf, C.J. The use of multi-target drugs in the treatment of neurodegenerative diseases. Expert Rev. Clin. Pharmacol. 2011, 4, 293–298. [Google Scholar] [CrossRef]
- Benek, O.; Korabecny, J.; Soukup, O. A Perspective on multi-target drugs for Alzheimer’s disease. Trends Pharmacol. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef]
- Golde, T.E.; DeKosky, S.T.; Galasko, D. Alzheimer’s disease: The right drug, the right time. Science 2018, 362, 1250–1251. [Google Scholar] [CrossRef]
- Hsu, D.; Marshall, G.A. Primary and secondary prevention trials in Alzheimer disease: Looking back, moving forward. Curr. Alzheimer Res. 2017, 14, 426–440. [Google Scholar] [CrossRef] [Green Version]
- Carrillo, M.C.; Brashear, H.R.; Logovinsky, V.; Ryan, J.M.; Feldman, H.H.; Siemers, E.R.; Abushakra, S.; Hartley, D.M.; Petersen, R.C.; Khachaturian, A.S.; et al. Can we prevent Alzheimer’s disease? Secondary “prevention” trials in Alzheimer’s disease. Alzheimers Dement. 2013, 9, 123–131.e1. [Google Scholar] [CrossRef]
- Suridjan, I.; Pollock, B.G.; Verhoeff, N.P.; Voineskos, A.N.; Chow, T.; Rusjan, P.M.; Lobaugh, N.J.; Houle, S.; Mulsant, B.H.; Mizrahi, R. In-vivo imaging of grey and white matter neuroinflammation in Alzheimer’s disease: A positron emission tomography study with a novel radioligand, [18F]-FEPPA. Mol. Psychiatry 2015, 20, 1579–1587. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Reddy, P.H. Therapeutics of neurotransmitters in Alzheimer’s disease. J. Alzheimers Dis. 2017, 57, 1049–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morphy, R.; Rankovic, Z. The physicochemical challenges of designing multiple ligands. J. Med. Chem. 2006, 49, 4961–4970. [Google Scholar] [CrossRef] [PubMed]
- González, J.F.; Alcántara, A.R.; Doadrio, A.L.; Sánchez-Montero, J.M. Developments with multi-target drugs for Alzheimer’s disease: An overview of the current discovery approaches. Expert Opin. Drug Discov. 2019, 14, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Xu, S.; Zhu, Z.; Xu, J. Multi-target design strategies for the improved treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 176, 228–247. [Google Scholar] [CrossRef]
- Li, Q.; He, S.; Chen, Y.; Feng, F.; Qu, W.; Sun, H. Donepezil-based multi-functional cholinesterase inhibitors for treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 158, 463–477. [Google Scholar] [CrossRef]
- Musial, A.; Bajda, M.; Malawska, B. Recent developments in cholinesterases inhibitors for Alzheimers disease treatment. Curr. Med. Chem. 2007, 14, 2654–2679. [Google Scholar] [CrossRef]
- Cai, Z. Monoamine oxidase inhibitors: Promising therapeutic agents for Alzheimer’s disease (Review). Mol. Med. Rep. 2014, 9, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Youdim, M.B.; Bakhle, Y.S. Monoamine oxidase: Isoforms and inhibitors in Parkinson’s disease and depressive illness. Br. J. Pharmacol. 2006, 147, S287–S296. [Google Scholar] [CrossRef] [Green Version]
- Claeysen, S.; Bockaert, J.; Giannoni, P. Serotonin: A new hope in Alzheimer’s disease? ACS Chem. Neurosci. 2015, 6, 940–943. [Google Scholar] [CrossRef] [Green Version]
- Lalut, J.; Karila, D.; Dallemagne, P.; Rochais, C. Modulating 5-HT4 and 5-HT6 receptors in Alzheimer’s disease treatment. Future Med. Chem. 2017, 9, 781–795. [Google Scholar] [CrossRef]
- Mössner, R.; Schmitt, A.; Syagailo, Y.; Gerlach, M.; Riederer, P.; Lesch, K.P. The serotonin transporter in Alzheimer’s and Parkinson’s disease. J. Neural Transm. Suppl. 2000, 60, 345–350. [Google Scholar]
- García-Osta, A.; Cuadrado-Tejedor, M.; García-Barroso, C.; Oyarzábal, J.; Franco, R. Phosphodiesterases as therapeutic targets for Alzheimer’s disease. ACS Chem. Neurosci. 2012, 3, 832–844. [Google Scholar] [CrossRef] [Green Version]
- García-Barroso, C.; Ricobaraza, A.; Pascual-Lucas, M.; Unceta, N.; Rico, A.J.; Goicolea, M.A.; Sallés, J.; Lanciego, J.L.; Oyarzabal, J.; Franco, R.; et al. Tadalafil crosses the blood-brain barrier and reverses cognitive dysfunction in a mouse model of AD. Neuropharmacology 2013, 64, 114–123. [Google Scholar] [CrossRef]
- Benito, C.; Núñez, E.; Tolón, R.M.; Carrier, E.J.; Rábano, A.; Hillard, C.J.; Romero, J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J. Neurosci. 2003, 23, 11136–11141. [Google Scholar] [CrossRef] [Green Version]
- Ehrhart, J.; Obregon, D.; Mori, T.; Hou, H.; Sun, N.; Bai, Y.; Klein, T.; Fernandez, F.; Tan, J.; Shytle, R.D. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J. Neuroinflamm. 2005, 2, 29. [Google Scholar] [CrossRef] [Green Version]
- Aso, E.; Ferrer, I. CB2 Cannabinoid receptor as potential target against Alzheimer’s disease. Front. Neurosci. 2016, 10, 243. [Google Scholar] [CrossRef]
- Bembenek, S.D.; Keith, J.M.; Letavic, M.A.; Apodaca, R.; Barbier, A.J.; Dvorak, L.; Aluisio, L.; Miller, K.L.; Lovenberg, T.W.; Carruthers, N.I. Lead identification of acetylcholinesterase inhibitors-histamine H3 receptor antagonists from molecular modeling. Bioorg. Med. Chem. 2008, 16, 2968–2973. [Google Scholar] [CrossRef]
- Pákáski, M.; Kálmán, J. Interactions between the amyloid and cholinergic mechanisms in Alzheimer’s disease. Neurochem. Int. 2008, 53, 103–111. [Google Scholar] [CrossRef]
- Ko, S.Y.; Ko, H.A.; Chu, K.H.; Shieh, T.M.; Chi, T.C.; Chen, H.I.; Chang, W.C.; Chang, S.S. The possible mechanism of advanced glycation end products (AGEs) for Alzheimer’s disease. PLoS ONE 2015, 10, e0143345. [Google Scholar] [CrossRef]
- Münch, G.; Thome, J.; Foley, P.; Schinzel, R.; Riederer, P. Advanced glycation endproducts in ageing and Alzheimer’s disease. Brain Res. Brain Res. Rev. 1997, 23, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Leuci, R.; Brunetti, L.; Laghezza, A.; Piemontese, L.; Carrieri, A.; Pisani, L.; Tortorella, P.; Catto, M.; Loiodice, F. A new series of aryloxyacetic acids endowed with multi-target activity towards peroxisome proliferator-activated receptors (PPARs), fatty acid amide hydrolase (FAAH), and acetylcholinesterase (AChE). Molecules 2022, 27, 958. [Google Scholar] [CrossRef] [PubMed]
- Ulasov, A.V.; Rosenkranz, A.A.; Georgiev, G.P.; Sobolev, A.S. Nrf2/Keap1/ARE signaling: Towards specific regulation. Life Sci. 2022, 291, 120111. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, H.; Wang, Y.; Liu, W.; Yin, G.; Wang, D.; Li, J.; Shi, T.; Wang, Z. Design, synthesis, and biological evaluation of carbamate derivatives of N-salicyloyl tryptamine as multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2022, 229, 114044. [Google Scholar] [CrossRef] [PubMed]
- Patrono, C. Cardiovascular effects of cyclooxygenase-2 inhibitors: A mechanistic and clinical perspective. Br. J. Clin. Pharmacol. 2016, 82, 957–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desale, S.E.; Chinnathambi, S. Role of dietary fatty acids in microglial polarization in Alzheimer’s disease. J. Neuroinflamm. 2020, 17, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kam, T.I.; Song, S.; Gwon, Y.; Park, H.; Yan, J.J.; Im, I.; Choi, J.W.; Choi, T.Y.; Kim, J.; Song, D.K.; et al. FcγRIIb mediates amyloid-β neurotoxicity and memory impairment in Alzheimer’s disease. J. Clin. Investig. 2013, 123, 2791–2802. [Google Scholar] [CrossRef]
- Kam, T.I.; Park, H.; Gwon, Y.; Song, S.; Kim, S.H.; Moon, S.W.; Jo, D.G.; Jung, Y.K. FcγRIIb-SHIP2 axis links Aβ to tau pathology by disrupting phosphoinositide metabolism in Alzheimer’s disease model. Elife 2016, 5, e18691. [Google Scholar] [CrossRef]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting new molecular targets for known drugs. Nature 2009, 462, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Rognan, D. Chemogenomic approaches to rational drug design. Br. J. Pharmacol. 2007, 152, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Viayna, E.; Sola, I.; Bartolini, M.; De Simone, A.; Tapia-Rojas, C.; Serrano, F.G.; Sabaté, R.; Juárez-Jiménez, J.; Pérez, B.; Luque, F.J.; et al. Synthesis and multitarget biological profiling of a novel family of rhein derivatives as disease-modifying anti-Alzheimer agents. J. Med. Chem. 2014, 57, 2549–2567. [Google Scholar] [CrossRef] [Green Version]
- Sivaprakasam, P.; Han, X.; Civiello, R.L.; Jacutin-Porte, S.; Kish, K.; Pokross, M.; Lewis, H.A.; Ahmed, N.; Szapiel, N.; Newitt, J.A.; et al. Discovery of new acylaminopyridines as GSK-3 inhibitors by a structure guided in-depth exploration of chemical space around a pyrrolopyridinone core. Bioorg. Med. Chem. Lett. 2015, 25, 1856–1863. [Google Scholar] [CrossRef]
- Rosini, M.; Simoni, E.; Bartolini, M.; Cavalli, A.; Ceccarini, L.; Pascu, N.; McClymont, D.W.; Tarozzi, A.; Bolognesi, M.L.; Minarini, A.; et al. Inhibition of acetylcholinesterase, beta-amyloid aggregation, and NMDA receptors in Alzheimer’s disease: A promising direction for the multi-target-directed ligands gold rush. J. Med. Chem. 2008, 51, 4381–4384. [Google Scholar] [CrossRef]
- Simoni, E.; Daniele, S.; Bottegoni, G.; Pizzirani, D.; Trincavelli, M.L.; Goldoni, L.; Tarozzo, G.; Reggiani, A.; Martini, C.; Piomelli, D.; et al. Combining galantamine and memantine in multitargeted, new chemical entities potentially useful in Alzheimer’s disease. J. Med. Chem. 2012, 55, 9708–9721. [Google Scholar] [CrossRef] [Green Version]
- Lecoutey, C.; Hedou, D.; Freret, T.; Giannoni, P.; Gaven, F.; Since, M.; Bouet, V.; Ballandonne, C.; Corvaisier, S.; Malzert Fréon, A.; et al. Design of donecopride, a dual serotonin subtype 4 receptor agonist/acetylcholinesterase inhibitor with potential interest for Alzheimer’s disease treatment. Proc. Natl. Acad. Sci. USA 2014, 111, E3825–E3830. [Google Scholar] [CrossRef] [Green Version]
- Ghanei-Nasab, S.; Khoobi, M.; Hadizadeh, F.; Marjani, A.; Moradi, A.; Nadri, H.; Emami, S.; Foroumadi, A.; Shafiee, A. Synthesis and anticholinesterase activity of coumarin-3-carboxamides bearing tryptamine moiety. Eur. J. Med. Chem. 2016, 121, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Anand, P.; Singh, B.; Singh, N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer’s disease. Bioorg. Med. Chem. 2012, 20, 1175–1180. [Google Scholar] [CrossRef]
- Luo, X.T.; Wang, C.M.; Liu, Y.; Huang, Z.G. New multifunctional melatonin-derived benzylpyridinium bromides with potent cholinergic, antioxidant, and neuroprotective properties as innovative drugs for Alzheimer’s disease. Eur. J. Med. Chem. 2015, 103, 302–311. [Google Scholar] [CrossRef]
- Cheng, S.; Zheng, W.; Gong, P.; Zhou, Q.; Xie, Q.; Yu, L.; Zhang, P.; Chen, L.; Li, J.; Chen, J.; et al. (-)-Meptazinol-melatonin hybrids as novel dual inhibitors of cholinesterases and amyloid-β aggregation with high antioxidant potency for Alzheimer’s therapy. Bioorg. Med. Chem. 2015, 23, 3110–3118. [Google Scholar] [CrossRef]
- Więckowska, A.; Kołaczkowski, M.; Bucki, A.; Godyń, J.; Marcinkowska, M.; Więckowski, K.; Zaręba, P.; Siwek, A.; Kazek, G.; Głuch-Lutwin, M.; et al. Novel multi-target-directed ligands for Alzheimer’s disease: Combining cholinesterase inhibitors and 5-HT6 receptor antagonists. Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2016, 124, 63–81. [Google Scholar] [CrossRef]
- Więckowska, A.; Wichur, T.; Godyń, J.; Bucki, A.; Marcinkowska, M.; Siwek, A.; Więckowski, K.; Zaręba, P.; Knez, D.; Głuch-Lutwin, M.; et al. Novel Multitarget-Directed Ligands Aiming at Symptoms and Causes of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Szałaj, N.; Godyń, J.; Jończyk, J.; Pasieka, A.; Panek, D.; Wichur, T.; Więckowski, K.; Zaręba, P.; Bajda, M.; Pislar, A.; et al. Multidirectional in vitro and in cellulo studies as a tool for identification of multi-target-directed ligands aiming at symptoms and causes of Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2020, 35, 1944–1952. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Lavado, J.; Gallardo-Garrido, C.; Mallea, M.; Bustos, V.; Osorio, R.; Hödar-Salazar, M.; Chung, H.; Araya-Maturana, R.; Lorca, M.; Pessoa-Mahana, C.D.; et al. Synthesis, in vitro evaluation and molecular docking of a new class of indolylpropyl benzamidopiperazines as dual AChE and SERT ligands for Alzheimer’s disease. Eur. J. Med. Chem. 2020, 198, 112368. [Google Scholar] [CrossRef] [PubMed]
- Modica, M.N.; Intagliata, S.; Pittalà, V.; Salerno, L.; Siracusa, M.A.; Cagnotto, A.; Salmona, M.; Romeo, G. Synthesis and binding properties of new long-chain 4-substituted piperazine derivatives as 5-HT₁A and 5-HT₇ receptor ligands. Bioorg. Med. Chem. Lett. 2015, 25, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Bagetta, D.; Cagide, F.; Teixeira, J.; Amorim, R.; Silva, T.; Garrido, J.; Remião, F.; Uriarte, E.; Oliveira, P.J.; et al. Benzoic Acid-Derived Nitrones: A new class of potential acetylcholinesterase inhibitors and neuroprotective agents. Eur. J. Med. Chem. 2019, 174, 116–129. [Google Scholar] [CrossRef]
- Rosini, M.; Andrisano, V.; Bartolini, M.; Bolognesi, M.L.; Hrelia, P.; Minarini, A.; Tarozzi, A.; Melchiorre, C. Rational approach to discover multipotent anti-Alzheimer drugs. J. Med. Chem. 2005, 48, 360–363. [Google Scholar] [CrossRef]
- Mao, F.; Wang, H.; Ni, W.; Zheng, X.; Wang, M.; Bao, K.; Ling, D.; Li, X.; Xu, Y.; Zhang, H.; et al. Design, synthesis, and biological evaluation of orally available first-generation dual-target selective inhibitors of acetylcholinesterase (AChE) and phosphodiesterase 5 (PDE5) for the treatment of Alzheimer’s disease. ACS Chem. Neurosci. 2018, 9, 328–345. [Google Scholar] [CrossRef]
- Singh, M.; Kaur, M.; Singh, N.; Silakari, O. Exploration of multi-target potential of chromen-4-one based compounds in Alzheimer’s disease: Design, synthesis and biological evaluations. Bioorg. Med. Chem. 2017, 25, 6273–6285. [Google Scholar] [CrossRef]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Scheiner, M.; Dolles, D.; Gunesch, S.; Hoffmann, M.; Nabissi, M.; Marinelli, O.; Naldi, M.; Bartolini, M.; Petralla, S.; Poeta, E.; et al. Dual-acting cholinesterase-human cannabinoid receptor 2 ligands show pronounced neuroprotection in vitro and overadditive and disease-modifying neuroprotective effects in vivo. J. Med. Chem. 2019, 62, 9078–9102. [Google Scholar] [CrossRef]
- Xu, Y.-X.; Wang, H.; Li, X.-K.; Dong, S.-N.; Liu, W.-W.; Gong, Q.; Wang, T.-D.-Y.; Tang, Y.; Zhu, J.; Li, J.; et al. Discovery of novel propargylamine-modified 4-aminoalkyl imidazole substituted pyrimidinylthiourea derivatives as multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 143, 33–47. [Google Scholar] [CrossRef]
- Marco-Contelles, J.; Unzeta, M.; Bolea, I.; Esteban, G.; Ramsay, R.R.; Romero, A.; Martínez-Murillo, R.; Carreiras, M.C.; Ismaili, L. ASS234, as a new multi-target directed propargylamine for Alzheimer’s disease therapy. Front. Neurosci. 2016, 10, 294. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.; Dwivedi, A.R.; Arora, T.; Raj, K.; Prashar, V.; Kumar, V.; Singh, S.; Prakash, J.; Kumar, V. Design, synthesis, and pharmacological evaluation of N-propargylated diphenylpyrimidines as multitarget directed ligands for the treatment of Alzheimer’s disease. ACS Chem. Neurosci. 2022, 13, 2122–2139. [Google Scholar] [CrossRef]
- Bolea, I.; Gella, A.; Unzeta, M. Propargylamine-derived multitarget-directed ligands: Fighting Alzheimer’s disease with monoamine oxidase inhibitors. J. Neural. Transm. 2013, 120, 893–902. [Google Scholar] [CrossRef]
- Weinreb, O.; Amit, T.; Bar-Am, O.; Youdim, M.B.H. Rasagiline: A novel anti-Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity. Prog. Neurobiol. 2010, 92, 330–344. [Google Scholar] [CrossRef]
- Carocci, A.; Barbarossa, A.; Leuci, R.; Carrieri, A.; Brunetti, L.; Laghezza, A.; Catto, M.; Limongelli, F.; Chaves, S.; Tortorella, P.; et al. Novel phenothiazine/donepezil-like hybrids endowed with antioxidant activity for a multi-target approach to the therapy of Alzheimer’s disease. Antioxidants 2022, 11, 1631. [Google Scholar] [CrossRef]
- Murphy, C.M.; Ravner, H.; Smith, N.L. Mode of action of phenothiazine-type antioxidants. Ind. Eng. Chem. 1950, 42, 2479–2489. [Google Scholar] [CrossRef]
- O’Leary, J.C.; Li, Q.; Marinec, P.; Blair, L.J.; Congdon, E.E.; Johnson, A.G.; Jinwal, U.K.; Koren, J.; Jones, J.R.; Kraft, C.; et al. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol. Neurodegener. 2010, 5, 45. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Cheng, M.; Liu, P.; Cao, D.; Luo, J.; Wan, Y.; Fang, Y.; Jin, Y.; Xie, S.-S.; Liu, J. A multi-target directed ligands strategy for the treatment of Alzheimer’s disease: Dimethyl fumarate plus Tranilast modified Dithiocarbate as AChE inhibitor and Nrf2 activator. Eur. J. Med. Chem. 2022, 242, 114630. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Ak, M.; Mrowietz, U. Reactivity of dimethyl fumarate and methylhydrogen fumarate towards glutathione and N-acetyl-L-cysteine--preparation of S-substituted thiosuccinic acid esters. Bioorg. Med. Chem. 2007, 15, 333–342. [Google Scholar] [CrossRef]
- Benchekroun, M.; Romero, A.; Egea, J.; León, R.; Michalska, P.; Buendía, I.; Jimeno, M.L.; Jun, D.; Janockova, J.; Sepsova, V.; et al. The antioxidant additive approach for Alzheimer’s disease therapy: New ferulic (Lipoic) acid plus melatonin modified Tacrines as cholinesterases inhibitors, direct antioxidants, and nuclear factor (erythroid-derived 2)-like 2 activators. J. Med. Chem. 2016, 59, 9967–9973. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Y.; Zhuo, J. Tranilast treatment attenuates cerebral ischemia-reperfusion injury in rats through the inhibition of inflammatory responses mediated by NF-kB and PPARs. Clin. Transl. Sci. 2019, 12, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, N.; Ding, J.; Liu, J.; Sun, X.; Zhang, Z.; Mo, Z.; Li, X.; Yin, H.; Tang, W.; Xie, S.-S. Novel chromanone-dithiocarbamate hybrids as multifunctional AChE inhibitors with β-amyloid anti-aggregation properties for the treatment of Alzheimer’s disease. Bioorg. Chem. 2019, 89, 103027. [Google Scholar] [CrossRef] [PubMed]
- Javed, M.A.; Bibi, S.; Jan, M.S.; Ikram, M.; Zaidi, A.; Farooq, U.; Sadiq, A.; Rashid, U. Diclofenac derivatives as concomitant inhibitors of cholinesterase, monoamine oxidase, cyclooxygenase-2 and 5-lipoxygenase for the treatment of Alzheimer’s disease: Synthesis, pharmacology, toxicity and docking studies. RSC Adv. 2022, 12, 22503–22517. [Google Scholar] [CrossRef]
- Javed, M.A.; Ashraf, N.; Saeed Jan, M.; Mahnashi, M.H.; Alqahtani, Y.S.; Alyami, B.A.; Alqarni, A.O.; Asiri, Y.I.; Ikram, M.; Sadiq, A.; et al. Structural modification, in vitro, in vivo, ex vivo, and in silico exploration of pyrimidine and pyrrolidine cores for targeting enzymes associated with neuroinflammation and cholinergic deficit in Alzheimer’s disease. ACS Chem. Neurosci. 2021, 12, 4123–4143. [Google Scholar] [CrossRef]
- Xie, S.; Chen, J.; Li, X.; Su, T.; Wang, Y.; Wang, Z.; Huang, L.; Li, X. Synthesis and evaluation of selegiline derivatives as monoamine oxidase inhibitor, antioxidant and metal chelator against Alzheimer’s disease. Bioorg. Med. Chem. 2015, 23, 3722–3729. [Google Scholar] [CrossRef]
- Gabr, M.T.; Abdel-Raziq, M.S. Design and synthesis of donepezil analogues as dual AChE and BACE-1 inhibitors. Bioorg. Chem. 2018, 80, 245–252. [Google Scholar] [CrossRef]
- Liu, W.; Wang, H.; Li, X.; Xu, Y.; Zhang, J.; Wang, W.; Gong, Q.; Qiu, X.; Zhu, J.; Mao, F.; et al. Design, synthesis and evaluation of vilazodone-tacrine hybrids as multitarget-directed ligands against depression with cognitive impairment. Bioorg. Med. Chem. 2018, 26, 3117–3125. [Google Scholar] [CrossRef]
- Zhou, L.-Y.; Zhu, Y.; Jiang, Y.-R.; Zhao, X.-J.; Guo, D. Design, synthesis and biological evaluation of dual acetylcholinesterase and phosphodiesterase 5A inhibitors in treatment for Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2017, 27, 4180–4184. [Google Scholar] [CrossRef]
- Weinstock, M.; Bejar, C.; Wang, R.H.; Poltyrev, T.; Gross, A.; Finberg, J.P.; Youdim, M.B. TV3326, a novel neuroprotective drug with cholinesterase and monoamine oxidase inhibitory activities for the treatment of Alzheimer’s disease. J. Neural Transm. Suppl. 2000, 60, 157–169. [Google Scholar]
- Weinreb, O.; Amit, T.; Bar-Am, O.; Youdim, M.B.H. Neuroprotective effects of multifaceted hybrid agents targeting MAO, cholinesterase, iron and β-amyloid in ageing and Alzheimer’s disease. Br. J. Pharmacol. 2016, 173, 2080–2094. [Google Scholar] [CrossRef] [Green Version]
- Safety and Efficacy Study of Ladostigil in Mild to Moderate Probable Alzheimer’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT01354691 (accessed on 26 August 2022).
- Schneider, L.S.; Geffen, Y.; Rabinowitz, J.; Thomas, R.G.; Schmidt, R.; Ropele, S.; Weinstock, M.; on behalf of the Ladostigil Study Group. Low-dose ladostigil for mild cognitive impairment: A phase 2 placebo-controlled clinical trial. Neurology 2019, 93, e1474–e1484. [Google Scholar] [CrossRef]
- Venkidath, A.; Oh, J.M.; Dev, S.; Amin, E.; Rasheed, S.P.; Vengamthodi, A.; Gambacorta, N.; Khames, A.; Abdelgawad, M.A.; George, G.; et al. Selected class of enamides bearing nitro functionality as dual-acting with highly selective monoamine oxidase-B and BACE1 inhibitors. Molecules 2021, 26, 6004. [Google Scholar] [CrossRef]
- Maliyakkal, N.; Eom, B.H.; Heo, J.H.; Abdullah Almoyad, M.A.; Thomas Parambi, D.G.; Gambacorta, N.; Nicolotti, O.; Beeran, A.A.; Kim, H.; Mathew, B. A new potent and selective monoamine oxidase-B Inhibitor with extended conjugation in a chalcone framework: 1-[4-(Morpholin-4-yl)phenyl]-5-phenylpenta-2,4-dien-1-one. ChemMedChem 2020, 15, 1629–1633. [Google Scholar] [CrossRef]
- Kavully, F.S.; Oh, J.M.; Dev, S.; Kaipakasseri, S.; Palakkathondi, A.; Vengamthodi, A.; Abdul Azeez, R.F.; Tondo, A.R.; Nicolotti, O.; Kim, H.; et al. Design of enamides as new selective monoamine oxidase-B inhibitors. J. Pharm. Pharmacol. 2020, 72, 916–926. [Google Scholar] [CrossRef]
- Moussa-Pacha, N.M.; Abdin, S.M.; Omar, H.A.; Alniss, H.; Al-Tel, T.H. BACE1 inhibitors: Current status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 2020, 40, 339–384. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Y.; Wang, B.; Li, W.; Huang, L.; Li, X. Design, synthesis, and evaluation of orally available clioquinol-moracin M hybrids as multitarget-directed ligands for cognitive improvement in a rat model of neurodegeneration in Alzheimer’s disease. J. Med. Chem. 2015, 58, 8616–8637. [Google Scholar] [CrossRef]
- Mao, F.; Yan, J.; Li, J.; Jia, X.; Miao, H.; Sun, Y.; Huang, L.; Li, X. New multi-target-directed small molecules against Alzheimer’s disease: A combination of resveratrol and clioquinol. Org. Biomol. Chem. 2014, 12, 5936–5944. [Google Scholar] [CrossRef]
- Koeberle, A.; Werz, O. Multi-target approach for natural products in inflammation. Drug Discov. Today 2014, 19, 1871–1882. [Google Scholar] [CrossRef]
- Peña-Altamira, E.; Prati, F.; Massenzio, F.; Virgili, M.; Contestabile, A.; Bolognesi, M.L.; Monti, B. Changing paradigm to target microglia in neurodegenerative diseases: From anti-inflammatory strategy to active immunomodulation. Expert Opin. Ther. Targets 2016, 20, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Bai, P.; Wang, K.; Mi, J.; Yang, J.; Hu, J.; Ban, Y.; Xu, R.; Chen, R.; Wang, C.; et al. Design, synthesis, and evaluation of novel O-alkyl ferulamide derivatives as multifunctional ligands for treating Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2022, 37, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.F.; Devi, K.P.; Malar, D.S.; Sureda, A.; Daglia, M.; Nabavi, S.M. Ferulic acid and Alzheimer’s disease: Promises and pitfalls. Mini Rev. Med. Chem. 2015, 15, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Legoabe, L.J.; Petzer, A.; Petzer, J. 2-Acetylphenol analogs as potent reversible monoamine oxidase inhibitors. Drug Des. Devel. Ther. 2015, 9, 3635–3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeřábek, J.; Uliassi, E.; Guidotti, L.; Korábečný, J.; Soukup, O.; Sepsova, V.; Hrabinova, M.; Kuča, K.; Bartolini, M.; Peña-Altamira, L.E.; et al. Tacrine-resveratrol fused hybrids as multi-target-directed ligands against Alzheimer’s disease. Eur. J. Med. Chem. 2017, 127, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Simoni, E.; Serafini, M.M.; Bartolini, M.; Caporaso, R.; Pinto, A.; Necchi, D.; Fiori, J.; Andrisano, V.; Minarini, A.; Lanni, C.; et al. Nature-inspired multifunctional ligands: Focusing on amyloid-based molecular mechanisms of Alzheimer’s disease. ChemMedChem 2016, 11, 1309–1317. [Google Scholar] [CrossRef]
- Rossi, M.; Freschi, M.; de Camargo Nascente, L.; Salerno, A.; de Melo Viana Teixeira, S.; Nachon, F.; Chantegreil, F.; Soukup, O.; Prchal, L.; Malaguti, M.; et al. Sustainable drug discovery of multi-target-directed ligands for Alzheimer’s disease. J. Med. Chem. 2021, 64, 4972–4990. [Google Scholar] [CrossRef]
- Sasidharan, R.; Eom, B.H.; Heo, J.H.; Park, J.E.; Abdelgawad, M.A.; Musa, A.; Gambacorta, N.; Nicolotti, O.; Manju, S.L.; Mathew, B.; et al. Morpholine-based chalcones as dual-acting monoamine oxidase-B and acetylcholinesterase inhibitors: Synthesis and biochemical investigations. J. Enzyme Inhib. Med. Chem. 2021, 36, 188–197. [Google Scholar] [CrossRef]
- Mathew, B.; Haridas, A.; Suresh, J.; Mathew, G.E.; Uçar, G.; Jayaprakash, V. Monoamine oxidase inhibitory action of chalcones: A mini review. Cent. Nerv. Syst. Agents Med. Chem. 2016, 16, 120–136. [Google Scholar] [CrossRef]
- Tian, C.; Qiang, X.; Song, Q.; Cao, Z.; Ye, C.; He, Y.; Deng, Y.; Zhang, L. Flurbiprofen-chalcone hybrid Mannich base derivatives as balanced multifunctional agents against Alzheimer’s disease: Design, synthesis and biological evaluation. Bioorg. Chem. 2020, 94, 103477. [Google Scholar] [CrossRef]
- Mathew, B.; Baek, S.C.; Thomas Parambi, D.G.; Lee, J.P.; Mathew, G.E.; Jayanthi, S.; Vinod, D.; Rapheal, C.; Devikrishna, V.; Kondarath, S.S.; et al. Potent and highly selective dual-targeting monoamine oxidase-B inhibitors: Fluorinated chalcones of morpholine versus imidazole. Arch. Pharm. 2019, 352, e1800309. [Google Scholar] [CrossRef]
- Rehuman, N.A.; Oh, J.M.; Nath, L.R.; Khames, A.; Abdelgawad, M.A.; Gambacorta, N.; Nicolotti, O.; Jat, R.K.; Kim, H.; Mathew, B. Halogenated coumarin-chalcones as multifunctional monoamine oxidase-B and butyrylcholinesterase inhibitors. ACS Omega 2021, 6, 28182–28193. [Google Scholar] [CrossRef]
- Chimenti, F.; Secci, D.; Bolasco, A.; Chimenti, P.; Bizzarri, B.; Granese, A.; Carradori, S.; Yáñez, M.; Orallo, F.; Ortuso, F.; et al. Synthesis, molecular modeling, and selective inhibitory activity against human monoamine oxidases of 3-carboxamido-7-substituted coumarins. J. Med. Chem. 2009, 52, 1935–1942. [Google Scholar] [CrossRef]
- Hammuda, A.; Shalaby, R.; Rovida, S.; Edmondson, D.E.; Binda, C.; Khalil, A. Design and synthesis of novel chalcones as potent selective monoamine oxidase-B inhibitors. Eur. J. Med. Chem. 2016, 114, 162–169. [Google Scholar] [CrossRef]
- Sang, Z.; Wang, K.; Zhang, P.; Shi, J.; Liu, W.; Tan, Z. Design, synthesis, in-silico and biological evaluation of novel chalcone derivatives as multi-function agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 180, 238–252. [Google Scholar] [CrossRef]
- Bai, P.; Wang, K.; Zhang, P.; Shi, J.; Cheng, X.; Zhang, Q.; Zheng, C.; Cheng, Y.; Yang, J.; Lu, X.; et al. Development of chalcone-O-alkylamine derivatives as multifunctional agents against Alzheimer’s disease. Eur. J. Med. Chem. 2019, 183, 111737. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Lu, Z.; Zheng, X.; Ni, W.; Zhu, J.; Fu, Y.; Lian, F.; Zhang, N.; Li, J.; et al. Development of multifunctional pyrimidinylthiourea derivatives as potential anti-Alzheimer agents. J. Med. Chem. 2016, 59, 8326–8344. [Google Scholar] [CrossRef]
- Oh, J.M.; Jang, H.-J.; Kang, M.-G.; Song, S.; Kim, D.-Y.; Kim, J.-H.; Noh, J.-I.; Park, J.E.; Park, D.; Yee, S.-T.; et al. Acetylcholinesterase and monoamine oxidase-B inhibitory activities by ellagic acid derivatives isolated from Castanopsis cuspidata var. sieboldii. Sci. Rep. 2021, 11, 13953. [Google Scholar] [CrossRef]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K.; Aggarwal, B.B. Curcumin, a component of golden spice: From bedside to bench and back. Biotechnol Adv. Biotechnol. Adv. 2014, 32, 1053–1064. [Google Scholar] [CrossRef]
- Mas-Bargues, C.; Borrás, C.; Viña, J. The multimodal action of genistein in Alzheimer’s and other age-related diseases. Free Radic. Biol. Med. 2022, 183, 127–137. [Google Scholar] [CrossRef]
- Prietsch, R.F.; Monte, L.G.; da Silva, F.A.; Beira, F.T.; Del Pino, F.A.B.; Campos, V.F.; Collares, T.; Pinto, L.S.; Spanevello, R.M.; Gamaro, G.D.; et al. Genistein induces apoptosis and autophagy in human breast MCF-7 cells by modulating the expression of proapoptotic factors and oxidative stress enzymes. Mol. Cell. Biochem. 2014, 390, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Foti, P.; Erba, D.; Riso, P.; Spadafranca, A.; Criscuoli, F.; Testolin, G. Comparison between daidzein and genistein antioxidant activity in primary and cancer lymphocytes. Arch. Biochem. Biophys. 2005, 433, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Mirahmadi, S.-M.-S.; Shahmohammadi, A.; Rousta, A.-M.; Azadi, M.-R.; Fahanik-Babaei, J.; Baluchnejadmojarad, T.; Roghani, M. Soy isoflavone genistein attenuates lipopolysaccharide-induced cognitive impairments in the rat via exerting anti-oxidative and anti-inflammatory effects. Cytokine 2018, 104, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Blay, M.; Espinel, A.E.; Delgado, M.A.; Baiges, I.; Bladé, C.; Arola, L.; Salvadó, J. Isoflavone effect on gene expression profile and biomarkers of inflammation. J. Pharm. Biomed. Anal. 2010, 51, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Lau, T.Y.; Leung, L.K. Soya isoflavones suppress phorbol 12-myristate 13-acetate-induced COX-2 expression in MCF-7 cells. Br. J. Nutr. 2006, 96, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahesha, H.G.; Singh, S.A.; Rao, A.G.A. Inhibition of lipoxygenase by soy isoflavones: Evidence of isoflavones as redox inhibitors. Arch. Biochem. Biophys. 2007, 461, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Bonet-Costa, V.; Herranz-Pérez, V.; Blanco-Gandía, M.; Mas-Bargues, C.; Inglés, M.; Garcia-Tarraga, P.; Rodriguez-Arias, M.; Miñarro, J.; Borras, C.; Garcia-Verdugo, J.M.; et al. Clearing amyloid-β through PPARγ/ApoE activation by genistein is a treatment of experimental Alzheimer’s disease. J. Alzheimers Dis. 2016, 51, 701–711. [Google Scholar] [CrossRef]
- Pierzynowska, K.; Podlacha, M.; Gaffke, L.; Majkutewicz, I.; Mantej, J.; Węgrzyn, A.; Osiadły, M.; Myślińska, D.; Węgrzyn, G. Autophagy-dependent mechanism of genistein-mediated elimination of behavioral and biochemical defects in the rat model of sporadic Alzheimer’s disease. Neuropharmacology 2019, 148, 332–346. [Google Scholar] [CrossRef]
- Liao, W.; Jin, G.; Zhao, M.; Yang, H. The effect of genistein on the content and activity of α- and β-secretase and protein kinase C in Aβ-injured hippocampal neurons. Basic Clin. Pharmacol. Toxicol. 2013, 112, 182–185. [Google Scholar] [CrossRef]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.-P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef]
- Viernes, D.R.; Choi, L.B.; Kerr, W.G.; Chisholm, J.D. Discovery and development of small molecule SHIP phosphatase modulators. Med. Res. Rev. 2014, 34, 795–824. [Google Scholar] [CrossRef] [Green Version]
- Mostafavi, S.; Gaiteri, C.; Sullivan, S.E.; White, C.C.; Tasaki, S.; Xu, J.; Taga, M.; Klein, H.-U.; Patrick, E.; Komashko, V.; et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci. 2018, 21, 811–819. [Google Scholar] [CrossRef]
- Lim, J.W.; Kim, S.K.; Choi, S.Y.; Kim, D.H.; Gadhe, C.G.; Lee, H.N.; Kim, H.-J.; Kim, J.; Cho, S.J.; Hwang, H.; et al. Identification of crizotinib derivatives as potent SHIP2 inhibitors for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 157, 405–422. [Google Scholar] [CrossRef]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef]
- Brown, D.G.; Wobst, H.J. Opportunities and challenges in phenotypic screening for neurodegenerative disease research. J. Med. Chem. 2020, 63, 1823–1840. [Google Scholar] [CrossRef]
- Daugherty, D.; Goldberg, J.; Fischer, W.; Dargusch, R.; Maher, P.; Schubert, D. A novel Alzheimer’s disease drug candidate targeting inflammation and fatty acid metabolism. Alzheimers. Res. Ther. 2017, 9, 50. [Google Scholar] [CrossRef] [Green Version]
- Bolós, M.; Perea, J.R.; Avila, J. Alzheimer’s disease as an inflammatory disease. Biomol. Concepts 2017, 8, 37–43. [Google Scholar] [CrossRef]
- Huang, W.; Tang, L.; Shi, Y.; Huang, S.; Xu, L.; Sheng, R.; Wu, P.; Li, J.; Zhou, N.; Hu, Y. Searching for the Multi-Target-Directed Ligands against Alzheimer’s disease: Discovery of quinoxaline-based hybrid compounds with AChE, H3R and BACE 1 inhibitory activities. Bioorg. Med. Chem. 2011, 19, 7158–7167. [Google Scholar] [CrossRef]
- Atwood, B.K.; MacKie, K. CB2: A cannabinoid receptor with an identity crisis. Br. J. Pharmacol. 2010, 160, 467–479. [Google Scholar] [CrossRef] [Green Version]
- González-Naranjo, P.; Pérez-Macias, N.; Campillo, N.E.; Pérez, C.; Arán, V.J.; Girón, R.; Sánchez-Robles, E.; Martín, M.I.; Gómez-Cañas, M.; García-Arencibia, M.; et al. Cannabinoid agonists showing BuChE inhibition as potential therapeutic agents for Alzheimer’s disease. Eur. J. Med. Chem. 2014, 73, 56–72. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Tripathi, A.; Tripathi, P.N.; Prajapati, S.K.; Seth, A.; Tripathi, M.K.; Srivastava, P.; Tiwari, V.; Krishnamurthy, S.; Shrivastava, S.K. Design and development of multitarget-directed N-Benzylpiperidine analogs as potential candidates for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 167, 510–524. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, L.; Karelson, M.; Dobchev, D.A. Multitarget approach to drug candidates against Alzheimer’s disease related to AChE, SERT, BACE1 and GSK3β protein targets. Molecules 2020, 25, 1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, S.K.; Nivrutti, A.A.; Bhardwaj, B.; Waiker, D.K.; Verma, A.; Tripathi, P.N.; Tripathi, M.; Saraf, P. Drug reposition-based design, synthesis, and biological evaluation of dual inhibitors of acetylcholinesterase and β-Secretase for treatment of Alzheimer’s disease. J. Mol. Struct. 2022, 1262, 132979. [Google Scholar] [CrossRef]
- Hemonnot, A.-L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer disease: Well-known targets and new opportunities. Front. Aging Neurosci. 2019, 11, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, Z.; Taylor, J.M.; Crack, P.J. The involvement of microglia in Alzheimer’s disease: A new dog in the fight. Br. J. Pharmacol. 2019, 176, 3533–3543. [Google Scholar] [CrossRef]
- Jin, J.-L.; Fang, M.; Zhao, Y.-X.; Liu, X.-Y. Roles of sigma-1 receptors in Alzheimer’s disease. Int. J. Clin. Exp. Med. 2015, 8, 4808–4820. [Google Scholar]
- Brimson, J.M.; Brimson, S.; Chomchoei, C.; Tencomnao, T. Using sigma-ligands as part of a multi-receptor approach to target diseases of the brain. Expert Opin. Ther. Targets 2020, 24, 1009–1028. [Google Scholar] [CrossRef]
- Sampietro, A.; Pérez-Areales, F.J.; Martínez, P.; Arce, E.M.; Galdeano, C.; Muñoz-Torrero, D. Unveiling the multitarget anti-Alzheimer drug discovery landscape: A bibliometric analysis. Pharmaceuticals 2022, 15, 545. [Google Scholar] [CrossRef]
- Shukla, S.; Tekwani, B.L. Histone deacetylases inhibitors in neurodegenerative diseases, neuroprotection and neuronal differentiation. Front. Pharmacol. 2020, 11, 537. [Google Scholar] [CrossRef]
- Rodrigues, D.A.; Pinheiro, P.D.S.M.; Sagrillo, F.S.; Bolognesi, M.L.; Fraga, C.A.M. Histone deacetylases as targets for the treatment of neurodegenerative disorders: Challenges and future opportunities. Med. Res. Rev. 2020, 40, 2177–2211. [Google Scholar] [CrossRef]
- Schmidt, J.; Rotter, M.; Weiser, T.; Wittmann, S.; Weizel, L.; Kaiser, A.; Heering, J.; Goebel, T.; Angioni, C.; Wurglics, M.; et al. A dual modulator of farnesoid X receptor and soluble epoxide hydrolase to counter nonalcoholic steatohepatitis. J. Med. Chem. 2017, 60, 7703–7724. [Google Scholar] [CrossRef]
- Griñán-Ferré, C.; Codony, S.; Pujol, E.; Yang, J.; Leiva, R.; Escolano, C.; Puigoriol-Illamola, D.; Companys-Alemany, J.; Corpas, R.; Sanfeliu, C.; et al. Pharmacological inhibition of soluble epoxide hydrolase as a new therapy for Alzheimer’s disease. Neurotherapeutics 2020, 17, 1825–1835. [Google Scholar] [CrossRef]
- Jiang, Q.; Heneka, M.; Landreth, G.E. The role of peroxisome proliferator-activated receptor-gamma (PPARgamma) in Alzheimer’s disease: Therapeutic implications. CNS Drugs 2008, 22, 1–14. [Google Scholar] [CrossRef]
- Villapol, S. Roles of peroxisome proliferator-activated receptor gamma on brain and peripheral inflammation. Cell. Mol. Neurobiol. 2018, 38, 121–132. [Google Scholar] [CrossRef]
- Demuro, S.; Di Martino, R.M.C.; Ortega, J.A.; Cavalli, A. GSK-3β, FYN, and DYRK1A: Master regulators in neurodegenerative pathways. Int. J. Mol. Sci. 2021, 22, 9098. [Google Scholar] [CrossRef]
- Martín-Cámara, O.; Cores, Á.; López-Alvarado, P.; Menéndez, J.C. Emerging targets in drug discovery against neurodegenerative diseases: Control of synapsis disfunction by the RhoA/ROCK Pathway. Eur. J. Med. Chem. 2021, 225, 113742. [Google Scholar] [CrossRef]
- Prins, N.D.; Harrison, J.E.; Chu, H.-M.; Blackburn, K.; Alam, J.J.; Scheltens, P. A phase 2 double-blind placebo-controlled 24-week treatment clinical study of the p38 alpha kinase inhibitor neflamapimod in mild Alzheimer’s disease. Alzheimer’s Res. Ther. 2021, 13, 106. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.-L.; Yardin, C.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef]
- Asih, P.R.; Prikas, E.; Stefanoska, K.; Tan, A.R.P.; Ahel, H.I.; Ittner, A. Functions of p38 MAP kinases in the central nervous system. Front. Mol. Neurosci. 2020, 13, 570586. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Biological Activities | Chemical Moiety |
|---|---|---|
| Flavonoid |
| Polyphenol with chroman-4-one or chromone core system |
| Coumarin |
| 2H-chromen-2-one heterocycle |
| Tacrine |
| 9-amino-1,2,3,4-tetrahydroacridine (THA) |
| Donepezil |
| Indanone and N-benzylpiperidine |
| Clioquinol |
| 5-chloro-7-iodoquinoline-8-ol |
| Rasagiline and Selegiline |
| Propargylamine |
| Serotonin and Dopamine |
| Indolamine and phenethylamine fragments |
| Lipoic acid |
| Alpha-lipoic acid (ALA) |
| Resveratrol |
| Polyphenolic phytoalexin |
| Ferulic acid and caffeic acid |
| 3,4-dihydroxycinnamic acid |
| Therapeutic Target | Role in AD Pathogenesis | References |
|---|---|---|
| AChE |
| [25,28] |
| BuChE |
| [71] |
| BACE-1 |
| [15] |
| GSK-3β |
| [16,19] |
| MAO: MAO-A and MAO-B |
| [72,73] |
| Metal ions |
| |
| NMDA receptor |
| [23,24] |
| 5-HT receptor (serotonergic receptor) |
| [74,75] |
| SERT |
| [76] |
| PDE |
| [77,78] |
| CB2 receptor |
| [79,80,81] |
| H3 receptor |
| [82,83] |
| AGEs |
| [84,85] |
| FAHH |
| [86] |
| Nrf2 |
| [87,88] |
| COX-2 |
| [89] |
| 5-LOX |
| [90] |
| SHIP2 |
| [91,92] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheong, S.L.; Tiew, J.K.; Fong, Y.H.; Leong, H.W.; Chan, Y.M.; Chan, Z.L.; Kong, E.W.J. Current Pharmacotherapy and Multi-Target Approaches for Alzheimer’s Disease. Pharmaceuticals 2022, 15, 1560. https://doi.org/10.3390/ph15121560
Cheong SL, Tiew JK, Fong YH, Leong HW, Chan YM, Chan ZL, Kong EWJ. Current Pharmacotherapy and Multi-Target Approaches for Alzheimer’s Disease. Pharmaceuticals. 2022; 15(12):1560. https://doi.org/10.3390/ph15121560
Chicago/Turabian StyleCheong, Siew Lee, Jian Kai Tiew, Yi Hang Fong, How Wan Leong, Yew Mun Chan, Zhi Ling Chan, and Ethan Wei Jie Kong. 2022. "Current Pharmacotherapy and Multi-Target Approaches for Alzheimer’s Disease" Pharmaceuticals 15, no. 12: 1560. https://doi.org/10.3390/ph15121560
APA StyleCheong, S. L., Tiew, J. K., Fong, Y. H., Leong, H. W., Chan, Y. M., Chan, Z. L., & Kong, E. W. J. (2022). Current Pharmacotherapy and Multi-Target Approaches for Alzheimer’s Disease. Pharmaceuticals, 15(12), 1560. https://doi.org/10.3390/ph15121560