
Impact of Linker Modification and PEGylation of Vancomycin Conjugates on Structure-Activity Relationships and Pharmacokinetics

 ,
, 
Abstract
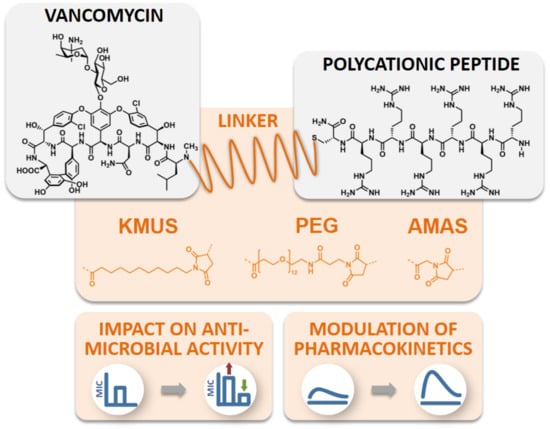
:
1. Introduction
2. Results
2.1. Conjugate Synthesis and Verification of the Site of Conjugation
2.2. Antimicrobial Activity Testing of Polycationic Peptide-Vancomycin Derivatives
2.3. Hemolysis Studies
2.4. Molecular Imaging and Biodistribution Studies
3. Discussion
4. Materials and Methods
4.1. General
4.2. Experimental Section
4.2.1. Chromatographic Analytics
4.2.2. Peptide Synthesis
4.2.3. Synthesis of the Conjugates
4.2.4. Structural Analysis by Deglycosylation
4.2.5. NMR
4.2.6. Antimicrobial Testing: Microdilution
4.2.7. Hemoglobin Release Assay
4.2.8. Biodistribution and Pharmacokinetic Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; HM Government and Welcome Trust: London, UK, 2016. [Google Scholar]
- Livermore, D. The need for new antibiotics. Clin. Microbio. Infect. 2004, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, C. Antibiotics at the crossroads. Nature 2004, 431, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Miethke, M.; Pieroni, M.; Weber, T.; Brönstrup, M.; Hammann, P.; Halby, L.; Arimondo, P.B.; Glaser, P.; Aigle, B.; Bode, H.B.; et al. Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem. 2021, 5, 726–749. [Google Scholar] [CrossRef] [PubMed]
- Antonoplis, A.; Zang, X.; Huttner, M.A.; Chong, K.K.; Lee, Y.B.; Co, J.Y.; Amieva, M.R.; Kline, K.A.; Wender, P.A.; Cegelski, L. A dual-function antibiotic-transporter conjugate exhibits superior activity in sterilizing MRSA biofilms and killing persister cells. J. Am. Chem. Soc. 2018, 140, 16140–16151. [Google Scholar] [CrossRef] [PubMed]
- Antonoplis, A.; Zang, X.; Wegner, T.; Wender, P.A.; Cegelski, L. Vancomycin–arginine conjugate inhibits growth of carbapenem-resistant E. coli and targets cell-wall synthesis. ACS Chem. Biol. 2019, 14, 2065–2070. [Google Scholar] [CrossRef] [PubMed]
- Blaskovich, M.A.; Hansford, K.A.; Gong, Y.; Butler, M.S.; Muldoon, C.; Huang, J.X.; Ramu, S.; Silva, A.B.; Cheng, M.; Kavanagh, A.M. Protein-inspired antibiotics active against vancomycin-and daptomycin-resistant bacteria. Nat. Commun. 2018, 9, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarlagadda, V.; Sarkar, P.; Samaddar, S.; Haldar, J. A Vancomycin Derivative with a Pyrophosphate-Binding Group: A Strategy to Combat Vancomycin-Resistant Bacteria. Angew. Chem. Int. Ed. 2016, 55, 7836–7840. [Google Scholar] [CrossRef]
- Okano, A.; Isley, N.A.; Boger, D.L. Peripheral modifications of [Ψ[CH2NH]Tpg4] vancomycin with added synergistic mechanisms of action provide durable and potent antibiotics. Proc. Natl. Acad. Sci. USA 2017, 114, E5052–E5061. [Google Scholar] [CrossRef] [Green Version]
- Okano, A.; Isley, N.A.; Boger, D.L. Total syntheses of vancomycin-related glycopeptide antibiotics and key analogues. Chem. Rev. 2017, 117, 11952–11993. [Google Scholar] [CrossRef]
- Umstätter, F.; Domhan, C.; Hertlein, T.; Ohlsen, K.; Mühlberg, E.; Kleist, C.; Zimmermann, S.; Beijer, B.; Klika, K.D.; Haberkorn, U.; et al. Vancomycin resistance is overcome by conjugation of polycationic peptides. Angew. Chem. Int. Ed. 2020, 59, 8823–8827. [Google Scholar] [CrossRef] [Green Version]
- Bailon, P.; Palleroni, A.; Schaffer, C.A.; Spence, C.L.; Fung, W.-J.; Porter, J.E.; Ehrlich, G.K.; Pan, W.; Xu, Z.-X.; Modi, M.W. Rational design of a potent, long-lasting form of interferon: A 40 kDa branched polyethylene glycol-conjugated interferon α-2a for the treatment of hepatitis C. Bioconjug. Chem. 2001, 12, 195–202. [Google Scholar] [CrossRef]
- Veronese, F.M.; Mero, A. The impact of PEGylation on biological therapies. BioDrugs 2008, 22, 315–329. [Google Scholar] [CrossRef]
- Basu, A.; Yang, K.; Wang, M.; Liu, S.; Chintala, R.; Palm, T.; Zhao, H.; Peng, P.; Wu, D.; Zhang, Z. Structure− function engineering of interferon-β-1b for improving stability, solubility, potency, immunogenicity, and pharmacokinetic properties by site-selective mono-PEGylation. Bioconjug. Chem. 2006, 17, 618–630. [Google Scholar] [CrossRef]
- Wang, M.; Basu, A.; Palm, T.; Hua, J.; Youngster, S.; Hwang, L.; Liu, H.-C.; Li, X.; Peng, P.; Zhang, Y. Engineering an arginine catabolizing bioconjugate: Biochemical and pharmacological characterization of PEGylated derivatives of arginine deiminase from Mycoplasma arthritidis. Bioconjug. Chem. 2006, 17, 1447–1459. [Google Scholar] [CrossRef]
- Greenwald, R.B.; Yang, K.; Zhao, H.; Conover, C.D.; Lee, S.; Filpula, D. Controlled release of proteins from their poly (ethylene glycol) conjugates: Drug delivery systems employing 1, 6-elimination. Bioconjug. Chem. 2003, 14, 395–403. [Google Scholar] [CrossRef]
- CLSI M100-S24, Performance Standards for Antimicrobial Susceptibility Testing: Twenty-Fourth Informational Supplement; Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2014.
- CLSI Document M07-A9, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Ninth Edition; Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2012.
- European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID). Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by broth dilution. Clin. Microbiol. Infect. 2003, 9, ix–xv. [Google Scholar] [CrossRef] [Green Version]
- Vicente, M.; Hodgson, J.; Massidda, O.; Tonjum, T.; Henriques-Normark, B.; Ron, E.Z. The fallacies of hope: Will we discover new antibiotics to combat pathogenic bacteria in time? FEMS Microbiol. Rev. 2006, 30, 841–852. [Google Scholar] [CrossRef] [Green Version]
- Courvalin, P.; Davies, J. Antimicrobials-Antimicrobials: Time to act! Curr. Opin. Microbiol. 2003, 5, 425–426. [Google Scholar] [CrossRef]
- Hughes, D.; Karlén, A. Discovery and preclinical development of new antibiotics. Upsala J. Med. Sci. 2014, 119, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Filippone, E.J.; Kraft, W.K.; Farber, J.L. The nephrotoxicity of vancomycin. Clin. Pharmacol. Ther. 2017, 102, 459–469. [Google Scholar] [CrossRef]
- Elyasi, S.; Khalili, H.; Dashti-Khavidaki, S.; Mohammadpour, A. Vancomycin-induced nephrotoxicity: Mechanism, incidence, risk factors and special populations. A literature review. Eur. J. Clin. Pharmacol. 2012, 68, 1243–1255. [Google Scholar] [CrossRef]
- Mouton, J.W.; Ambrose, P.G.; Canton, R.; Drusano, G.L.; Harbarth, S.; MacGowan, A.; Theuretzbacher, U.; Turnidge, J. Conserving antibiotics for the future: New ways to use old and new drugs from a pharmacokinetic and pharmacodynamic perspective. Drug Resist. Updates 2011, 14, 107–117. [Google Scholar] [CrossRef]
- Guan, D.; Chen, F.; Qiu, Y.; Jiang, B.; Gong, L.; Lan, L.; Huang, W. Sulfonium, an Underestimated Moiety for Structural Modification, Alters the Antibacterial Profile of Vancomycin Against Multidrug-Resistant Bacteria. Angew. Chem. Int. Ed. 2019, 58, 6678–6682. [Google Scholar] [CrossRef]
- Katip, W.; Oberdorfer, P. A monocentric retrospective study of AUC/MIC ratio of vancomycin associated with clinical outcomes and nephrotoxicity in patients with enterococcal infections. Pharmaceutics 2021, 13, 1378. [Google Scholar] [CrossRef]
- Witzigmann, D.; Uhl, P.; Sieber, S.; Kaufman, C.; Einfalt, T.; Schöneweis, K.; Grossen, P.; Buck, J.; Ni, Y.; Schenk, S.H. Optimization-by-design of hepatotropic lipid nanoparticles targeting the sodium-taurocholate cotransporting polypeptide. eLife 2019, 8, e42276. [Google Scholar] [CrossRef]
- Barenholz, Y.C. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Klika, K.D. The Application of simple and easy to implement decoupling pulse scheme combinations to effect decoupling of large J values with reduced artifacts. Int. J. Spectrosc. 2014, 2014, 289638. [Google Scholar] [CrossRef] [Green Version]
- Virta, P.; Koch, A.; Roslund, M.U.; Mattjus, P.; Kleinpeter, E.; Kronberg, L.; Sjöholm, R.; Klika, K.D. Synthesis, characterisation and theoretical calculations of 2,6-diaminopurine etheno derivatives. Org. Biomol. Chem. 2005, 3, 2924–2929. [Google Scholar] [CrossRef]
- Klika, K.D.; Bernát, J.; Imrich, J.; Chomča, I.; Sillanpää, R.; Pihlaja, K. Unexpected formation of a spiro acridine and fused ring system from the reaction between an N-acridinylmethyl-substituted thiourea and bromoacetonitrile under basic conditions. J. Org. Chem. 2001, 66, 4416–4418. [Google Scholar] [CrossRef]
- Balentová, E.; Imrich, J.; Bernát, J.; Suchá, L.; Vilková, M.; Prónayová, N.; Kristian, P.; Pihlaja, K.; Klika, K.D. Stereochemistry, tautomerism, and reactions of acridinyl thiosemicarbazides in the synthesis of 1,3-thiazolidines. J. Heterocycl. Chem. 2006, 43, 645–656. [Google Scholar] [CrossRef]
- Mäki, J.; Tähtinen, P.; Kronberg, L.; Klika, K.D. Restricted rotation/tautomeric equilibrium and determination of the site and extent of protonation in bi-imidazole nucleosides by multinuclear NMR and GIAO-DFT calculations. J. Phys. Org. Chem. 2005, 18, 240–249. [Google Scholar] [CrossRef]
- Crim, J.W.; Garczynski, S.F.; Brown, M.R. Approaches to radioiodination of insect neuropeptides. Peptides 2002, 23, 2045–2051. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | Spacer [Å] | [g/mol] |
|---|---|---|---|
| FU002 (SMCC linker) |  | 8.3 | 2725.81 |
| VAN1 (KMUS linker) |  | 16.3 | 2769.91 |
| VAN2 (PEG linker) |  | 53.4 | 3257.41 |
| VAN3 (AMAS linker) |  | 4.4 | 2643.69 |
| Compound | PEG Derivative | Average Mn of PEG Unit [g/mol] | Calculated Final Molar Mass [g/mol] |
|---|---|---|---|
| VAN:PEG1 | mPEG-thiol | 800 | 3943.3 |
| VAN:PEG2 | mPEG-thiol | 2000 | 5094.6 |
| VAN:PEG3 | Mercapto-PEG-monomethyl ether | 5000 | 8108.2 |
| Compound | Ratio Liver/Kidney | Ratio Liver/Blood | Ratio Kidney/Blood |
|---|---|---|---|
| Vancomycin | 0.17 | 0.36 | 2.16 |
| VAN1 | 4.47 | 10.13 | 2.22 |
| VAN2 | 0.92 | 9.25 | 10.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umstätter, F.; Werner, J.; Zerlin, L.; Mühlberg, E.; Kleist, C.; Klika, K.D.; Hertlein, T.; Beijer, B.; Domhan, C.; Zimmermann, S.; et al. Impact of Linker Modification and PEGylation of Vancomycin Conjugates on Structure-Activity Relationships and Pharmacokinetics. Pharmaceuticals 2022, 15, 159. https://doi.org/10.3390/ph15020159
Umstätter F, Werner J, Zerlin L, Mühlberg E, Kleist C, Klika KD, Hertlein T, Beijer B, Domhan C, Zimmermann S, et al. Impact of Linker Modification and PEGylation of Vancomycin Conjugates on Structure-Activity Relationships and Pharmacokinetics. Pharmaceuticals. 2022; 15(2):159. https://doi.org/10.3390/ph15020159
Chicago/Turabian StyleUmstätter, Florian, Julia Werner, Leah Zerlin, Eric Mühlberg, Christian Kleist, Karel D. Klika, Tobias Hertlein, Barbro Beijer, Cornelius Domhan, Stefan Zimmermann, and et al. 2022. "Impact of Linker Modification and PEGylation of Vancomycin Conjugates on Structure-Activity Relationships and Pharmacokinetics" Pharmaceuticals 15, no. 2: 159. https://doi.org/10.3390/ph15020159
APA StyleUmstätter, F., Werner, J., Zerlin, L., Mühlberg, E., Kleist, C., Klika, K. D., Hertlein, T., Beijer, B., Domhan, C., Zimmermann, S., Ohlsen, K., Haberkorn, U., Mier, W., & Uhl, P. (2022). Impact of Linker Modification and PEGylation of Vancomycin Conjugates on Structure-Activity Relationships and Pharmacokinetics. Pharmaceuticals, 15(2), 159. https://doi.org/10.3390/ph15020159