
Predicting Anticancer Drug Resistance Mediated by Mutations

 , and
, and
Abstract
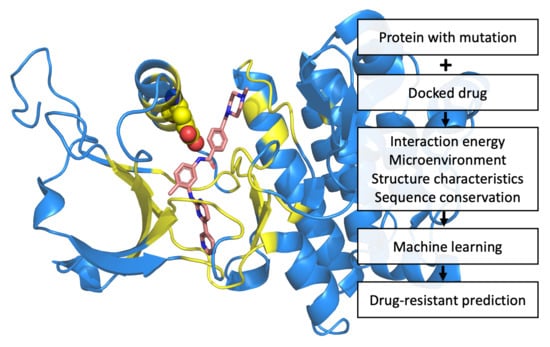
:
1. Introduction
2. Results
2.1. Performance Evaluation of the Training Set
2.2. Performance Evaluation of the Testing Set
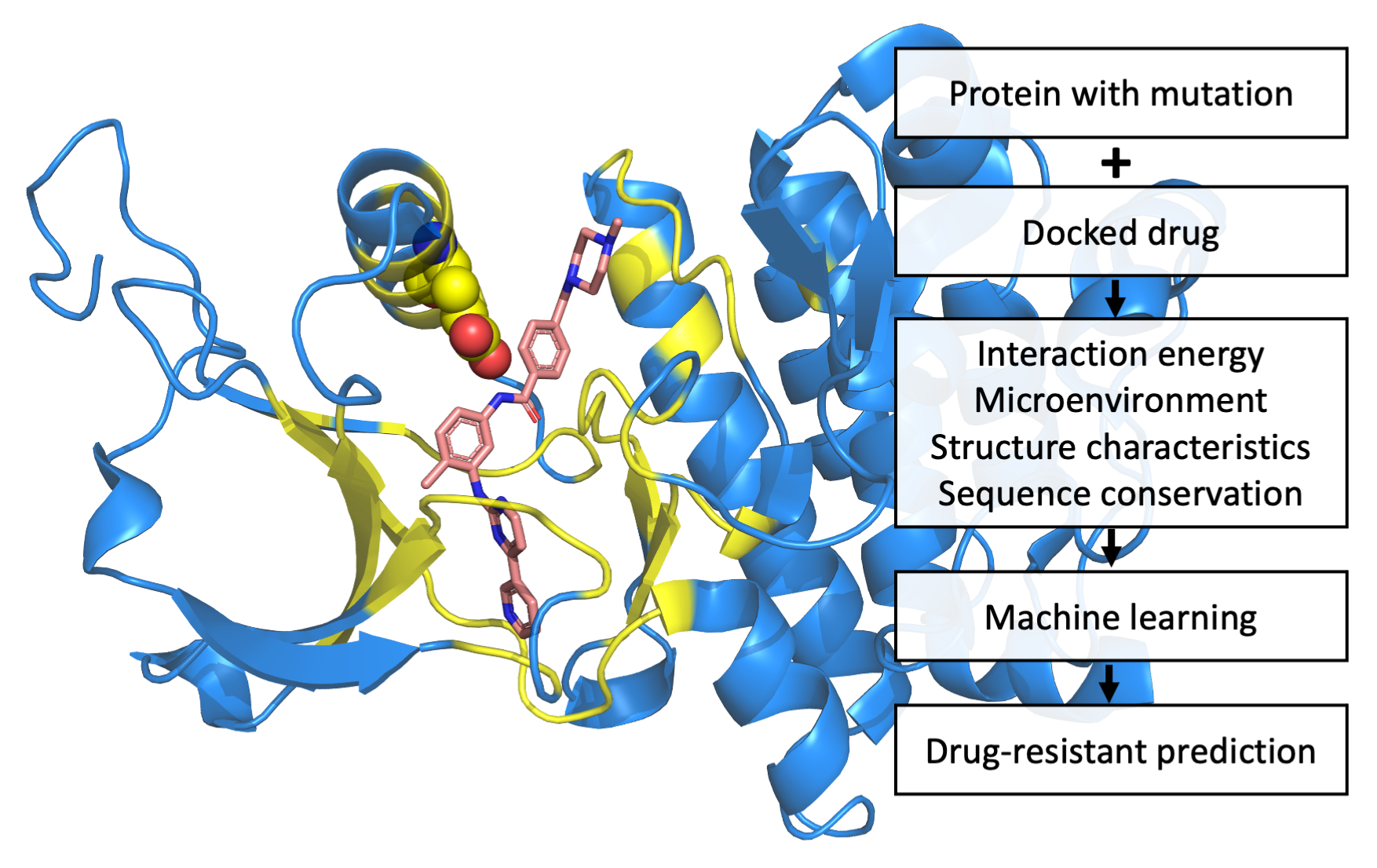
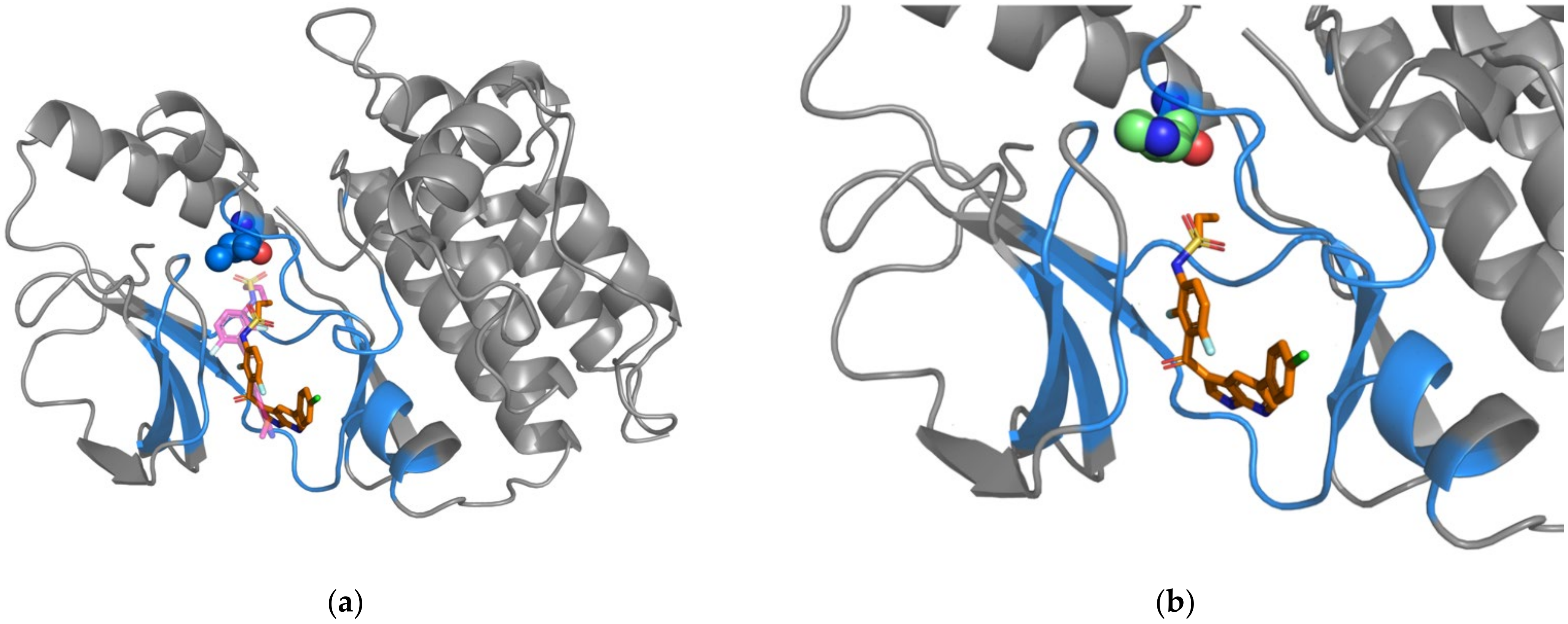
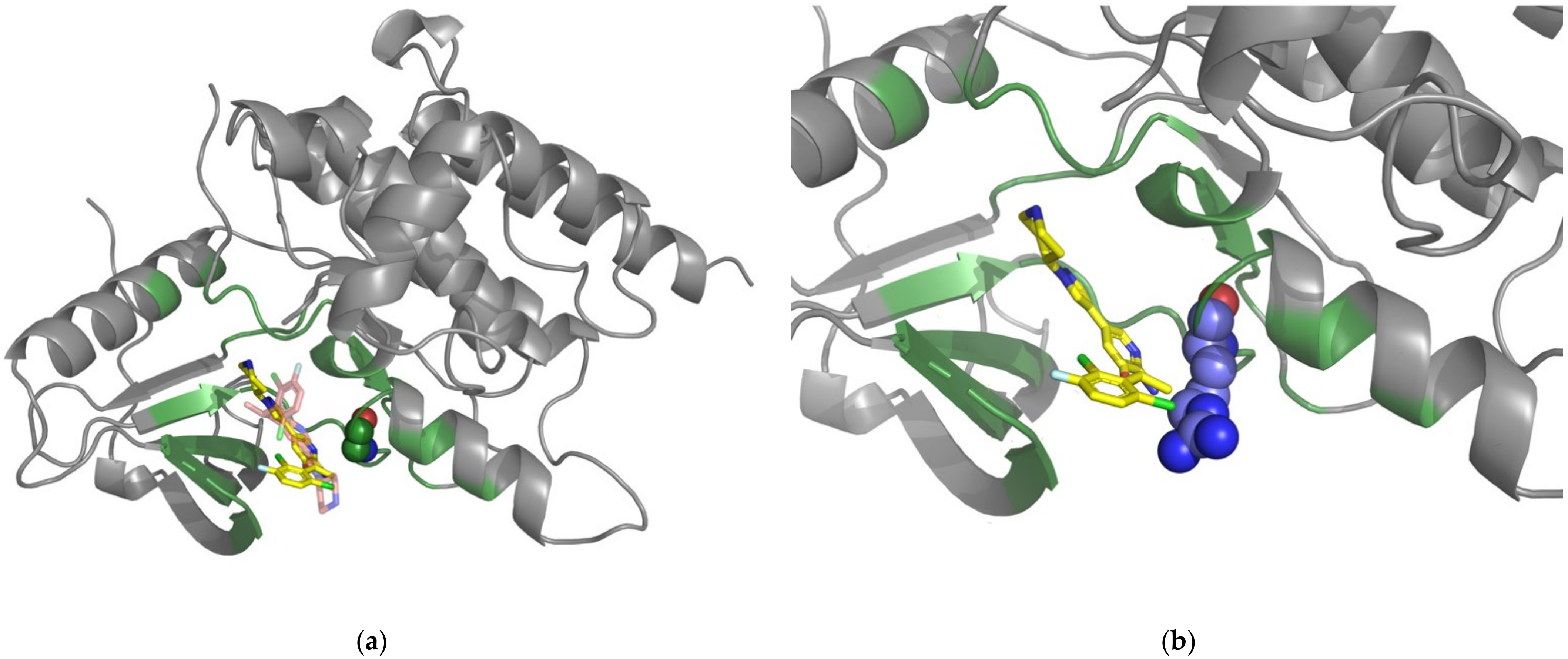
2.3. Case Study: L505 in BRAF and V215E in MAP2K2
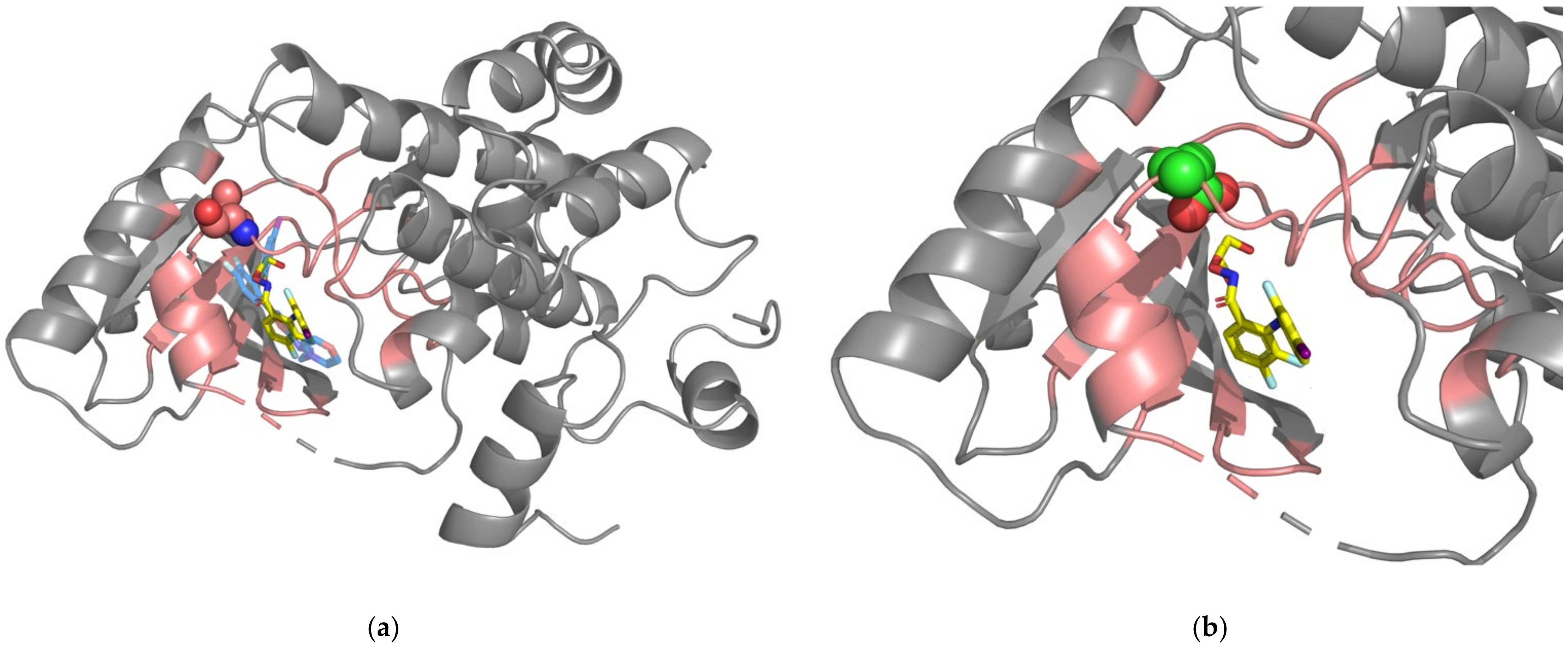
2.4. Case Study: The ROS1-G2032R Mutation
3. Discussion
4. Materials and Methods
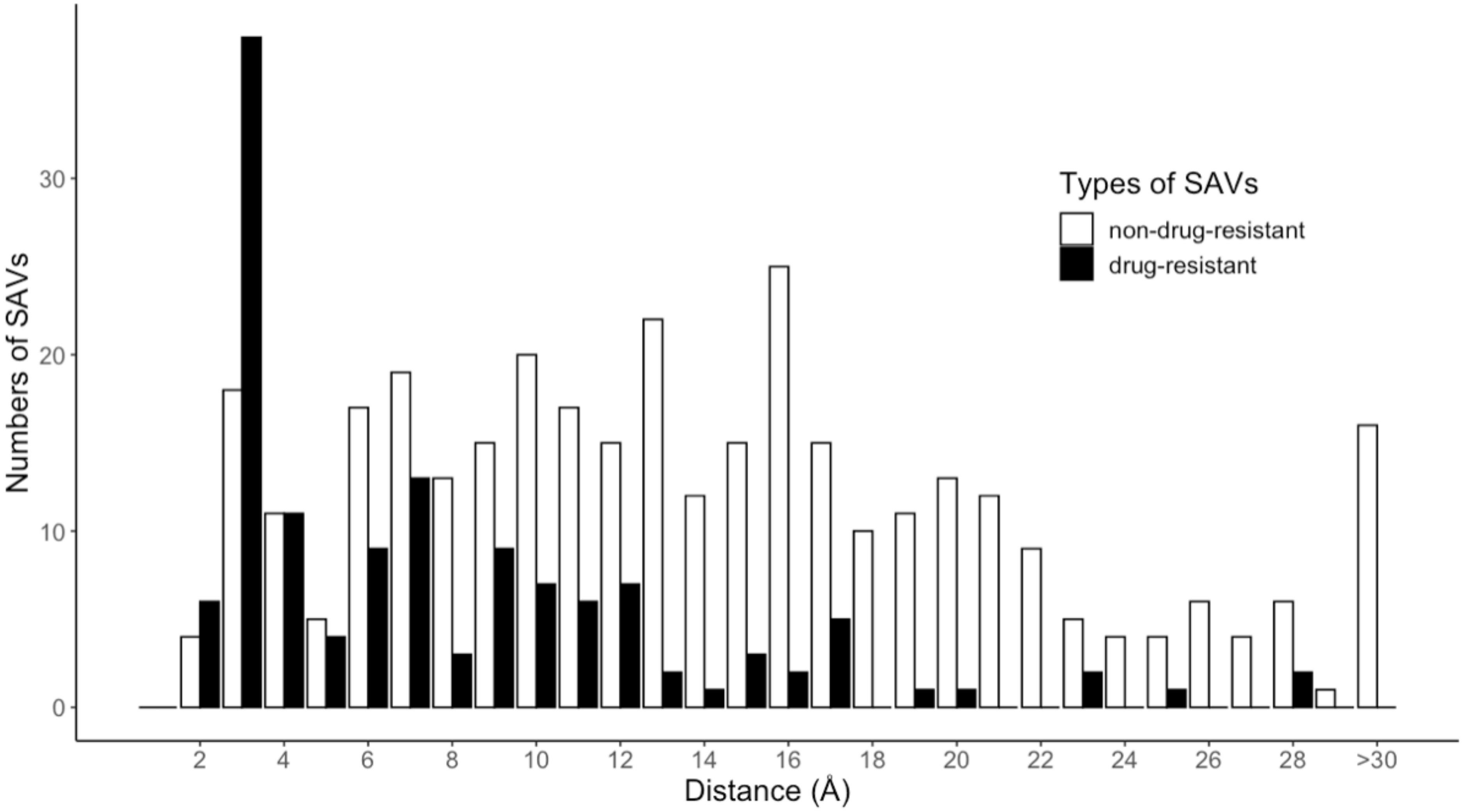
4.1. Dataset Preparation
4.2. Construction of Prediction Systems
4.3. Machine Learning Method
4.4. Feature Selection
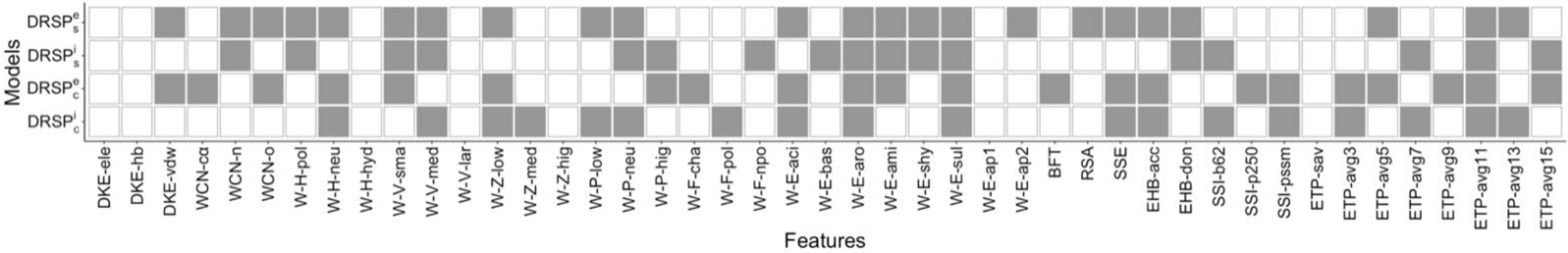
4.5. Generation of Feature Sets
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef]
- Camidge, D.R.; Pao, W.; Sequist, L.V. Acquired resistance to TKIs in solid tumours: Learning from lung cancer. Nat. Rev. Clin. Oncol. 2014, 11, 473–481. [Google Scholar] [CrossRef]
- Tukagoshi, S. Cancer chemotherapy; past, present and future—From the aspect of fundamental studies. Gan Kagaku Ryoho 2003, 30, 1398–1403. [Google Scholar]
- Asano, T. Drug Resistance in Cancer Therapy and the Role of Epigenetics. J. Nippon Med. Sch. 2020, 87, 244–251. [Google Scholar] [CrossRef]
- Hinds, M.; Deisseroth, K.; Mayes, J.; Altschuler, E.; Jansen, R.; Ledley, F.D.; Zwelling, L.A. Identification of a point mutation in the topoisomerase II gene from a human leukemia cell line containing an amsacrine-resistant form of topoisomerase II. Cancer Res. 1991, 51, 4729–4731. [Google Scholar]
- Jhaveri, M.S.; Morrow, C.S. Methylation-mediated regulation of the glutathione S-transferase P1 gene in human breast cancer cells. Gene 1998, 210, 1–7. [Google Scholar] [CrossRef]
- Cabral, F.R.; Brady, R.C.; Schibler, M.J. A mechanism of cellular resistance to drugs that interfere with microtubule assembly. Ann. N. Y. Acad. Sci. 1986, 466, 745–756. [Google Scholar] [CrossRef]
- Moscow, J.A.; Cowan, K.H. Multidrug resistance. J. Natl. Cancer Inst. 1988, 80, 14–20. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, S.H.; Guo, X.L. New insights into Vinca alkaloids resistance mechanism and circumvention in lung cancer. Biomed. Pharmacother. 2017, 96, 659–666. [Google Scholar] [CrossRef]
- Tsuruo, T. Mechanisms of multidrug resistance and implications for therapy. Jpn. J. Cancer Res. 1988, 79, 285–296. [Google Scholar] [CrossRef]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Mirzaei, S.; Gholami, M.H.; Hashemi, F.; Zabolian, A.; Raei, M.; Hushmandi, K.; Zarrabi, A.; Voelcker, N.H.; Aref, A.R.; et al. Hyaluronic acid-based nanoplatforms for Doxorubicin: A review of stimuli-responsive carriers, co-delivery and resistance suppression. Carbohydr. Polym. 2021, 272, 118491. [Google Scholar] [CrossRef]
- Mirzaei, S.; Gholami, M.H.; Hashemi, F.; Zabolian, A.; Farahani, M.V.; Hushmandi, K.; Zarrabi, A.; Goldman, A.; Ashrafizadeh, M.; Orive, G. Advances in understanding the role of P-gp in doxorubicin resistance: Molecular pathways, therapeutic strategies, and prospects. Drug Discov. Today 2021. [Google Scholar] [CrossRef]
- Maleki Dana, P.; Sadoughi, F.; Asemi, Z.; Yousefi, B. The role of polyphenols in overcoming cancer drug resistance: A comprehensive review. Cell Mol. Biol. Lett. 2022, 27, 1–26. [Google Scholar] [CrossRef]
- Villanueva, J.; Vultur, A.; Herlyn, M. Resistance to BRAF inhibitors: Unraveling mechanisms and future treatment options. Cancer Res. 2011, 71, 7137–7140. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Lee, S.M.; Khan, I.; Upadhyay, S.; Lewanski, C.; Falk, S.; Skailes, G.; Marshall, E.; Woll, P.J.; Hatton, M.; Lal, R.; et al. First-line erlotinib in patients with advanced non-small-cell lung cancer unsuitable for chemotherapy (TOPICAL): A double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2012, 13, 1161–1170. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Wang, Y. Role of the EGFR-KDD mutation as a possible mechanism of acquired resistance of non-small cell lung cancer to EGFR tyrosine kinase inhibitors: A case report. Mol. Clin. Oncol. 2022, 16, 30. [Google Scholar] [CrossRef]
- Jackman, D.; Pao, W.; Riely, G.J.; Engelman, J.A.; Kris, M.G.; Janne, P.A.; Lynch, T.; Johnson, B.E.; Miller, V.A. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 357–360. [Google Scholar] [CrossRef]
- West, H.; Oxnard, G.R.; Doebele, R.C. Acquired resistance to targeted therapies in advanced non-small cell lung cancer: New strategies and new agents. Am. Soc. Clin. Oncol. Educ. Book 2013, 33, e272–e278. [Google Scholar] [CrossRef]
- Neel, D.S.; Bivona, T.G. Resistance is futile: Overcoming resistance to targeted therapies in lung adenocarcinoma. NPJ Precis. Oncol. 2017, 1, 1–6. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Lavi, O.; Hall, M.D.; Gillet, J.P. Toward a Better Understanding of the Complexity of Cancer Drug Resistance. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 85–102. [Google Scholar] [CrossRef]
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–372. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Nayar, U.; Cohen, O.; Kapstad, C.; Cuoco, M.S.; Waks, A.G.; Wander, S.A.; Painter, C.; Freeman, S.; Persky, N.S.; Marini, L.; et al. Acquired HER2 mutations in ER(+) metastatic breast cancer confer resistance to estrogen receptor-directed therapies. Nat. Genet. 2019, 51, 207–216. [Google Scholar] [CrossRef]
- Mao, X.G.; Wang, C.; Liu, D.Y.; Zhang, X.; Wang, L.; Yan, M.; Zhang, W.; Zhu, J.; Li, Z.C.; Mi, C.; et al. Hypoxia upregulates HIG2 expression and contributes to bevacizumab resistance in glioblastoma. Oncotarget 2016, 7, 47808–47820. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Nan, F.; Guo, Q.; Guan, D.; Zhou, C. Resistance to bevacizumab in ovarian cancer SKOV3 xenograft due to EphB4 overexpression. J. Cancer Res. Ther. 2019, 15, 1282–1287. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef] [Green Version]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Cokol, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef] [Green Version]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Goncalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Ciriano, I.; van Westen, G.J.; Bouvier, G.; Nilges, M.; Overington, J.P.; Bender, A.; Malliavin, T.E. Improved large-scale prediction of growth inhibition patterns using the NCI60 cancer cell line panel. Bioinformatics 2016, 32, 85–95. [Google Scholar] [CrossRef]
- Naulaerts, S.; Dang, C.C.; Ballester, P.J. Precision and recall oncology: Combining multiple gene mutations for improved identification of drug-sensitive tumours. Oncotarget 2017, 8, 97025–97040. [Google Scholar] [CrossRef] [Green Version]
- Gayvert, K.M.; Aly, O.; Platt, J.; Bosenberg, M.W.; Stern, D.F.; Elemento, O. A Computational Approach for Identifying Synergistic Drug Combinations. PLoS Comput. Biol. 2017, 13, e1005308. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Mezencev, R.; McDonald, J.F.; Vannberg, F. Open source machine-learning algorithms for the prediction of optimal cancer drug therapies. PLoS ONE 2017, 12, e0186906. [Google Scholar] [CrossRef] [Green Version]
- Menden, M.P.; Iorio, F.; Garnett, M.; McDermott, U.; Benes, C.H.; Ballester, P.J.; Saez-Rodriguez, J. Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties. PLoS ONE 2013, 8, e61318. [Google Scholar] [CrossRef] [Green Version]
- Costello, J.C.; Heiser, L.M.; Georgii, E.; Gonen, M.; Menden, M.P.; Wang, N.J.; Bansal, M.; Ammad-ud-din, M.; Hintsanen, P.; Khan, S.A.; et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat. Biotechnol. 2014, 32, 1202–1212. [Google Scholar] [CrossRef]
- Gonen, M.; Margolin, A.A. Drug susceptibility prediction against a panel of drugs using kernelized Bayesian multitask learning. Bioinformatics 2014, 30, i556–i563. [Google Scholar] [CrossRef] [Green Version]
- Komarova, N.L.; Wodarz, D. Drug resistance in cancer: Principles of emergence and prevention. Proc. Natl. Acad. Sci. USA 2005, 102, 9714–9719. [Google Scholar] [CrossRef] [Green Version]
- Faratian, D.; Goltsov, A.; Lebedeva, G.; Sorokin, A.; Moodie, S.; Mullen, P.; Kay, C.; Um, I.H.; Langdon, S.; Goryanin, I.; et al. Systems biology reveals new strategies for personalizing cancer medicine and confirms the role of PTEN in resistance to trastuzumab. Cancer Res. 2009, 69, 6713–6720. [Google Scholar] [CrossRef] [Green Version]
- Tomasetti, C.; Levy, D. An elementary approach to modeling drug resistance in cancer. Math. Biosci. Eng. 2010, 7, 905–918. [Google Scholar] [CrossRef]
- Sun, X.; Bao, J.; Nelson, K.C.; Li, K.C.; Kulik, G.; Zhou, X. Systems modeling of anti-apoptotic pathways in prostate cancer: Psychological stress triggers a synergism pattern switch in drug combination therapy. PLoS Comput. Biol. 2013, 9, e1003358. [Google Scholar] [CrossRef]
- Choi, J.; Park, S.; Ahn, J. RefDNN: A reference drug based neural network for more accurate prediction of anticancer drug resistance. Sci. Rep. 2020, 10, 1861. [Google Scholar] [CrossRef]
- Jimenez, C.R.; Zhang, H.; Kinsinger, C.R.; Nice, E.C. The cancer proteomic landscape and the HUPO Cancer Proteome Project. Clin. Proteom. 2018, 15, 4. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Han, S.Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef] [Green Version]
- Soskine, M.; Tawfik, D.S. Mutational effects and the evolution of new protein functions. Nat. Rev. Genet. 2010, 11, 572–582. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Wagenaar, T.R.; Ma, L.; Roscoe, B.; Park, S.M.; Bolon, D.N.; Green, M.R. Resistance to vemurafenib resulting from a novel mutation in the BRAFV600E kinase domain. Pigment. Cell Melanoma Res. 2014, 27, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Gou, W.; Zhou, X.; Liu, Z.; Wang, L.; Shen, J.; Xu, X.; Li, Z.; Zhai, X.; Zuo, D.; Wu, Y. CD74-ROS1 G2032R mutation transcriptionally up-regulates Twist1 in non-small cell lung cancer cells leading to increased migration, invasion, and resistance to crizotinib. Cancer Lett. 2018, 422, 19–28. [Google Scholar] [CrossRef]
- Pratilas, C.A.; Xing, F.; Solit, D.B. Targeting oncogenic BRAF in human cancer. Curr. Top. Microbiol. Immunol. 2012, 355, 83–98. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [Green Version]
- Barras, D. BRAF Mutation in Colorectal Cancer: An Update. Biomark Cancer 2015, 7 (Suppl. S1), 9–12. [Google Scholar] [CrossRef]
- Brose, M.S.; Volpe, P.; Feldman, M.; Kumar, M.; Rishi, I.; Gerrero, R.; Einhorn, E.; Herlyn, M.; Minna, J.; Nicholson, A.; et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002, 62, 6997–7000. [Google Scholar]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Wagle, N.; Van Allen, E.M.; Treacy, D.J.; Frederick, D.T.; Cooper, Z.A.; Taylor-Weiner, A.; Rosenberg, M.; Goetz, E.M.; Sullivan, R.J.; Farlow, D.N.; et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014, 4, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Luebker, S.A.; Koepsell, S.A. Diverse Mechanisms of BRAF Inhibitor Resistance in Melanoma Identified in Clinical and Preclinical Studies. Front. Oncol. 2019, 9, 268. [Google Scholar] [CrossRef] [Green Version]
- Hoogstraat, M.; Gadellaa-van Hooijdonk, C.G.; Ubink, I.; Besselink, N.J.; Pieterse, M.; Veldhuis, W.; van Stralen, M.; Meijer, E.F.; Willems, S.M.; Hadders, M.A.; et al. Detailed imaging and genetic analysis reveal a secondary BRAF(L505H) resistance mutation and extensive intrapatient heterogeneity in metastatic BRAF mutant melanoma patients treated with vemurafenib. Pigment. Cell Melanoma Res. 2015, 28, 318–323. [Google Scholar] [CrossRef]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [Green Version]
- Hatzivassiliou, G.; Liu, B.; O’Brien, C.; Spoerke, J.M.; Hoeflich, K.P.; Haverty, P.M.; Soriano, R.; Forrest, W.F.; Heldens, S.; Chen, H.; et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol. Cancer Ther. 2012, 11, 1143–1154. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.; Hwang, J.K.; Yang, J.M. (PS)2-v2: Template-based protein structure prediction server. BMC Bioinform. 2009, 10, 366. [Google Scholar] [CrossRef] [Green Version]
- Karoulia, Z.; Wu, Y.; Ahmed, T.A.; Xin, Q.; Bollard, J.; Krepler, C.; Wu, X.; Zhang, C.; Bollag, G.; Herlyn, M.; et al. An Integrated Model of RAF Inhibitor Action Predicts Inhibitor Activity against Oncogenic BRAF Signaling. Cancer Cell 2016, 30, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Wenglowsky, S.; Ren, L.; Ahrendt, K.A.; Laird, E.R.; Aliagas, I.; Alicke, B.; Buckmelter, A.J.; Choo, E.F.; Dinkel, V.; Feng, B.; et al. Pyrazolopyridine Inhibitors of B-Raf(V600E). Part 1: The Development of Selective, Orally Bioavailable, and Efficacious Inhibitors. ACS Med. Chem. Lett. 2011, 2, 342–347. [Google Scholar] [CrossRef] [Green Version]
- Ohren, J.F.; Chen, H.; Pavlovsky, A.; Whitehead, C.; Zhang, E.; Kuffa, P.; Yan, C.; McConnell, P.; Spessard, C.; Banotai, C.; et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat. Struct. Mol. Biol. 2004, 11, 1192–1197. [Google Scholar] [CrossRef]
- Awad, M.M.; Katayama, R.; McTigue, M.; Liu, W.; Deng, Y.L.; Brooun, A.; Friboulet, L.; Huang, D.; Falk, M.D.; Timofeevski, S.; et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N. Engl. J. Med. 2013, 368, 2395–2401. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.D.; Doebele, R.C. Molecular pathways: ROS1 fusion proteins in cancer. Clin. Cancer Res. 2013, 19, 4040–4045. [Google Scholar] [CrossRef] [Green Version]
- Gainor, J.F.; Tseng, D.; Yoda, S.; Dagogo-Jack, I.; Friboulet, L.; Lin, J.J.; Hubbeling, H.G.; Dardaei, L.; Farago, A.F.; Schultz, K.R.; et al. Patterns of Metastatic Spread and Mechanisms of Resistance to Crizotinib in ROS1-Positive Non-Small-Cell Lung Cancer. JCO Precis. Oncol. 2017, 1, 1–13. [Google Scholar] [CrossRef]
- Song, A.; Kim, T.M.; Kim, D.W.; Kim, S.; Keam, B.; Lee, S.H.; Heo, D.S. Molecular Changes Associated with Acquired Resistance to Crizotinib in ROS1-Rearranged Non-Small Cell Lung Cancer. Clin. Cancer Res. 2015, 21, 2379–2387. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.J.; Shaw, A.T. Recent Advances in Targeting ROS1 in Lung Cancer. J. Thorac. Oncol. 2017, 12, 1611–1625. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Jenkins, C.; Iyer, S.; Schoenfeld, A.; Keddy, C.; Davare, M.A. ROS1-dependent cancers—Biology, diagnostics and therapeutics. Nat. Rev. Clin. Oncol. 2021, 18, 35–55. [Google Scholar] [CrossRef]
- Lee, J.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Park, K.; Choi, Y.; Ahn, M.J. Efficacy and Safety of Lorlatinib in Korean Non-Small-Cell Lung Cancer Patients With ALK or ROS1 Rearrangement Whose Disease Failed to Respond to a Previous Tyrosine Kinase Inhibitor. Clin. Lung Cancer 2019, 20, 215–221. [Google Scholar] [CrossRef] [Green Version]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [Green Version]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Garraway, L.A. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell 2018, 33, 801–815. [Google Scholar] [CrossRef] [Green Version]
- Wilmott, J.S.; Tembe, V.; Howle, J.R.; Sharma, R.; Thompson, J.F.; Rizos, H.; Lo, R.S.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Intratumoral molecular heterogeneity in a BRAF-mutant, BRAF inhibitor-resistant melanoma: A case illustrating the challenges for personalized medicine. Mol. Cancer Ther. 2012, 11, 2704–2708. [Google Scholar] [CrossRef] [Green Version]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Stank, A.; Kokh, D.B.; Fuller, J.C.; Wade, R.C. Protein Binding Pocket Dynamics. Acc. Chem. Res. 2016, 49, 809–815. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, V.; Gherardini, P.F.; Helmer-Citterich, M.; Ausiello, G. Identification of binding pockets in protein structures using a knowledge-based potential derived from local structural similarities. BMC Bioinform. 2012, 13 (Suppl. S4), S17. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.K.; Park, J.I. MEK1/2 Inhibitors: Molecular Activity and Resistance Mechanisms. Semin. Oncol. 2015, 42, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Hua, S.; Sun, Z. A novel method of protein secondary structure prediction with high segment overlap measure: Support vector machine approach. J. Mol. Biol. 2001, 308, 397–407. [Google Scholar] [CrossRef]
- Yu, C.S.; Wang, J.Y.; Yang, J.M.; Lyu, P.C.; Lin, C.J.; Hwang, J.K. Fine-grained protein fold assignment by support vector machines using generalized npeptide coding schemes and jury voting from multiple-parameter sets. Proteins 2003, 50, 531–536. [Google Scholar] [CrossRef]
- Yu, C.S.; Lin, C.J.; Hwang, J.K. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 2004, 13, 1402–1406. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Lin, Y.S.; Lin, C.J.; Hwang, J.K. Prediction of the bonding states of cysteines using the support vector machines based on multiple feature vectors and cysteine state sequences. Proteins 2004, 55, 1036–1042. [Google Scholar] [CrossRef]
- Lei, Z.; Dai, Y. An SVM-based system for predicting protein subnuclear localizations. BMC Bioinform. 2005, 6, 291. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.J.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Secondary structure prediction with support vector machines. Bioinformatics 2003, 19, 1650–1655. [Google Scholar]
- Chang, C.C.; Lin, C.J. LIBSVM: A Library for Support Vector Machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 1–27. [Google Scholar] [CrossRef]
- Lin, C.-J. Formulations of support vector machines: A note from an optimization point of view. Neural Comput. 2001, 13, 307–317. [Google Scholar]
- Lu, C.H.; Chen, Y.C.; Yu, C.S.; Hwang, J.K. Predicting disulfide connectivity patterns. Proteins 2007, 67, 262–270. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.S.; Lu, C.H. Identification of antifreeze proteins and their functional residues by support vector machine and genetic algorithms based on n-peptide compositions. PLoS ONE 2011, 6, e20445. [Google Scholar] [CrossRef]
- Liu, J.J.; Yu, C.S.; Wu, H.W.; Chang, Y.J.; Lin, C.P.; Lu, C.H. The structure-based cancer-related single amino acid variation prediction. Sci. Rep. 2021, 11, 13599. [Google Scholar] [CrossRef]
- Yang, J.-M.; Chen, C.-C. GEMDOCK: A generic evolutionary method for molecular docking. Proteins Struct. Funct. Bioinform. 2004, 55, 288–304. [Google Scholar] [CrossRef]
- Lin, C.P.; Huang, S.W.; Lai, Y.L.; Yen, S.C.; Shih, C.H.; Lu, C.H.; Huang, C.C.; Hwang, J.K. Deriving protein dynamical properties from weighted protein contact number. Proteins 2008, 72, 929–935. [Google Scholar] [CrossRef]
- Shih, C.H.; Chang, C.M.; Lin, Y.S.; Lo, W.C.; Hwang, J.K. Evolutionary information hidden in a single protein structure. Proteins 2012, 80, 1647–1657. [Google Scholar] [CrossRef]
- Yu, C.S.; Chen, Y.C.; Lu, C.H.; Hwang, J.K. Prediction of protein subcellular localization. Proteins 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Accuracy | Sensitivity | Specificity | MCC | Precision | F1 Score |
|---|---|---|---|---|---|---|
| 0.8377 | 0.5338 | 0.9224 | 0.4936 | 0.6574 | 0.5892 | |
| 0.8508 | 0.5188 | 0.9434 | 0.5241 | 0.7188 | 0.6026 | |
| 0.8886 | 0.5385 | 0.9378 | 0.4803 | 0.5490 | 0.5437 | |
| 0.7819 | 0.7284 | 0.8224 | 0.5536 | 0.7564 | 0.7421 | |
| 0.8886 | 0.5577 | 0.9351 | 0.4888 | 0.5472 | 0.5524 | |
| 0.7660 | 0.6914 | 0.8224 | 0.5196 | 0.7467 | 0.7179 | |
| 0.8557 | 0.6541 | 0.9119 | 0.5724 | 0.6744 | 0.6641 | |
| 0.8508 | 0.6391 | 0.9099 | 0.5567 | 0.6641 | 0.6513 |
| Protein | Drug-Resistant SAV | Distance 1 | Model | Predicted Result |
|---|---|---|---|---|
| BRAF | L505H | 5.41 | TP | |
| FN | ||||
| MAP2K2 | V215E | 4.27 | TP | |
| FN | ||||
| ROS1 | G2032R | 3.30 | TP | |
| TP |
| Protein | Drug | PDB ID | Drug-Resistant 1 | Non-Drug-Resistant 2 |
|---|---|---|---|---|
| ABL1 | Imatinib | 1OPJ | 31 | 36 |
| ALK | Alectinib | 3AOX | 24 | 50 |
| BTK | Ibrutinib | 5P9I | 4 | 36 |
| EGFR | Osimertinib | 4ZAU | 15 | 54 |
| ESR1 | Raloxifene | 1ERR | 6 | 23 |
| FLT3 | Quizartinib | 4RT7 | 5 | 48 |
| KIT | Imatinib | 1T46 | 21 | 51 |
| MAP2K1 | PD0325901 | 3VVH | 2 | 31 |
| PDGFRA | Sunitinib | 6JOK | 1 | 65 |
| SMO | Vismodegib | 5L7I | 17 | 42 |
| MET | Crizotinib | 2WGJ | 7 | 41 |
| TOTAL | 133 | 477 |
| Protein | Drug | PDB ID | Drug-Resistant 1 | Non-Drug-Resistant 2 |
|---|---|---|---|---|
| BRAF | Vemurafenib | 3TV6 | 1 | 48 |
| MAP2K2 | PD0325901 | 1S9I | 1 | 24 |
| ROS1 | Crizotinib | 3ZBF | 1 | 40 |
| TOTAL | 3 | 112 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.-F.; Liu, J.-J.; Chang, Y.-J.; Yu, C.-S.; Yi, W.; Lane, H.-Y.; Lu, C.-H. Predicting Anticancer Drug Resistance Mediated by Mutations. Pharmaceuticals 2022, 15, 136. https://doi.org/10.3390/ph15020136
Lin Y-F, Liu J-J, Chang Y-J, Yu C-S, Yi W, Lane H-Y, Lu C-H. Predicting Anticancer Drug Resistance Mediated by Mutations. Pharmaceuticals. 2022; 15(2):136. https://doi.org/10.3390/ph15020136
Chicago/Turabian StyleLin, Yu-Feng, Jia-Jun Liu, Yu-Jen Chang, Chin-Sheng Yu, Wei Yi, Hsien-Yuan Lane, and Chih-Hao Lu. 2022. "Predicting Anticancer Drug Resistance Mediated by Mutations" Pharmaceuticals 15, no. 2: 136. https://doi.org/10.3390/ph15020136
APA StyleLin, Y. -F., Liu, J. -J., Chang, Y. -J., Yu, C. -S., Yi, W., Lane, H. -Y., & Lu, C. -H. (2022). Predicting Anticancer Drug Resistance Mediated by Mutations. Pharmaceuticals, 15(2), 136. https://doi.org/10.3390/ph15020136