
In-Silico Screening of Novel Synthesized Thienopyrimidines Targeting Fms Related Receptor Tyrosine Kinase-3 and Their In-Vitro Biological Evaluation

 , ,
, ,
Abstract
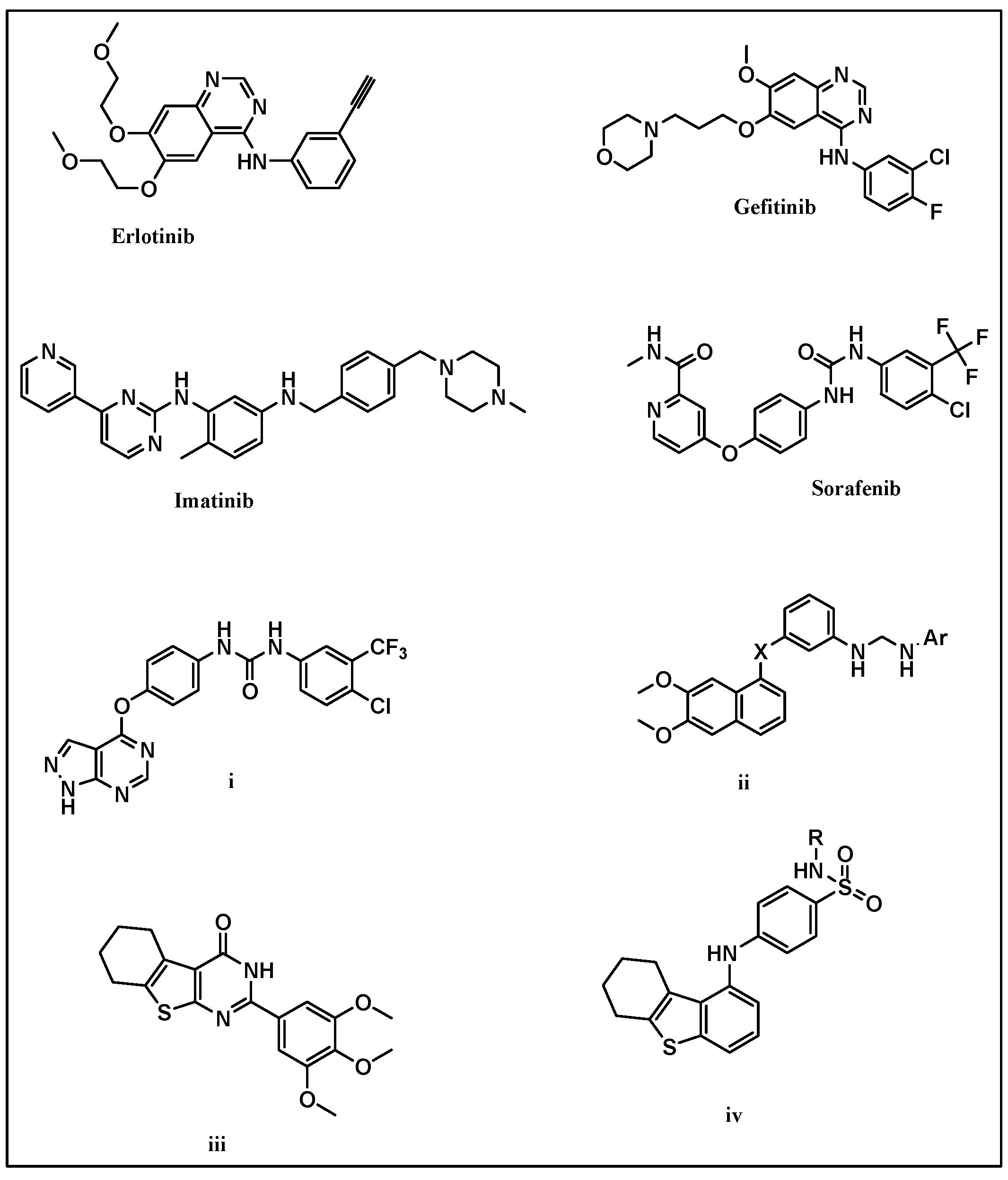
:1. Introduction
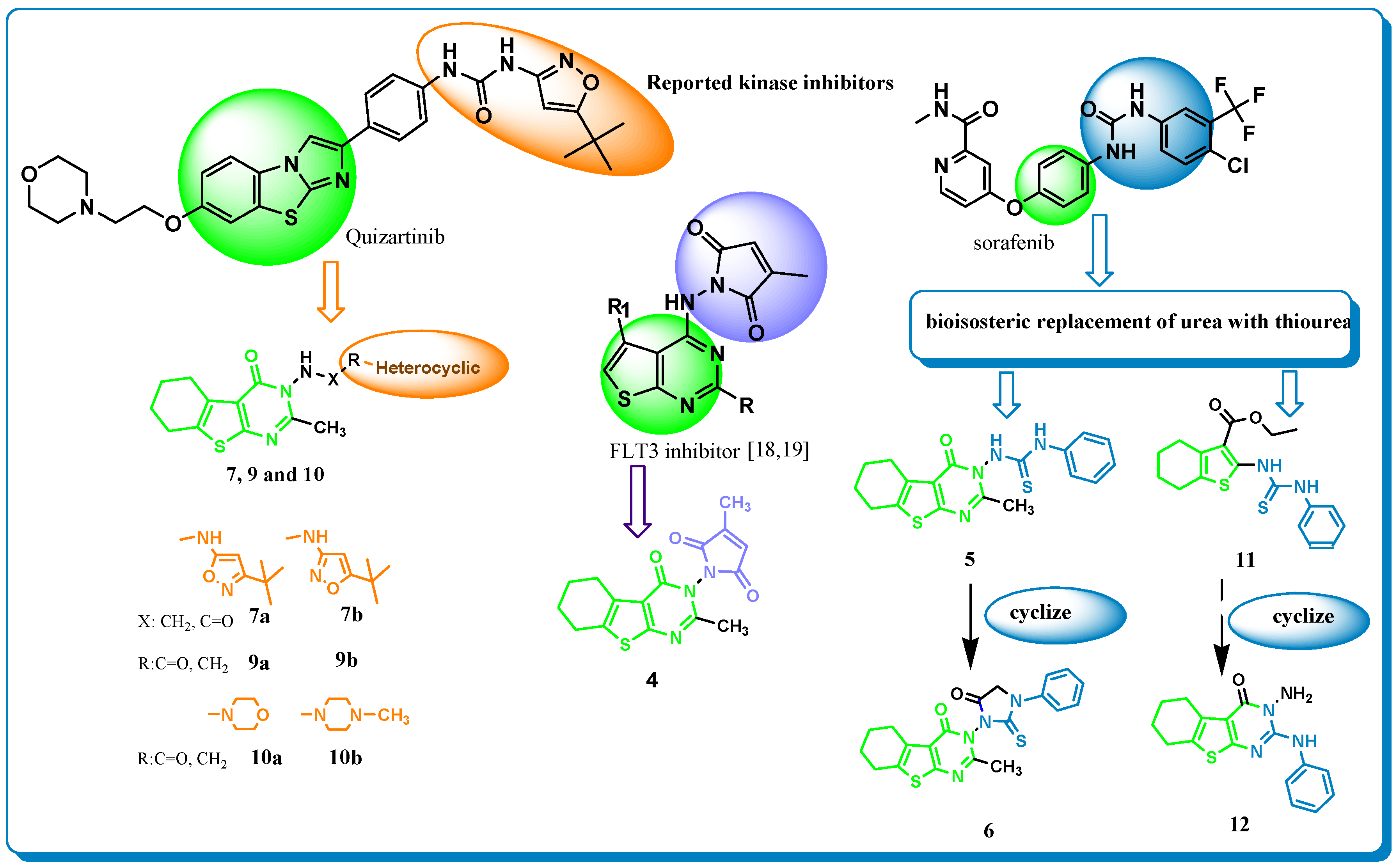
Design Strategy
2. Results and Discussion
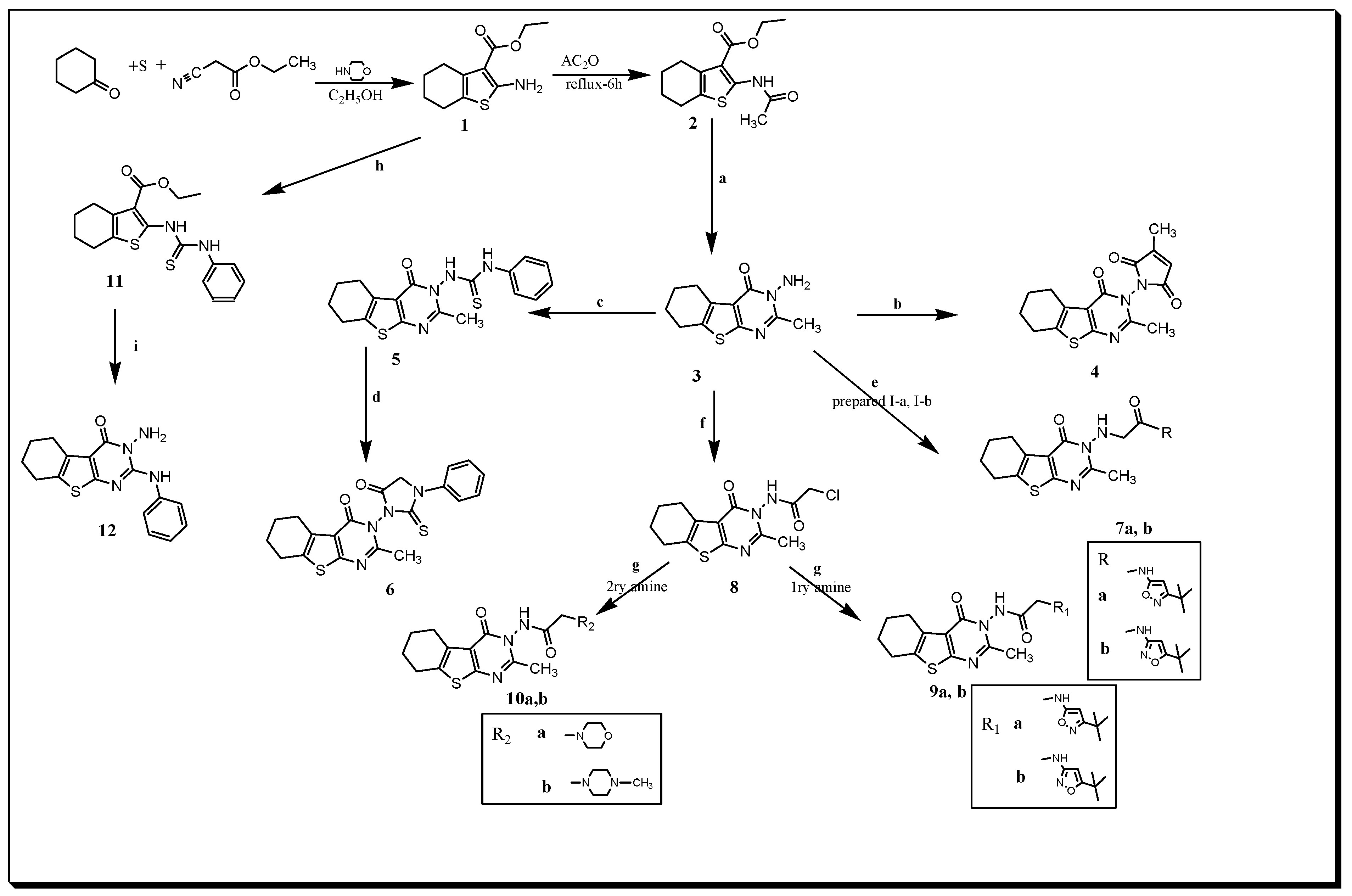
2.1. Chemistry
2.2. Biological Activity
2.2.1. Antiproliferative/Cytotoxicity Assessment
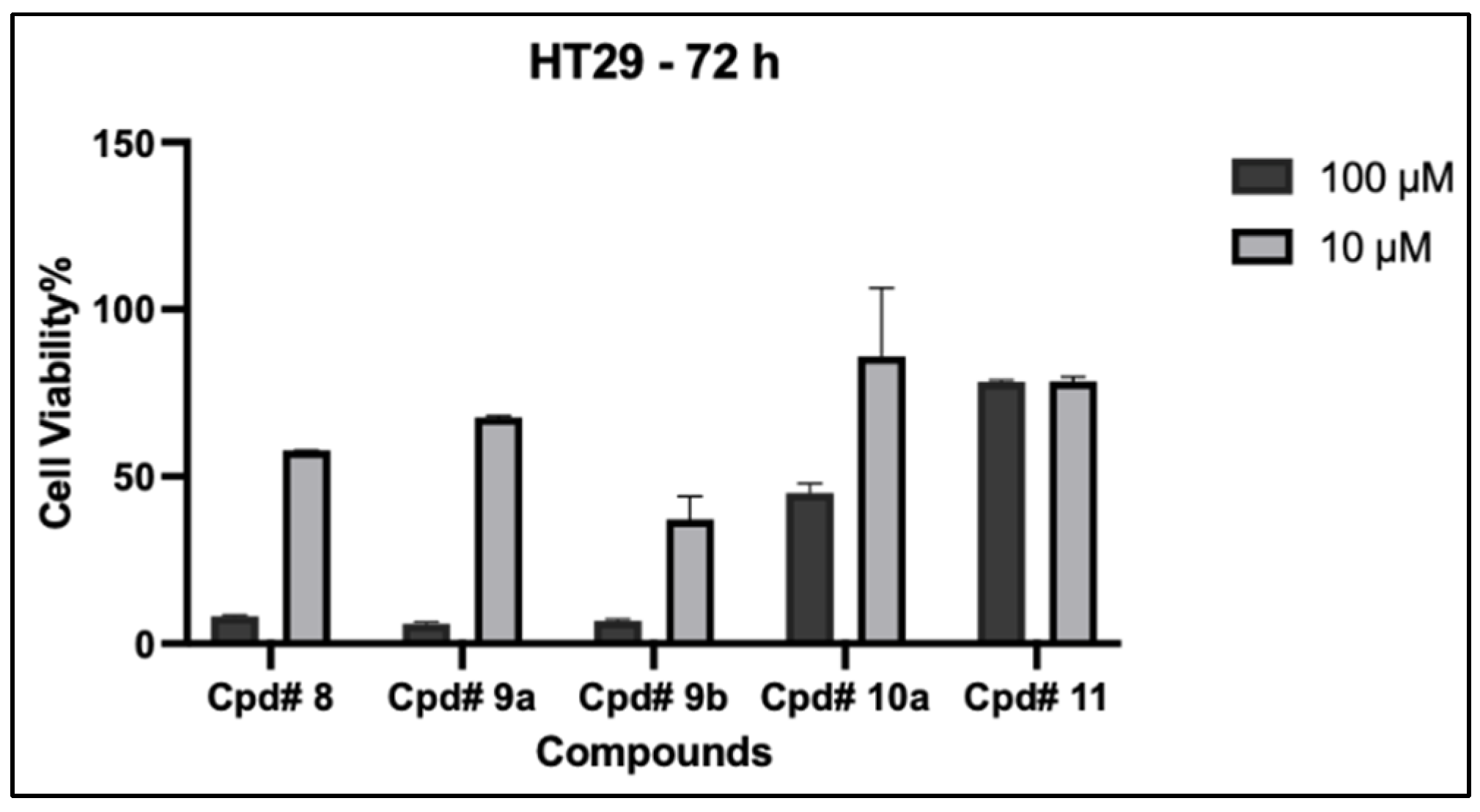
2.2.2. Detailed Dose–Response Relationship against Cancer Cells
2.2.3. Selectivity Assay
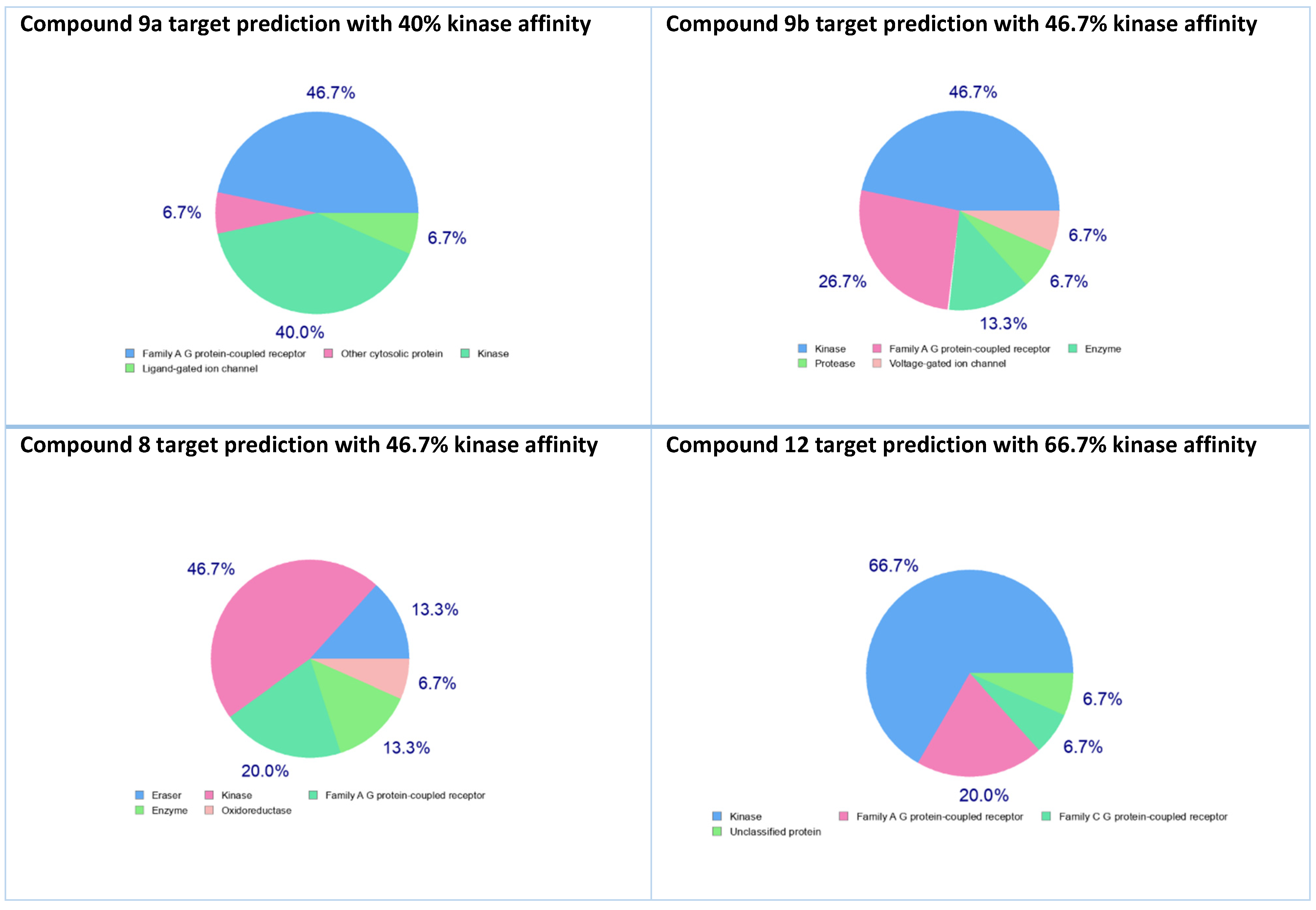
2.2.4. Target Prediction
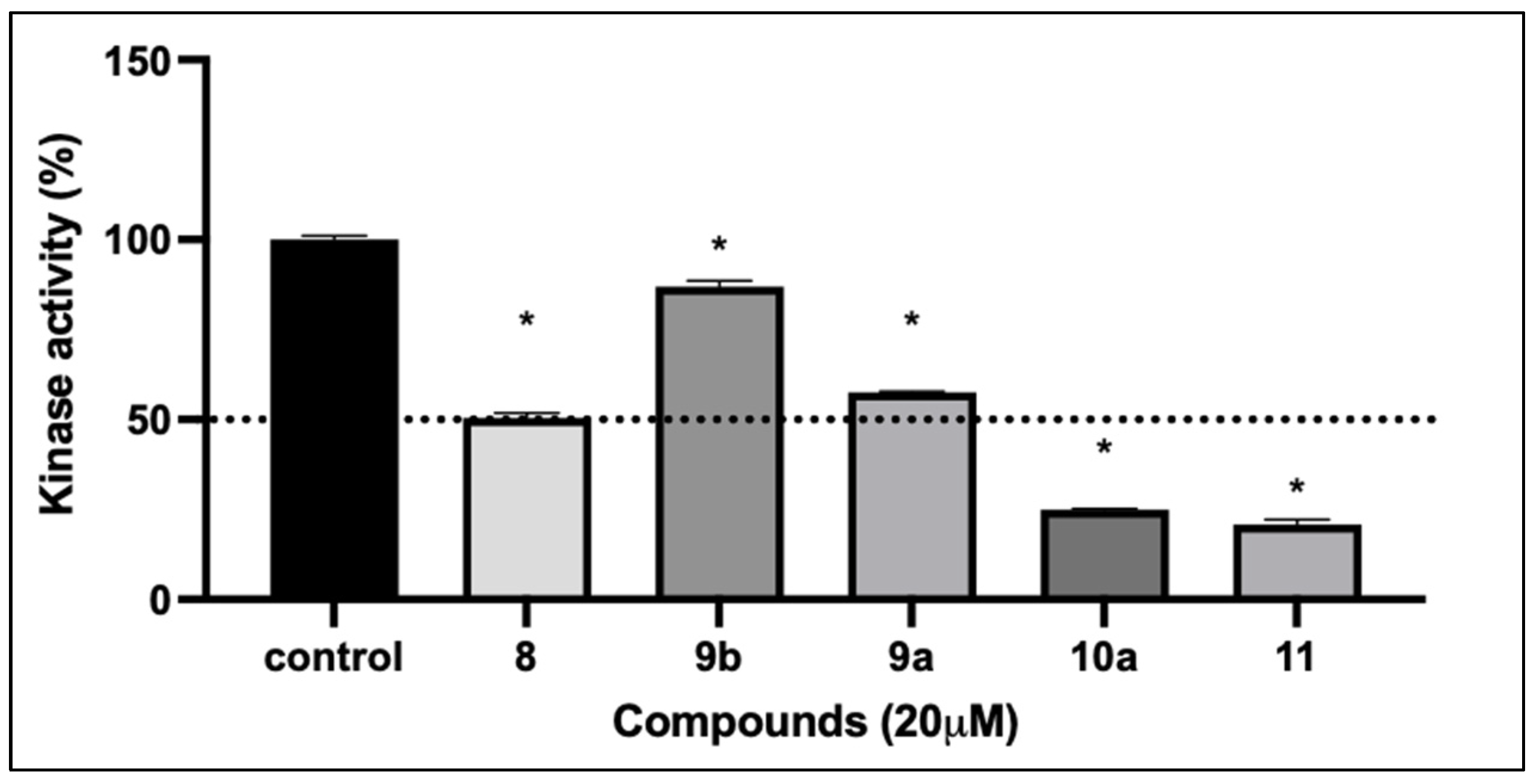
2.2.5. Kinase Inhibition Activity
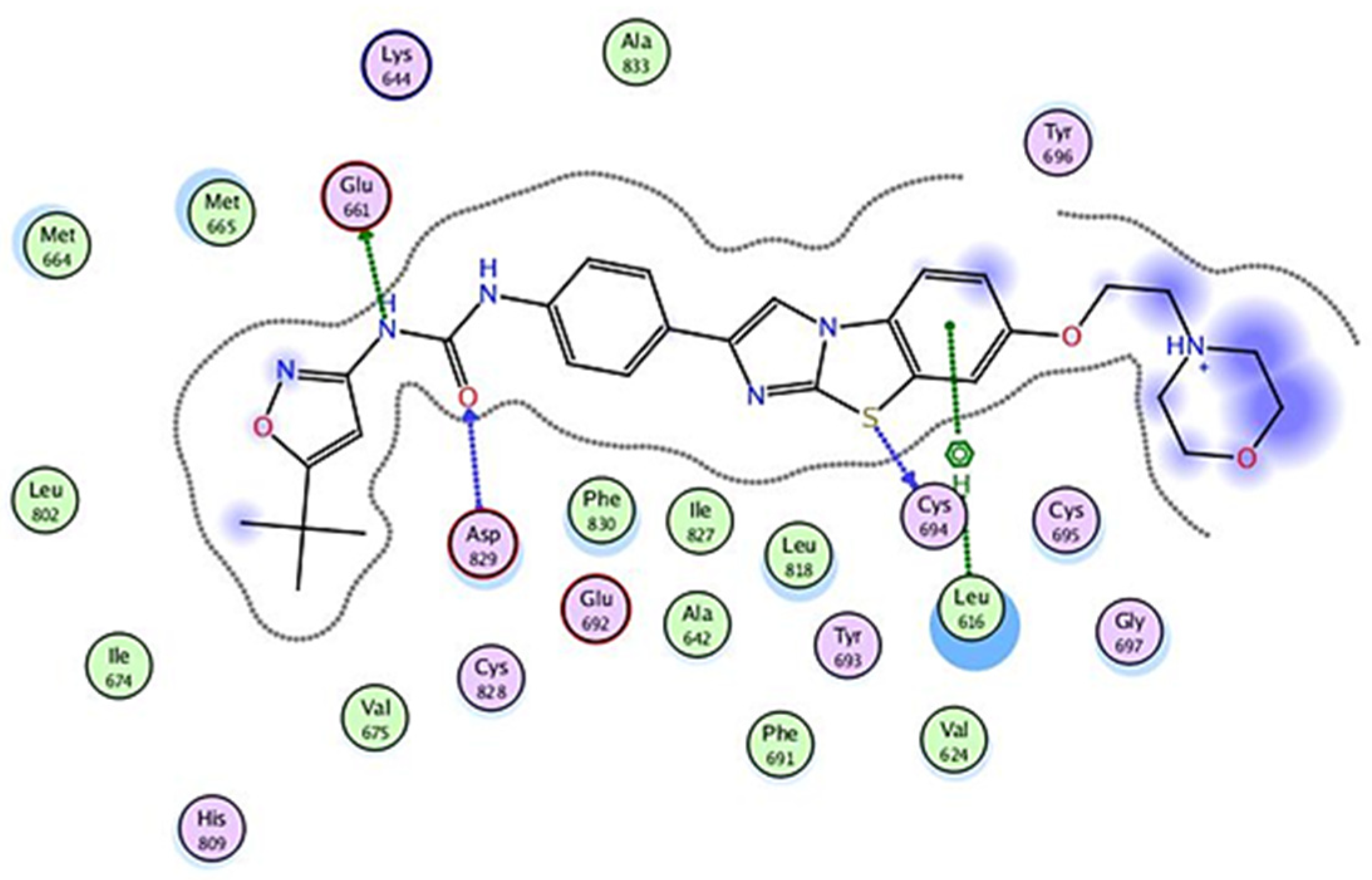
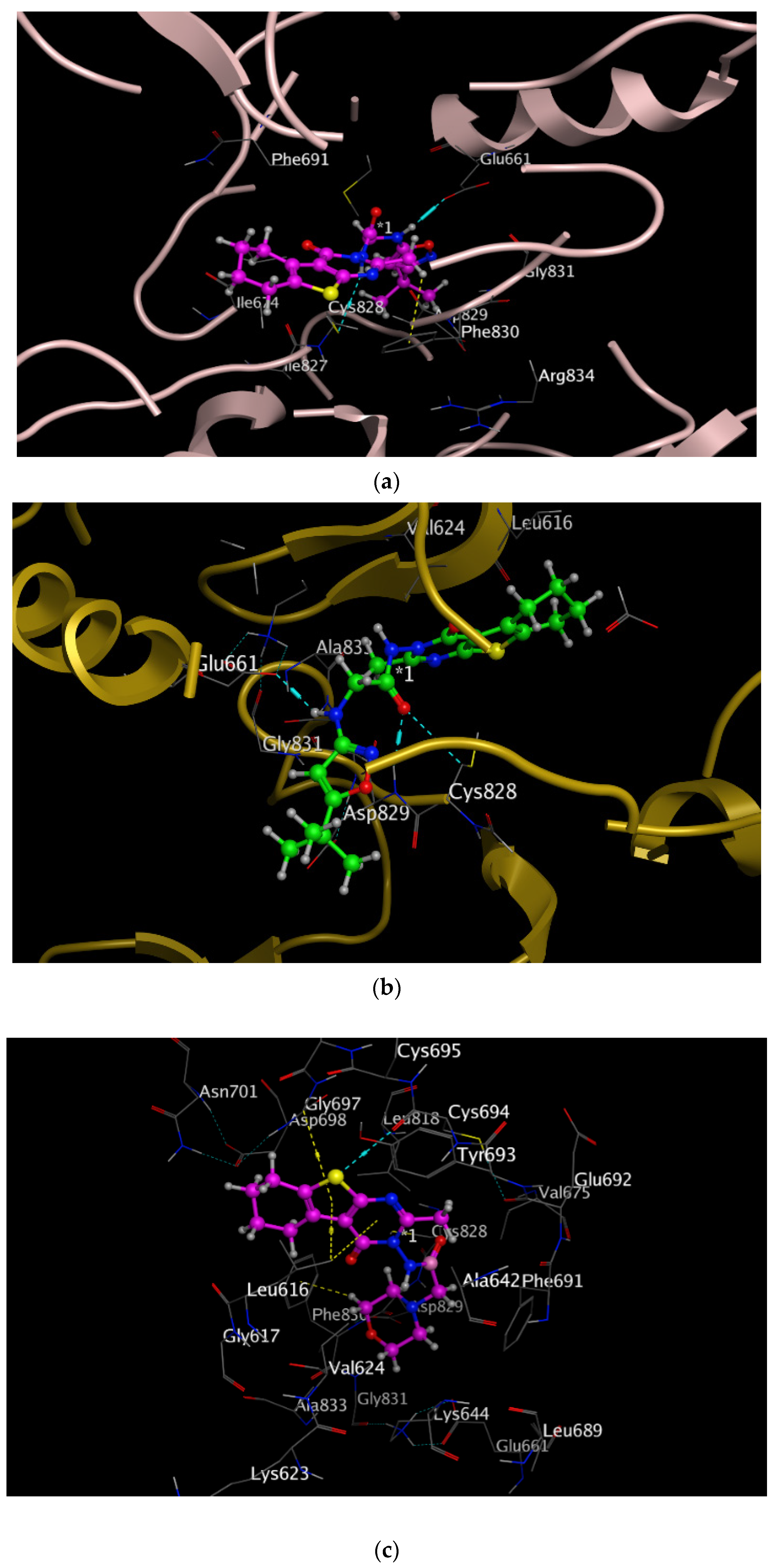
2.2.6. Molecular Docking
2.2.7. In-Vitro Enzyme Inhibition Assay
2.2.8. Pharmacokinetics Evaluation and Drug Likeness
3. Materials and Methods
3.1. Chemistry
3.2. Biology
3.2.1. In Vitro Anti-Proliferative Activities
3.2.2. In-Vitro Enzyme Inhibition Assay
3.3. Molecular Modeling Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 2018, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef] [PubMed]
- Elmongy, E.I. Thieno[2,3-d]pyrimidine derivatives: Synthetic approaches and their FLT3 kinase inhibition. J. Heterocycl. Chem. 2020, 57, 2067–2078. [Google Scholar] [CrossRef]
- Heng, H.; Zhi, Y.; Yuan, H.; Wang, Z.; Li, H.; Wang, S.; Tian, J.; Liu, H.; Chen, Y.; Lu, T.; et al. Discovery of a highly selective FLT3 inhibitor with specific proliferation inhibition against AML cells harboring FLT3-ITD mutation. Eur. J. Med. Chem. 2019, 163, 195–206. [Google Scholar] [CrossRef]
- Oh, C.; Kim, H.; Kang, J.S.; Yun, J.; Sim, J.; Kim, H.M.; Han, G. Synthetic strategy for increasing solubility of potential FLT3 inhibitor thieno[2,3-d]pyrimidine derivatives through structural modifications at the C2 and C6 positions. Bioorganic Med. Chem. Lett. 2017, 27, 496. [Google Scholar] [CrossRef]
- Adepu, R.; Rambabu, D.; Prasad, B.; Meda, C.L.T.; Kandale, A.; Krishna, G.R.; Reddy, C.M.; Chennuru, L.N.; Parsa, K.V.; Pal, M. Novel thieno[2,3-d]pyrimidines: Their design, synthesis, crystal structure analysis and pharmacological evaluation. Org. Biomol. Chem. 2012, 10, 5554–5569. [Google Scholar] [CrossRef] [Green Version]
- Modica, M.; Santagati, M.; Guccione, S.; Russo, F.; Cagnotto, A.; Goegan, M.; Mennini, T. Design, synthesis and binding properties of novel and selective 5-HT 3 and 5-HT 4 receptor ligands. Eur. J. Med. Chem. 2001, 36, 287–301. [Google Scholar] [CrossRef]
- Modica, M.; Romeo, G.; Materia, L.; Russo, F.; Cagnotto, A.; Mennini, T.; Gáspár, R.; Falkay, G.; Fülöp, F. Synthesis and binding properties of novel selective 5-HT 3 receptor ligands. Bioorganic Med. Chem. 2004, 12, 3891–3901. [Google Scholar] [CrossRef] [Green Version]
- Gorja, D.R.; Kumar, K.S.; Mukkanti, K.; Pal, M. C–C (alkynylation) vs. C–O (ether) bond formation under pd/C–Cu catalysis: Synthesis and pharmacological evaluation of 4-alkynylthieno[2,3-d]pyrimidines. Beilstein J. Org. Chem. 2011, 7, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Guan, D.; Sun, S.; Chen, J.; He, J.; Kong, X.; Wang, N.; Yao, J.; Wang, H. Synthesis and evaluation of antitumor activity of sorafenib derivatives possessing diphenylamine and thiourea structures. Yŏujī Huàxué 2018, 38, 1414. [Google Scholar] [CrossRef]
- Catalano, A.; Iacopetta, D.; Sinicropi, M.S.; Franchini, C. Diarylureas as antitumor agents. Appl. Sci. 2021, 11, 374. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, J.; Wang, N.; Kong, X.; Fu, F.; Wang, H.; Yao, J. Design and discovery of quinazoline- and thiourea-containing sorafenib analogs as EGFR and VEGFR-2 dual TK inhibitors. Molecules 2017, 23, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.L.; Li, G.B.; Ma, S.; Zou, C.; Zhou, S.; Sun, Q.Z.; Cheng, C.; Chen, X.; Wang, L.J.; Feng, S.; et al. Structure–Activity relationship studies of pyrazolo[3,4-d]pyrimidine derivatives leading to the discovery of a novel multikinase inhibitor that potently inhibits FLT3 and VEGFR2 and evaluation of its activity against acute myeloid leukemia in vitro and in vivo. J. Med. Chem. 2013, 56, 1641–1655. [Google Scholar] [CrossRef] [PubMed]
- Ali, E.M.H.; Abdel-Maksoud, M.S.; Oh, C. Thieno[2,3-d]pyrimidine as a promising scaffold in medicinal chemistry: Recent advances. Bioorganic Med. Chem. 2019, 27, 1159–1194. [Google Scholar] [CrossRef]
- Kassab, A.E.; Gedawy, E.M.; El-Malah, A.A.; Abdelghany, T.M.; Abdel-Bakky, M.S. Synthesis, anticancer activity, effect on cell cycle profile, and apoptosis-inducing ability of novel hexahydrocyclooctathieno[2,3-d]pyrimidine derivatives. Chem. Pharm. Bull. 2016, 64, 490–496. [Google Scholar] [CrossRef] [Green Version]
- Mghwary, A.E.-; Gedawy, E.M.; Kamal, A.M.; Abuel-Maaty, S.M. Novel thienopyrimidine derivatives as dual EGFR and VEGFR-2 inhibitors: Design, synthesis, anticancer activity and effect on cell cycle profile. J. Enzym. Inhib. Med. Chem. 2019, 34, 838–852. [Google Scholar] [CrossRef] [Green Version]
- Park, C.H.; Lee, C.; Yang, J.S.; Joe, B.Y.; Chun, K.; Kim, H.; Kim, H.Y.; Kang, J.S.; Lee, J.I.; Kim, M.H.; et al. Discovery of thienopyrimidine-based FLT3 inhibitors from the structural modification of known IKKβ inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 2655–2660. [Google Scholar] [CrossRef]
- Yang, J.S.; Park, C.H.; Lee, C.; Kim, H.; Oh, C.; Choi, Y.; Kang, J.S.; Yun, J.; Jeong, J.H.; Kim, M.H.; et al. Synthesis and biological evaluation of novel thieno[2,3-d]pyrimidine-based FLT3 inhibitors as anti-leukemic agents. Eur. J. Med. Chem. 2014, 85, 399–407. [Google Scholar] [CrossRef]
- Gewald, K.; Schinke, E.; Böttcher, H. Heterocyclen aus CH-aciden nitrilen, VIII. 2-amino-thiophene aus methylenaktiven nitrilen, carbonylverbindungen und schwefel. Chem. Ber. 1966, 99, 94–100. Available online: https://api.istex.fr/ark:/67375/WNG-8C2ZSVNT-S/fulltext.pdf (accessed on 9 January 2022). [CrossRef]
- Elmongy, E.I.; Khedr, M.A.; Taleb, N.A.; Awad, H.M.; Abbas, S.E. Design, synthesis, and biological evaluation of some cyclohepta[b]thiophene and substituted pentahydrocycloheptathieno[2,3-d]pyrimidine derivatives. J. Heterocycl. Chem. 2017, 54, 1084–1093. [Google Scholar] [CrossRef]
- Elmongy, E.; Kedr, M.; Abotaleb, N.; Abbas, S. Design and synthesis of new thienopyrimidine derivatives along with their antioxidant activity. Egypt. J. Chem. 2021, 64, 6857–6867. [Google Scholar] [CrossRef]
- Indrayanto, G.; Putra, G.S.; Suhud, F. Validation of in-vitro bioassay methods: Application in herbal drug research. Profiles Drug Subst. Excip. Relat. Methodol. 2021, 46, 273–307. [Google Scholar] [CrossRef] [PubMed]
- Mahavorasirikul, W.; Viyanant, V.; Chaijaroenkul, W.; Itharat, A.; Na-Bangchang, K. Cytotoxic activity of thai medicinal plants against human cholangiocarcinoma, laryngeal and hepatocarcinoma cells in vitro. BMC Complementary Altern. Med. 2010, 10, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorn, J.A.; Wang, Q.; Fujimura, E.; Barros, T.; Kuriyan, J. Crystal structure of the FLT3 kinase domain bound to the inhibitor quizartinib (AC220). PLoS ONE 2015, 10, e0121177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.H.; Coumar, M.S.; Chu, C.Y.; Lin, W.H.; Chen, Y.R.; Chen, C.T.; Shiao, H.Y.; Rafi, S.; Wang, S.Y.; Hsu, H.; et al. Design and synthesis of tetrahydropyridothieno[2,3-d]pyrimidine scaffold based epidermal growth factor receptor (EGFR) kinase inhibitors: The role of side chain chirality and michael acceptor group for maximal potency. J. Med. Chem. 2010, 53, 7316–7326. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Lee, H.J.; Barden, C.J.; Weaver, D.F. The Blood–Brain barrier (BBB) score. J. Med. Chem. 2019, 62, 9824–9836. [Google Scholar] [CrossRef]
- Elmongy, E.I.; Attallah, N.G.M.; Altwaijry, N.; AlKahtani, M.M.; Henidi, H.A. Design and synthesis of new thiophene/thieno[2,3-d]pyrimidines along with their cytotoxic biological evaluation as tyrosine kinase inhibitors in addition to their apoptotic and autophagic induction. Molecules 2021, 27, 123. [Google Scholar] [CrossRef]
- Abdelhaleem, E.F.; Abdelhameid, M.K.; Kassab, A.E.; Kandeel, M.M. Design and synthesis of thienopyrimidine urea derivatives with potential cytotoxic and pro-apoptotic activity against breast cancer cell line MCF-7. Eur. J. Med. Chem. 2018, 143, 1807–1825. [Google Scholar] [CrossRef]
- Al-Rashood, S.T.; Hamed, A.R.; Hassan, G.S.; Alkahtani, H.M.; Almehizia, A.A.; Alharbi, A.; Al-Sanea, M.M.; Eldehna, W.M. Antitumor properties of certain spirooxindoles towards hepatocellular carcinoma endowed with antioxidant activity. J. Enzym. Inhib. Med. Chem. 2020, 35, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2012; p. 10. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | Anti-Proliferative Activities in Different Cancer Cell Lines (IC50 (μM)) | ||
|---|---|---|---|
| HT-29 | HepG-2 | MCF-7 | |
| 8 | 2.0 ± 0.62 | 13.47 ± 2.7 | 11.0 ± 0.8 |
| 9a | 1.21 ± 0.34 | 6.62 ± 0.7 | 7.2 ± 1.9 |
| 9b | 0.85 ± 0.16 | 9.11 ± 0.3 | 16.26 ± 2.3 |
| 10a | 167.4 ± 8.15 | 187.55 ± 28.2 | 104.83 ± 36.4 |
| 11 | 1.78 ± 0.6 | 16.38 ± 5.7 | 7.537 ± 0.07 |
| Dox | 1.4 ± 1.16 | 13.915 ± 2.2 | 8.43 ± 0.5 |
| Compound No. | IC50 of Non-Cancer Cells CCD 841 (μM) | Selectivity Index (SI) (IC50 of CCD 841/IC50 of HT29 (μM)) |
|---|---|---|
| 8 | 151.0 ± 20.2 | 75.5 |
| 9a | 11.3 ± 5.6 | 9.4 |
| 9b | 17.2 ± 1.4 | 20.2 |
| 11 | 177.5 ± 20.4 | 99.7 |
| Compound No. | Binding Affinity | Root Mean Square Deviation (RMSD) | Amino Acids Involved in Interactions at the Active Site |
|---|---|---|---|
| 9a | −7.785 | 1.754 | Glu 661, Cys 828. |
| 9b | −8.479 | 1.015 | Glu 661, Cys 828, Asp 829. |
| 10a | −6.831 | 1.760 | Leu 616, Cys 694, Asp 698. |
| 11 | −7.671 | 1.467 | Glu 692, Cys 694, Asp 829, Phe 691 |
| 12 | −6.632 | 1.778 | Glu 692, Asp 829, Phe 691 |
| P30 | −9.989 | 1.681 | Glu 661, Cys 694, Asp 829, Leu 616. |
| Compound No. | IC50 (µM) * |
|---|---|
| 9a | 20.4 ± 2.8 |
| 9b | 47.64 ± 9.3 |
| 10a | 17.83 ± 3.8 |
| 11 | 40.05 ± 5.5 |
| 12 | 27.22 ± 5.6 |
| Molecule | MW | #H-Bond Acceptors | #H-Bond donors | MR (Molar Refractivity) | TPSA | iLogP | BBB | Bioavailability | Drug Likeness |
|---|---|---|---|---|---|---|---|---|---|
| 8 | 311.79 | 3 | 1 | 81.12 | 92.23 | 1.85 | 4.17 | 0.55 | 0.77 |
| 9a | 415.51 | 5 | 2 | 114.49 | 130.29 | 3.11 | 3.09 | 0.55 | 0.90 |
| 9b | 415.51 | 5 | 2 | 114.49 | 130.29 | 2.88 | 3.10 | 0.55 | 0.90 |
| 10a | 362.45 | 5 | 1 | 101.34 | 104.7 | 2.45 | 3.60 | 0.55 | 1.06 |
| 11 | 360.49 | 2 | 2 | 103.76 | 110.69 | 3.46 | 4.29 | 0.55 | 0.52 |
| 12 | 312.39 | 2 | 2 | 91 | 101.18 | 2.63 | 4.01 | 0.55 | 0.57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elmongy, E.I.; Altwaijry, N.; Attallah, N.G.M.; AlKahtani, M.M.; Henidi, H.A. In-Silico Screening of Novel Synthesized Thienopyrimidines Targeting Fms Related Receptor Tyrosine Kinase-3 and Their In-Vitro Biological Evaluation. Pharmaceuticals 2022, 15, 170. https://doi.org/10.3390/ph15020170
Elmongy EI, Altwaijry N, Attallah NGM, AlKahtani MM, Henidi HA. In-Silico Screening of Novel Synthesized Thienopyrimidines Targeting Fms Related Receptor Tyrosine Kinase-3 and Their In-Vitro Biological Evaluation. Pharmaceuticals. 2022; 15(2):170. https://doi.org/10.3390/ph15020170
Chicago/Turabian StyleElmongy, Elshaymaa I., Najla Altwaijry, Nashwah G. M. Attallah, Manal Mubarak AlKahtani, and Hanan Ali Henidi. 2022. "In-Silico Screening of Novel Synthesized Thienopyrimidines Targeting Fms Related Receptor Tyrosine Kinase-3 and Their In-Vitro Biological Evaluation" Pharmaceuticals 15, no. 2: 170. https://doi.org/10.3390/ph15020170
APA StyleElmongy, E. I., Altwaijry, N., Attallah, N. G. M., AlKahtani, M. M., & Henidi, H. A. (2022). In-Silico Screening of Novel Synthesized Thienopyrimidines Targeting Fms Related Receptor Tyrosine Kinase-3 and Their In-Vitro Biological Evaluation. Pharmaceuticals, 15(2), 170. https://doi.org/10.3390/ph15020170