
Exploring the Natural Compounds in Flavonoids for Their Potential Inhibition of Cancer Therapeutic Target MEK1 Using Computational Methods


Abstract
:1. Introduction
2. Results and Discussion
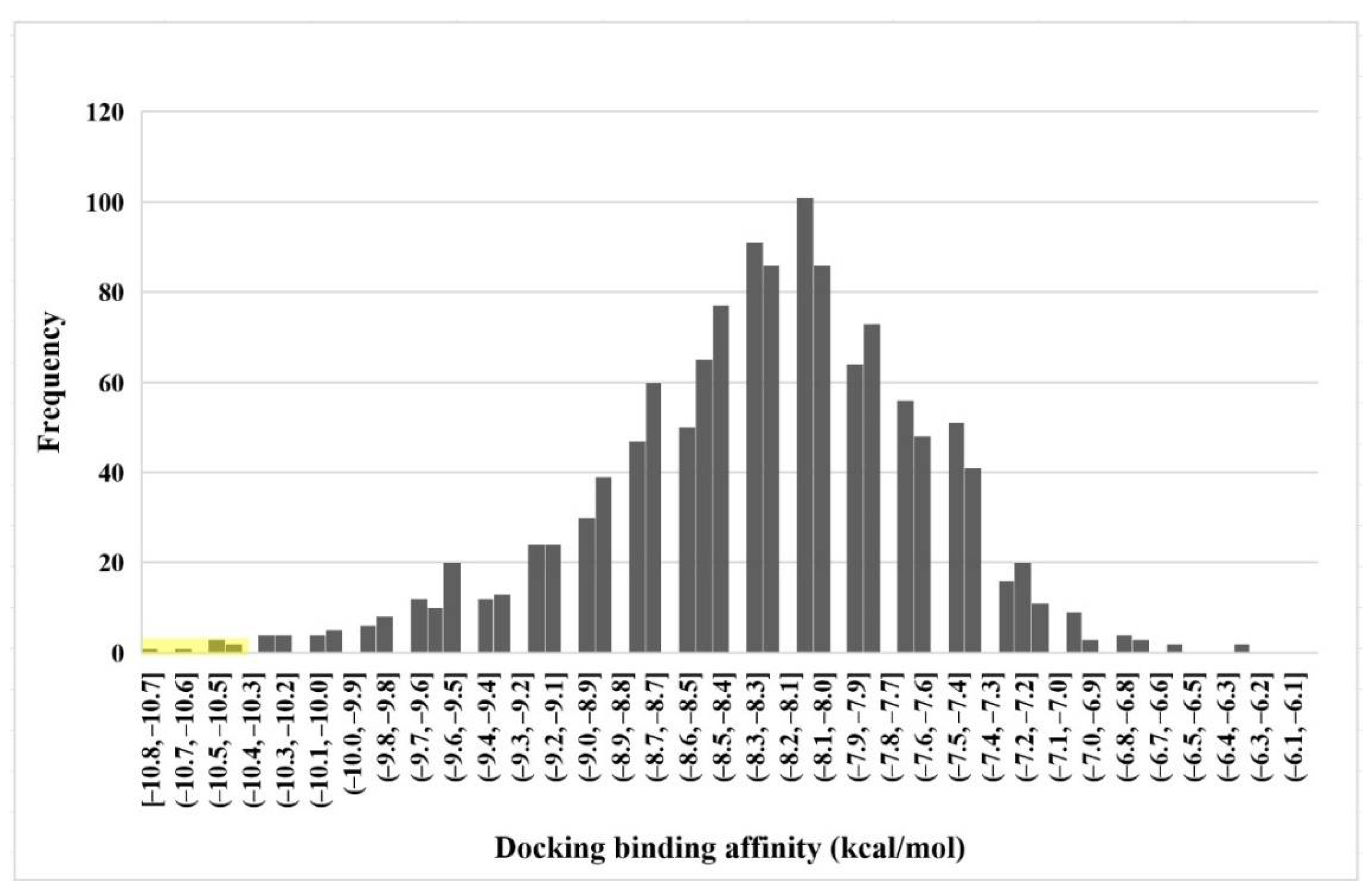
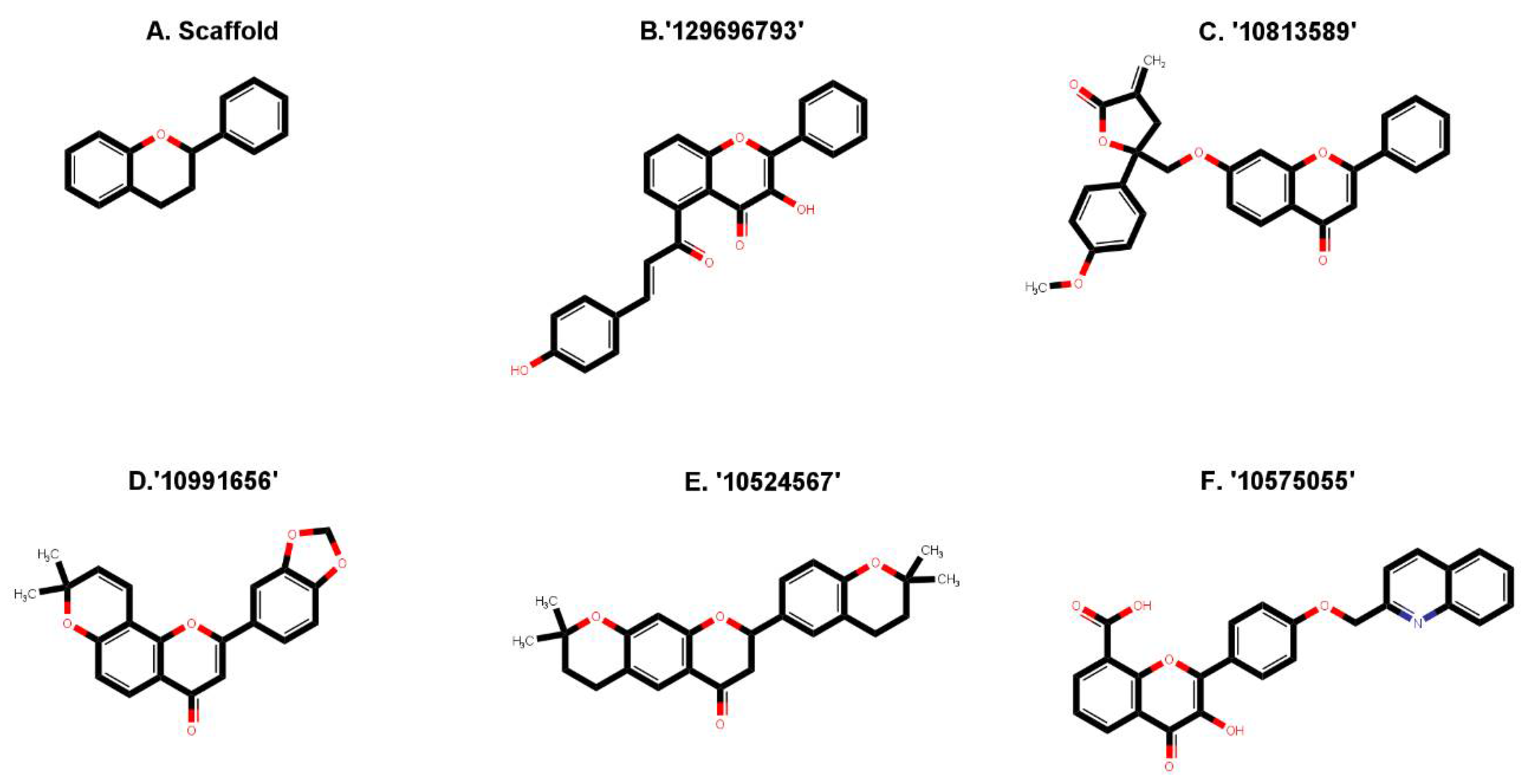
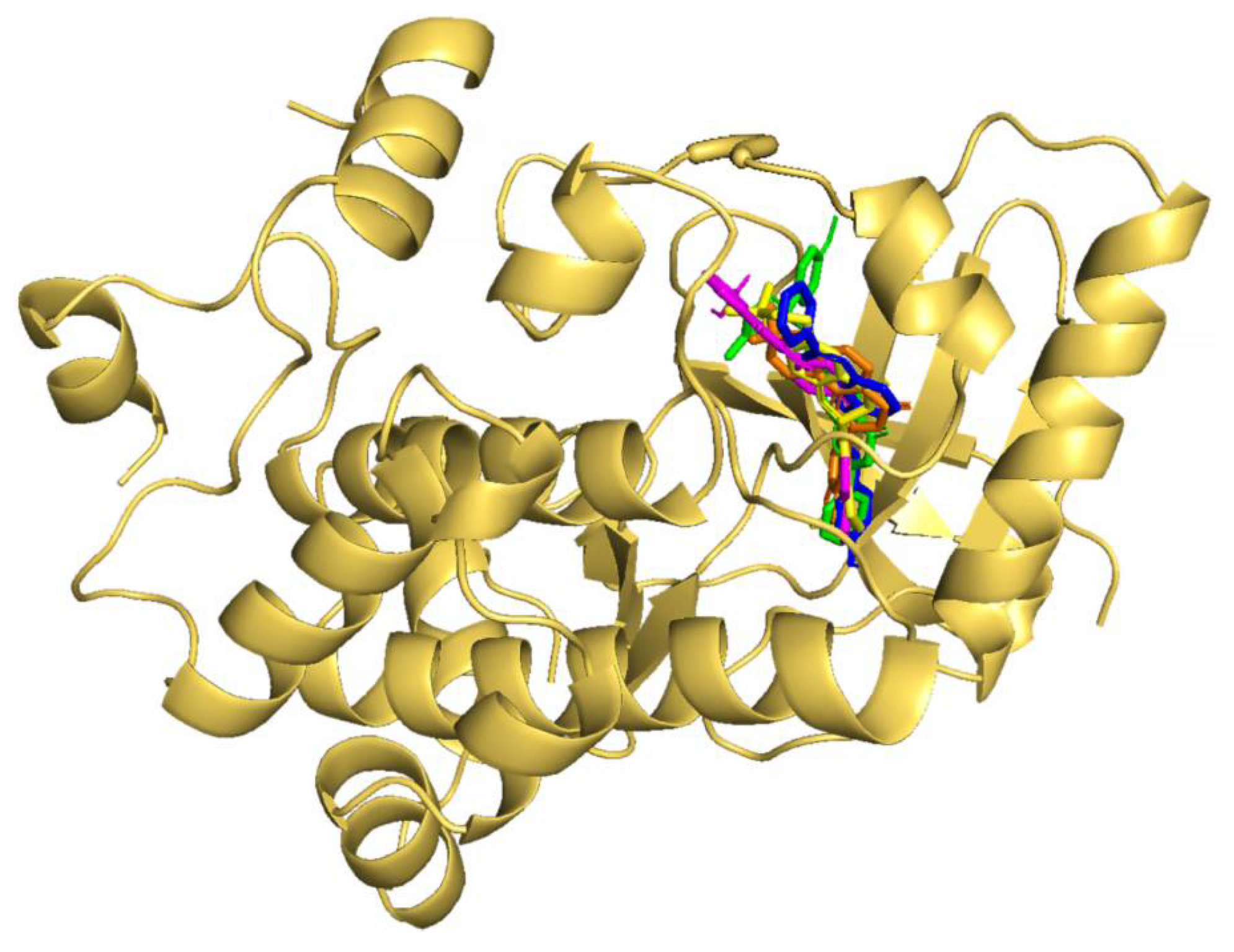
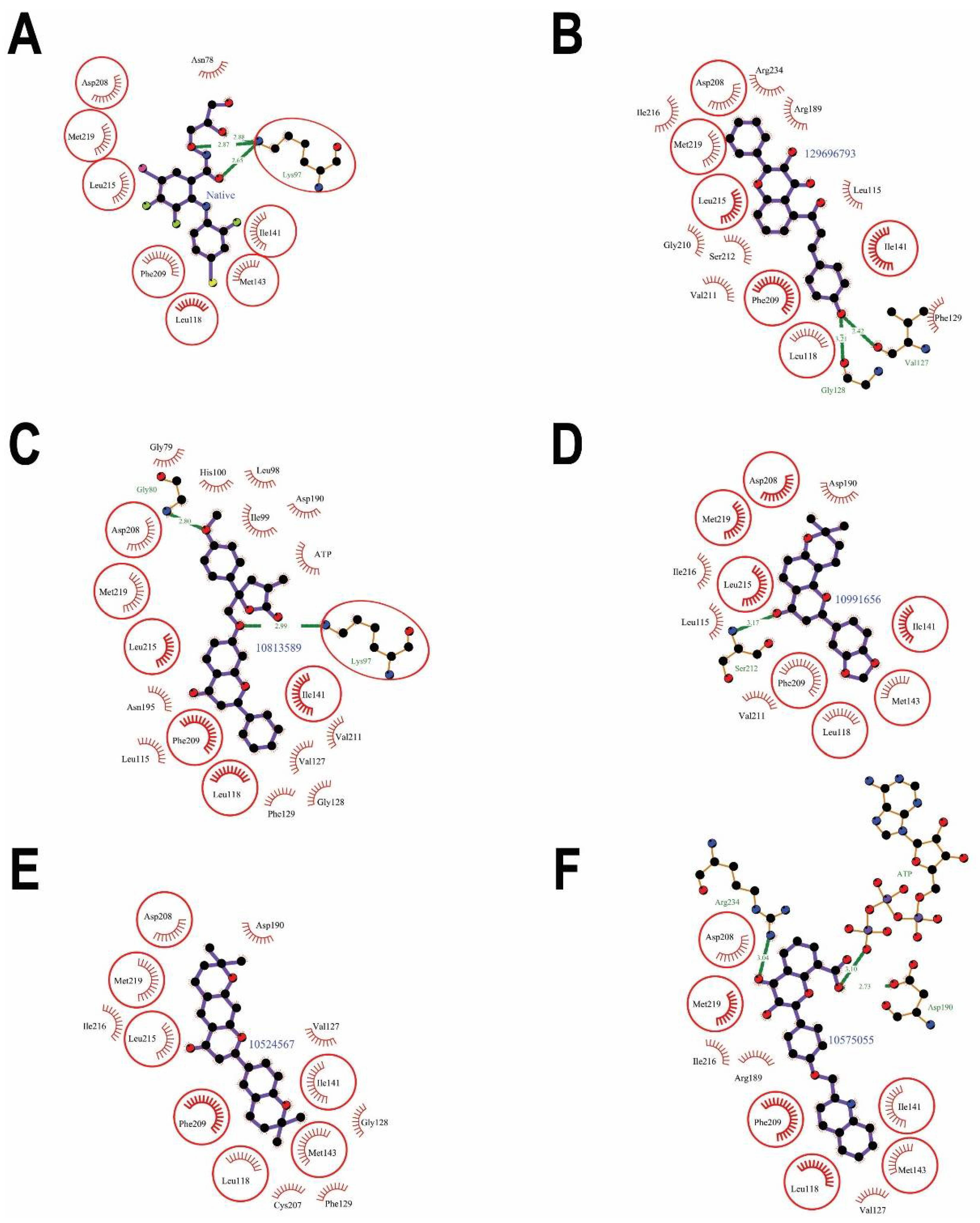
2.1. Virtual Screening of Natural Compound Class Flavonoids for MEK1 Inhibition
2.2. Drug-Likeness and Pharmacokinetics Prediction
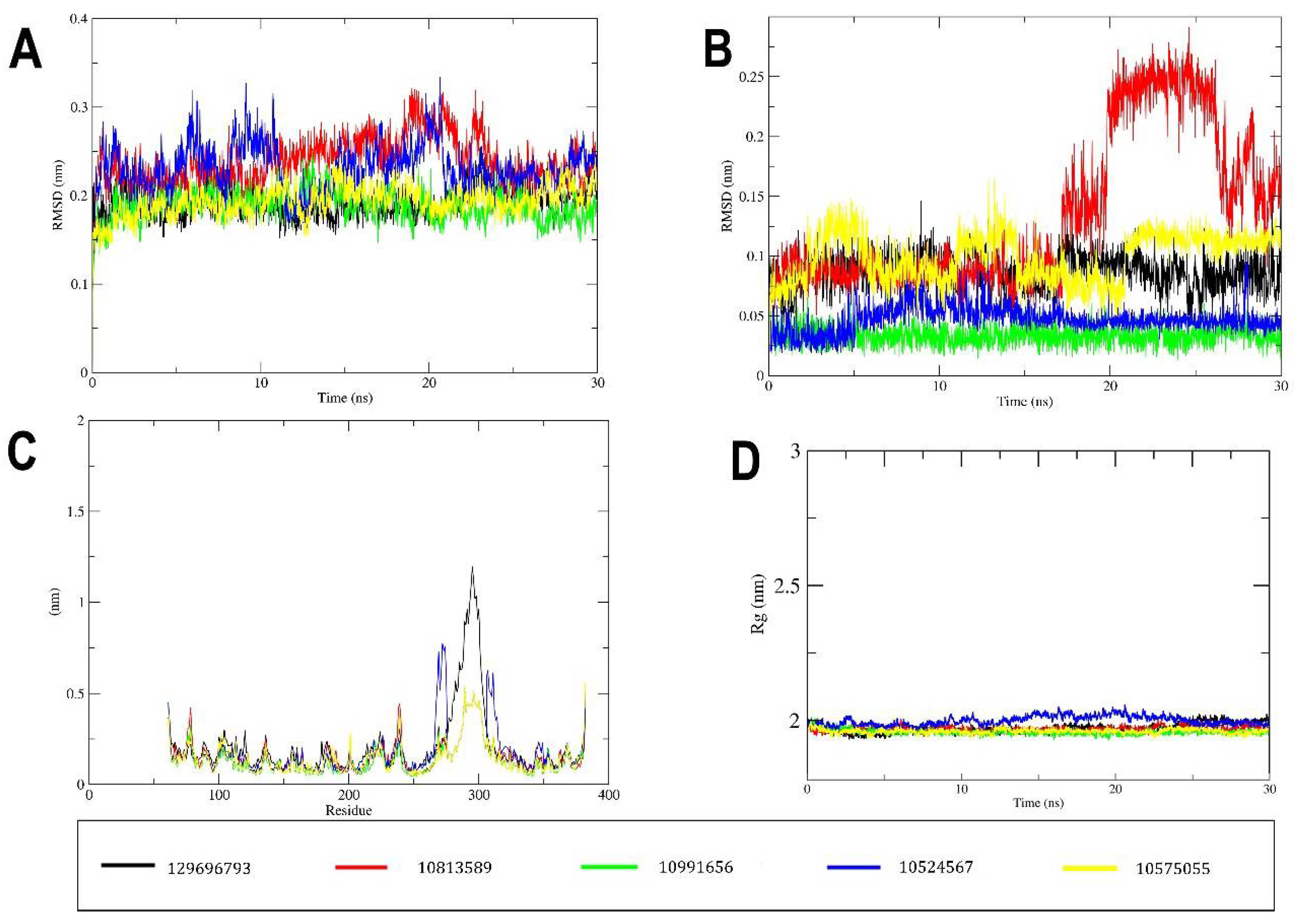
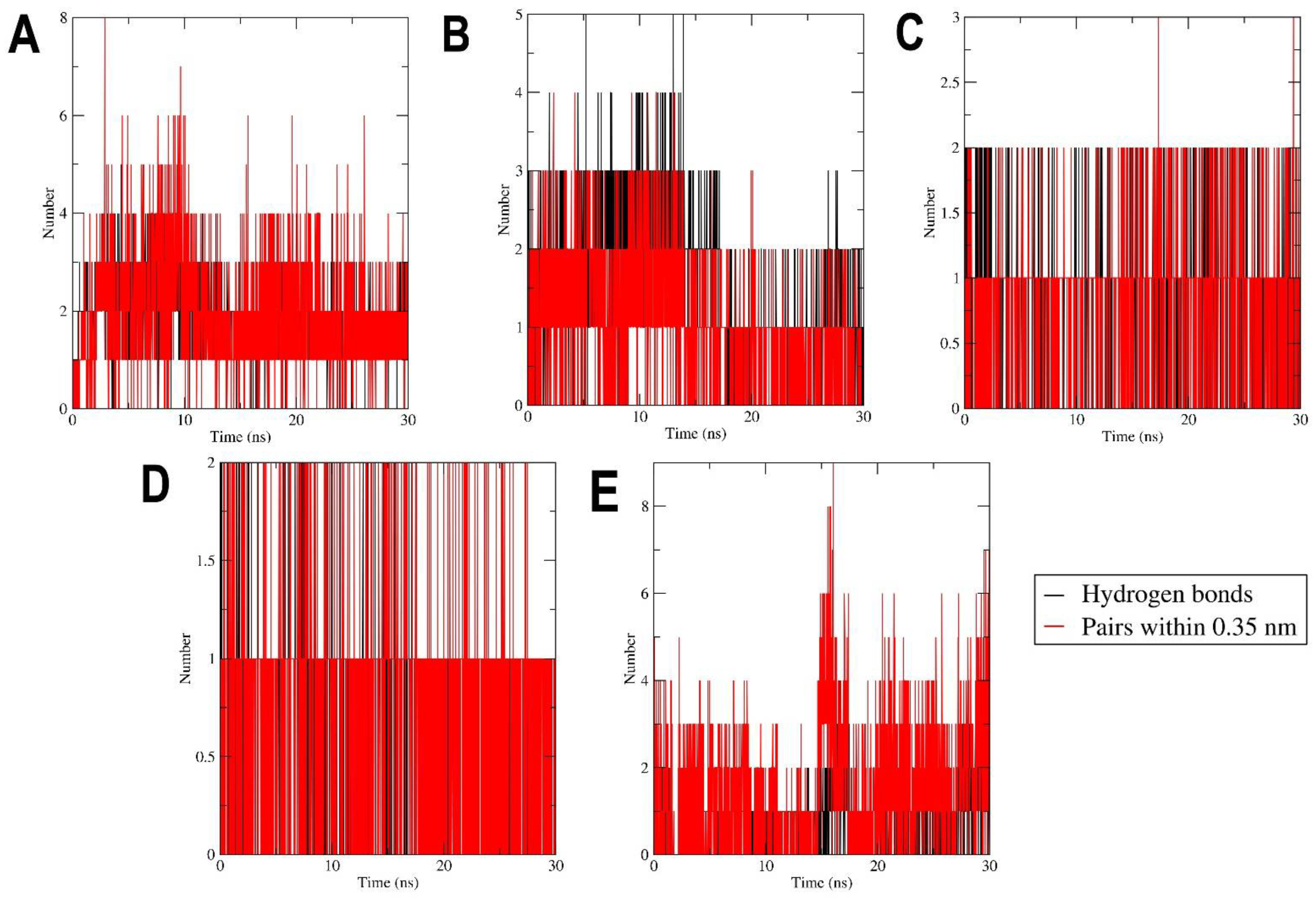
2.3. MD Simulation
3. Materials and Methods
3.1. Data Retrieval and Preparation
3.2. Molecular Docking
3.3. Drug-Likeness and Pharmacokinetics Prediction
3.4. Molecular Dynamic Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Haider, T.; Tiwari, R.; Vyas, S.P.; Soni, V. Pharmacology & Therapeutics Molecular Determinants as Therapeutic Targets in Cancer Chemotherapy: An Update. Pharmacol. Ther. 2019, 200, 85–109. [Google Scholar] [CrossRef] [PubMed]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, D.; Ji, H.; Liu, P.; Gasparian, A.; Gardiner, E.; Lee, S.; Zenteno, A.; Perinskaya, L.O.; Chen, M.; Buckhaults, P.; et al. Identification of Novel Cancer Therapeutic Targets Using a Designed and Pooled ShRNA Library Screen. Nat. Sci. Rep. 2017, 7, 43023. [Google Scholar] [CrossRef] [Green Version]
- Martino, S.D.I.; Rainone, A.; Troise, A.; Paolo, M.D.I.; Pugliese, S.; Zappavigna, S.; Grimaldi, A.; Valente, D. Overview of FDA-approved anti cancer drugs used for targeted therapy. World Cancer Res. J. 2015, 2, e553. [Google Scholar]
- Trotta, A.P.; Chipuk, J.E. Mitochondrial Dynamics As Regulators Of Cancer Biology. Physiol. Behav. 2018, 176, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Kidger, A.M.; Sipthorp, J.; Cook, S.J. ERK1/2 Inhibitors: New Weapons to Inhibit the RAS-Regulated RAF-MEK1/2-ERK1/2 Pathway. Pharmacol. Ther. 2018, 187, 45–60. [Google Scholar] [CrossRef]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [Green Version]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK Mitogen-Activated Protein Kinase Cascade for the Treatment of Cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courcelles, M.; Lemieux, S.; Voisin, L.; Meloche, S.; Thibault, P. ProteoConnections: A Bioinformatics Platform to Facilitate Proteome and Phosphoproteome Analyses. Proteomics 2011, 11, 2654–2671. [Google Scholar] [CrossRef]
- Roskoski, R. Targeting ERK1/2 Protein-Serine/Threonine Kinases in Human Cancers. Pharmacol. Res. 2019, 142, 151–168. [Google Scholar] [CrossRef]
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. MEK1 and MEK2 Inhibitors and Cancer Therapy: The Long and Winding Road. Nat. Rev. Cancer 2015, 15, 577–592. [Google Scholar] [CrossRef]
- Lian, T.; Li, C.; Wang, H. Trametinib in the Treatment of Multiple Malignancies Harboring MEK1 Mutations. Cancer Treat. Rev. 2019, 81, 101907. [Google Scholar] [CrossRef]
- Mccubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, W.T.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Stivala, F.; et al. Roles of The RAF/MEK/ERK Pathway in Cell Growth, Malignant Transforamtion and Drug Resistance. Biochim. Biophys. Acta 2009, 1773, 1263–1284. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Adjei, A.A. The Clinical Development of MEK Inhibitors. Nat. Rev. Clin. Oncol. 2014, 11, 385–400. [Google Scholar] [CrossRef]
- Ohren, J.F.; Chen, H.; Pavlovsky, A.; Whitehead, C.; Zhang, E.; Kuffa, P.; Yan, C.; McConnell, P.; Spessard, C.; Banotai, C.; et al. Structures of Human MAP Kinase Kinase 1 (MEK1) and MEK2 Describe Novel Noncompetitive Kinase Inhibition. Nat. Struct. Mol. Biol. 2004, 11, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. MEK1/2 Dual-Specificity Protein Kinases: Structure and Regulation. Biochem. Biophys. Res. Commun. 2012, 417, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Lu, S.; Jiang, Y.; Zhang, J. Allosteric Modulators of MEK1: Drug Design and Discovery. Chem. Biol. Drug Des. 2016, 88, 485–497. [Google Scholar] [CrossRef]
- Melagraki, G.; Afantitis, A.; Sarimveis, H.; Igglessi-Markopoulou, O.; Koutentis, P.A.; Kollias, G. In Silico Exploration for Identifying Structure-Activity Relationship of MEK Inhibition and Oral Bioavailability for Isothiazole Derivatives. Chem. Biol. Drug Des. 2010, 76, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Tian, H. Current Development Status of MEK Inhibitors. Molecules 2017, 22, 1551. [Google Scholar] [CrossRef] [Green Version]
- Desai, A.; Qazi, G.; Ganju, R.; El-Tamer, M.; Singh, J.; Saxena, A.; Bedi, Y.; Taneja, S.; Bhat, H. Medicinal Plants and Cancer Chemoprevention. Curr. Drug Metab. 2008, 9, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Jiang, W.; Xie, M. Flavonoids: Recent Advances as Anticancer Drugs. Recent Pat. AntiCancer Drug Discov. 2010, 5, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Last 25 Years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [Green Version]
- Block, G.; Patterson, B.; Subar, A. Fruit, Vegetables, and Cancer Prevention: A Review of the Epidemiological Evidence. Nutr. Cancer 1992, 18, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Garg, S. Flavonoids: Biosynthesis, Metabolism, Mechanism of Antioxidation and Clinical Implications: A Review. Agric. Rev. 2020, 41, 227–237. [Google Scholar] [CrossRef]
- Patil, V.M.; Masand, N. Anticancer Potential of Flavonoids: Chemistry, Biological Activities, and Future Perspectives, 1st ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2018; Volume 59. [Google Scholar] [CrossRef]
- Ververidis, F.; Trantas, E.; Douglas, C.; Vollmer, G.; Kretzschmar, G.; Panopoulos, N. Biotechnology of Flavonoids and Other Phenylpropanoid-Derived Natural Products. Part I: Chemical Diversity, Impacts on Plant Biology and Human Health. Biotechnol. J. 2007, 2, 1214–1234. [Google Scholar] [CrossRef]
- Abotaleb, M.; Samuel, S.M.; Varghese, E.; Varghese, S.; Kubatka, P.; Liskova, A.; Büsselberg, D. Flavonoids in Cancer and Apoptosis. Cancers 2019, 11, 28. [Google Scholar] [CrossRef] [Green Version]
- Bisol, Â.; de Campos, P.S.; Lamers, M.L. Flavonoids as Anticancer Therapies: A Systematic Review of Clinical Trials. Phyther. Res. 2020, 34, 568–582. [Google Scholar] [CrossRef]
- Dobrzynska, M.; Napierala, M.; Florek, E. Flavonoid Nanoparticles: A Promising Approach for Cancer Therapy. Biomolecules 2020, 10, 1268. [Google Scholar] [CrossRef] [PubMed]
- Stanisic, D.; Costa, A.F.; Cruz, G.; Durán, N.; Tasic, L. Applications of Flavonoids, With an Emphasis on Hesperidin, as Anticancer Prodrugs: Phytotherapy as an Alternative to Chemotherapy. Stud. Nat. Prod. Chem. 2018, 58, 161–212. [Google Scholar] [CrossRef]
- Guerra, B.; Issinger, O.G. Natural Compounds and Derivatives as Ser/Thr Protein Kinase Modulators and Inhibitors. Pharmaceuticals 2019, 12, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teillet, F.; Boumendjel, A.; Boutonnat, J.; Ronot, X. Flavonoids as RTK Inhibitors and Potential Anticancer Agents. Harv. Bus. Rev. 2008, 86, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Baby, B.; Antony, P.; Al Halabi, W.; Al Homedi, Z.; Vijayan, R. Structural Insights into the Polypharmacological Activity of Quercetin on Serine/Threonine Kinases. Drug Des. Dev. Ther. 2016, 10, 3109–3123. [Google Scholar] [CrossRef]
- Hsieh, M.H.; Tsai, J.P.; Yang, S.F.; Chiou, H.L.; Lin, C.L.; Hsieh, Y.H.; Chang, H.R. Fisetin Suppresses the Proliferation and Metastasis of Renal Cell Carcinoma through Upregulation of MEK/ERK-Targeting CTSS and ADAM9. Cells 2019, 8, 948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalaf, H.H.A.; El-Saadani, R.M.A.; El-Amry, H.G.; Abd El-Salam, R.S. Bioactive Compounds of Flaxseed as Natural Antioxidants and Anticancer. Ann. Agric. Sci. Moshtohor 2019, 57, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Tavsan, Z.; Kayali, H.A. Flavonoids Showed Anticancer Effects on the Ovarian Cancer Cells: Involvement of Reactive Oxygen Species, Apoptosis, Cell Cycle and Invasion. Biomed. Pharmacother. 2019, 116, 109004. [Google Scholar] [CrossRef] [PubMed]
- Mateeva, N.; Eyunni, S.V.K.; Redda, K.K.; Ononuju, U.; Hansberry, T.D.; Aikens, C.; Nag, A. Functional Evaluation of Synthetic Flavonoids and Chalcones for Potential Antiviral and Anticancer Properties. Bioorg. Med. Chem. Lett. 2017, 27, 2350–2356. [Google Scholar] [CrossRef] [PubMed]
- Rehan, M.; Mahmoud, M.M.; Tabrez, S.; Hassan, H.M.A.; Ashraf, G.M.D. Exploring Flavonoids for Potential Inhibitors of a Cancer Signaling Protein PI3Kγ Kinase Using Computational Methods. Anticancer Res. 2020, 40, 4547–4556. [Google Scholar] [CrossRef] [PubMed]
- Martínez Medina, J.J.; Naso, L.G.; Pérez, A.L.; Rizzi, A.; Ferrer, E.G.; Williams, P.A.M. Antioxidant and Anticancer Effects and Bioavailability Studies of the Flavonoid Baicalin and Its Oxidovanadium (IV) Complex. J. Inorg. Biochem. 2017, 166, 150–161. [Google Scholar] [CrossRef]
- Jarial, R.; Shard, A.; Thakur, S.; Sakinah, M.; Zularisam, A.W.; Rezania, S.; Kanwar, S.S.; Singh, L. Characterization of Flavonoids from Fern Cheilanthes Tenuifolia and Evaluation of Antioxidant, Antimicrobial and Anticancer Activities. J. King Saud Univ.-Sci. 2018, 30, 425–432. [Google Scholar] [CrossRef]
- de Souza, P.O.; Bianchi, S.E.; Figueiró, F.; Heimfarth, L.; Moresco, K.S.; Gonçalves, R.M.; Hoppe, J.B.; Klein, C.P.; Salbego, C.G.; Gelain, D.P.; et al. Anticancer Activity of Flavonoids Isolated from Achyrocline Satureioides in Gliomas Cell Lines. Toxicol. Vitr. 2018, 51, 23–33. [Google Scholar] [CrossRef]
- Hassan, A.H.E.; Lee, K.T.; Lee, Y.S. Flavone-Based Arylamides as Potential Anticancers: Design, Synthesis and in Vitro Cell-Based/Cell-Free Evaluations. Eur. J. Med. Chem. 2020, 187, 111965. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.H.E.; Choi, E.; Yoon, Y.M.; Lee, K.W.; Yoo, S.Y.; Cho, M.C.; Yang, J.S.; Kim, H.I.; Hong, J.Y.; Shin, J.S.; et al. Natural Products Hybrids: 3,5,4′-Trimethoxystilbene-5,6,7-Trimethoxyflavone Chimeric Analogs as Potential Cytotoxic Agents against Diverse Human Cancer Cells. Eur. J. Med. Chem. 2019, 161, 559–580. [Google Scholar] [CrossRef] [PubMed]
- Bektic, J.; Guggenberger, R.; Spengler, B.; Christoffel, V.; Pelzer, A.; Berger, A.P.; Ramoner, R.; Bartsch, G.; Klocker, H. The Flavonoid Apigenin Inhibits the Proliferation of Prostatic Stromal Cells via the MAPK-Pathway and Cell-Cycle Arrest in G1/S. Maturitas 2006, 55 (Suppl. 1), 37–46. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, J.G.; Kim, C.S.; Ihm, S.H.; Choi, M.G.; Yoo, H.J.; Lee, S.J. Suppression of AKT Potentiates Synergistic Cytotoxicity of Apigenin with TRAIL in Anaplastic Thyroid Carcinoma Cells. Anticancer Res. 2015, 35, 6529–6538. [Google Scholar]
- Kim, S.H.; Kang, J.G.; Kim, C.S.; Ihm, S.H.; Choi, M.G.; Yoo, H.J.; Lee, S.J. Akt Inhibition Enhances the Cytotoxic Effect of Apigenin in Combination with PLX4032 in Anaplastic Thyroid Carcinoma Cells Harboring BRAF V600E. J. Endocrinol. Investig. 2013, 36, 1099–1104. [Google Scholar] [CrossRef]
- Zhao, G.; Han, X.; Cheng, W.; Ni, J.; Zhang, Y.; Lin, J.; Song, Z. Apigenin Inhibits Proliferation and Invasion, and Induces Apoptosis and Cell Cycle Arrest in Human Melanoma Cells. Oncol. Rep. 2017, 37, 2277–2285. [Google Scholar] [CrossRef] [Green Version]
- Hua, F.; Li, C.H.; Chen, X.G.; Liu, X.P. Daidzein Exerts Anticancer Activity towards SKOV3 Human Ovarian Cancer Cells by Inducing Apoptosis and Cell Cycle Arrest, and Inhibiting the Raf/MEK/ERK Cascade. Int. J. Mol. Med. 2018, 41, 3485–3492. [Google Scholar] [CrossRef]
- Zheng, Z.P.; Yan, Y.; Xia, J.; Zhang, S.; Wang, M.; Chen, J.; Xu, Y. A Phenylacetaldehyde-Flavonoid Adduct, 8-C-(E-Phenylethenyl)-Norartocarpetin, Exhibits Intrinsic Apoptosis and MAPK Pathways-Related Anticancer Potential on HepG2, SMMC-7721 and QGY-7703. Food Chem. 2016, 197, 1085–1092. [Google Scholar] [CrossRef]
- Hseu, Y.C.; Lee, M.S.; Wu, C.R.; Cho, H.J.; Lin, K.Y.; Lai, G.H.; Wang, S.Y.; Kuo, Y.H.; Kumar, K.J.S.; Yang, H.L. The Chalcone Flavokawain b Induces G2/M Cell-Cycle Arrest and Apoptosis in Human Oral Carcinoma HSC-3 Cells through the Intracellular ROS Generation and Downregulation of the Akt/P38 MAPK Signaling Pathway. J. Agric. Food Chem. 2012, 60, 2385–2397. [Google Scholar] [CrossRef]
- Lee, K.W.; Kang, N.J.; Rogozin, E.A.; Kim, H.G.; Cho, Y.Y.; Bode, A.M.; Lee, H.J.; Surh, Y.J.; Bowden, G.T.; Dong, Z. Myricetin Is a Novel Natural Inhibitor of Neoplastic Cell Transformation and MEK1. Carcinogenesis 2007, 28, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Hou, D.X.; Kumamoto, T. Flavonoids as Protein Kinase Inhibitors for Cancer Chemoprevention: Direct Binding and Molecular Modeling. Antioxid. Redox Signal. 2010, 13, 691–719. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Sato, T.; Imada, K.; Dobashi, A.; Yano, M.; Ito, A. A Citrus Polymethoxyflavonoid, Nobiletin, Is a Novel MEK Inhibitor That Exhibits Antitumor Metastasis in Human Fibrosarcoma HT-1080 Cells. Biochem. Biophys. Res. Commun. 2008, 366, 168–173. [Google Scholar] [CrossRef]
- Chen, H.; Yao, K.; Nadas, J.; Bode, A.M.; Malakhova, M.; Oi, N.; Li, H.; Lubet, R.A.; Dong, Z. Prediction of Molecular Targets of Cancer Preventing Flavonoid Compounds Using Computational Methods. PLoS ONE 2012, 7, e38261. [Google Scholar] [CrossRef] [Green Version]
- Alessi, D.R.; Cuenda, A.; Cohen, P.; Dudley, D.T.; Saltiel, A.R. PD 098059 Is a Specific Inhibitor of the Activation of Mitogen-Activated Protein Kinase Kinase in Vitro and in Vivo. J. Biol. Chem. 1995, 270, 27489–27494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Suhail, M.; Parveen, A.; Husain, A.; Rehan, M. Exploring Inhibitory Mechanisms of Green Tea Catechins as Inhibitors of a Cancer Therapeutic Target, Nuclear Factor-ΚB (NF-ΚB). Biosci. Biotechnol. Res. Asia 2019, 16, 715–723. [Google Scholar] [CrossRef]
- Rehan, M. Anticancer Compound XL765 as PI3K/MTOR Dual Inhibitor: A Structural Insight into the Inhibitory Mechanism Using Computational Approaches. PLoS ONE 2019, 14, e0219180. [Google Scholar] [CrossRef]
- Rehan, M.; Mostafa, M. Virtual Screening of 1,4-Naphthoquinone Derivatives for Inhibition of a Key Cancer Signaling Protein, AKT1 Kinase. Anticancer Res. 2019, 39, 3823–3833. [Google Scholar] [CrossRef]
- Rehan, M.; Bajouh, O.S. Virtual Screening of Naphthoquinone Analogs for Potent Inhibitors against the Cancer-Signaling PI3K/AKT/MTOR Pathway. J. Cell. Biochem. 2018, 120, 1328–1339. [Google Scholar] [CrossRef]
- Rehan, M. An Anti-Cancer Drug Candidate OSI-027 and Its Analog as Inhibitors of MTOR: Computational Insights Into the Inhibitory Mechanisms. J. Cell. Biochem. 2017, 118, 4558–4567. [Google Scholar] [CrossRef] [PubMed]
- Rehan, M. A Structural Insight into the Inhibitory Mechanism of an Orally Active PI3K/MTOR Dual Inhibitor, PKI-179 Using Computational Approaches. J. Mol. Graph. Model. 2015, 62, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Rehan, M.; Beg, M.A.; Parveen, S.; Damanhouri, G.A.; Zaher, G.F. Computational Insights into the Inhibitory Mechanism of Human AKT1 by an Orally Active Inhibitor, MK-2206. PLoS ONE 2014, 9, e109705. [Google Scholar] [CrossRef] [PubMed]
- Jamal, M.S.; Parveen, S.; Beg, M.A.; Suhail, M.; Chaudhary, A.G.A.; Damanhouri, G.A.; Abuzenadah, A.M.; Rehan, M. Anticancer Compound Plumbagin and Its Molecular Targets: A Structural Insight into the Inhibitory Mechanisms Using Computational Approaches. PLoS ONE 2014, 9, e87309. [Google Scholar] [CrossRef] [Green Version]
- El-Demerdash, A.; Al-Karmalawy, A.A.; Abdel-Aziz, T.M.; Elhady, S.S.; Darwish, K.M.; Hassan, A.H.E. Investigating the Structure-Activity Relationship of Marine Natural Polyketides as Promising SARS-CoV-2 Main Protease Inhibitors. RSC Adv. 2021, 11, 31339–31363. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Kim, N.Y.; Hassan, A.H.E.; Park, J.E.; Paik, S.; Yang, J.E.; Oh, K.S.; Lee, B.H.; Lee, M.Y.; Shin, K.J.; et al. Thiazolidine-2,4-Dione-Based Irreversible Allosteric IKK-β Kinase Inhibitors: Optimization into in Vivo Active Anti-Inflammatory Agents. Eur. J. Med. Chem. 2020, 188, 111955. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Kim, N.Y.; Hassan, A.H.E.; Park, J.E.; Yang, J.E.; Elsherbeny, M.H.; Paik, S.; Oh, K.S.; Lee, B.H.; Lee, M.Y.; et al. Optimization Study towards More Potent Thiazolidine-2,4-Dione IKK-β Modulator: Synthesis, Biological Evaluation and in Silico Docking Simulation. Bioorg. Chem. 2019, 92, 103261. [Google Scholar] [CrossRef]
- Hashemzadeh, S.; Ramezani, F.; Rafii-Tabar, H. Study of Molecular Mechanism of the Interaction Between MEK1/2 and Trametinib with Docking and Molecular Dynamic Simulation. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 115–124. [Google Scholar] [CrossRef]
- Ya’u Ibrahim, Z.; Uzairu, A.; Shallangwa, G.; Abechi, S. Molecular Docking Studies, Drug-Likeness and in-Silico ADMET Prediction of Some Novel β-Amino Alcohol Grafted 1,4,5-Trisubstituted 1,2,3-Triazoles Derivatives as Elevators of P53 Protein Levels. Sci. Afr. 2020, 10, e00570. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Xie, L.; Bourne, P.E. Insights into the Binding Mode of MEK Type-III Inhibitors. A Step towards Discovering and Designing Allosteric Kinase Inhibitors across the Human Kinome. PLoS ONE 2017, 12, e0179936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software News and Updates AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1996, 64, 4–17. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Lua, R.C.; Lichtarge, O. PyETV: A PyMOL Evolutionary Trace Viewer to Analyze Functional Site Predictions in Protein Complexes. Bioinformatics 2010, 26, 2981–2982. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Lai, L.; Wang, S. Further Development and Validation of Empirical Scoring Functions for Structure-Based Binding Affinity Prediction. J. Comput. Aided Mol. Des. 2002, 16, 11–26. [Google Scholar] [CrossRef]
- Roy, A.S.; Tripathy, D.R.; Chatterjee, A.; Dasgupta, S. A Spectroscopic Study of the Interaction of the Antioxidant Naringin with Bovine Serum Albumin. J. Biophys. Chem. 2010, 1, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, S.J.; Thornton, J.M. ‘NACCESS’, Computer Program; Department of Biochemistry and Molecular Biology, University College London: London, UK, 1993. [Google Scholar]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Flavonoids | Docking Affinity (kcal/mol) | Binding Energy (Kcal/mol) | |
|---|---|---|---|---|
| 1 | 129696793 | −10.8 | −10.25 | 7.52 |
| 2 | 10813589 | −10.6 | −10.96 | 8.03 |
| 3 | 10991656 | −10.5 | −9.49 | 6.95 |
| 4 | 10524567 | −10.5 | −10.83 | 7.94 |
| 5 | 10575055 | −10.4 | −10.11 | 7.41 |
| Native | −9.0 | −8.95 | 6.56 |
| Interacting Residues | Hydrogen Bonds | Non-Bonding Interactions | |
|---|---|---|---|
| Leu-115 | 0 | 1 | 3.89 |
| Leu-118 | 0 | 5 | 17.12 |
| Val-127 | 1 | 2 | 10.27 |
| Gly-128 | 1 | 1 | 0.78 |
| Phe-129 | 0 | 2 | 6.45 |
| Ile-141 | 0 | 6 | 21.79 |
| Arg-189 | 0 | 3 | 11.93 |
| Asp-208 | 0 | 5 | 39.18 |
| Phe-209 | 0 | 9 | 25.06 |
| Gly-210 | 0 | 1 | 8.22 |
| Val-211 | 0 | 4 | 4.59 |
| Ser-212 | 0 | 4 | 2.41 |
| Leu-215 | 0 | 4 | 14.8 |
| Ile-216 | 0 | 9 | 25.95 |
| Met-219 | 0 | 4 | 42.37 |
| Arg-234 | 0 | 2 | 16.78 |
| Interacting Residues | Hydrogen Bonds | Non-Bonding Interactions | |
|---|---|---|---|
| Gly-79 | 0 | 3 | 17.63 |
| Gly-80 | 1 | 4 | 15.78 |
| Lys-97 | 1 | 8 | 31.09 |
| Leu-98 | 0 | 1 | 0.55 |
| Ile-99 | 0 | 15 | 27.12 |
| His-100 | 0 | 2 | 16.96 |
| Leu-115 | 0 | 2 | 3.89 |
| Leu-118 | 0 | 3 | 16.47 |
| Val-127 | 0 | 2 | 9.47 |
| Gly-128 | 0 | 1 | 0.78 |
| Phe-129 | 0 | 1 | 5.57 |
| Ile-141 | 0 | 2 | 21.79 |
| Asp-190 | 0 | 1 | 20.27 |
| Asn-195 | 0 | 2 | 4.05 |
| Asp-208 | 0 | 9 | 39.24 |
| Phe-209 | 0 | 6 | 22.18 |
| Val-211 | 0 | 3 | 4.59 |
| Leu-215 | 0 | 1 | 12.45 |
| Met-219 | 0 | 5 | 43.97 |
| ATP | 0 | 7 |
| Interacting Residues | Hydrogen Bonds | Non-Bonding Interactions | |
|---|---|---|---|
| Leu-115 | 0 | 2 | 3.89 |
| Leu-118 | 0 | 1 | 15.6 |
| Ile-141 | 0 | 2 | 21.79 |
| Met-143 | 0 | 1 | 9.92 |
| Asp-190 | 0 | 4 | 27.2 |
| Asp-208 | 0 | 13 | 40.1 |
| Phe-209 | 0 | 10 | 24.33 |
| Val-211 | 0 | 3 | 4.59 |
| Ser-212 | 1 | 3 | 2.41 |
| Leu-215 | 0 | 5 | 14.8 |
| Ile-216 | 0 | 5 | 13.79 |
| Met-219 | 0 | 5 | 46.4 |
| Interacting Residues | Hydrogen Bonds | Non-Bonding Interactions | |
|---|---|---|---|
| Leu-118 | 0 | 6 | 16.62 |
| Val-127 | 0 | 2 | 9.35 |
| Gly-128 | 0 | 1 | 0.78 |
| Phy-129 | 0 | 1 | 5.48 |
| Ile-141 | 0 | 5 | 21.79 |
| Met-143 | 0 | 6 | 9.92 |
| Asp-190 | 0 | 2 | 30.23 |
| Cys-207 | 0 | 2 | 4.33 |
| Asp-208 | 0 | 11 | 38.96 |
| Phe-209 | 0 | 14 | 24.77 |
| Leu-215 | 0 | 4 | 14.8 |
| Ile-216 | 0 | 3 | 21.97 |
| Met-219 | 0 | 4 | 52.28 |
| Interacting Residues | Hydrogen Bonds | Non-Bonding Interactions | |
|---|---|---|---|
| Leu-118 | 0 | 4 | 16.73 |
| Val-127 | 0 | 1 | 9.63 |
| Ile-141 | 0 | 4 | 21.79 |
| Met-143 | 0 | 3 | 9.92 |
| Arg-189 | 0 | 3 | 14.72 |
| Asp-190 | 1 | 4 | 41.14 |
| Asp-208 | 0 | 9 | 40.01 |
| Phe-209 | 0 | 11 | 25.06 |
| Ile-216 | 0 | 1 | 18.14 |
| Met-219 | 0 | 2 | 50.93 |
| Arg-234 | 1 | 2 | 13.09 |
| ATP | 1 | 0 |
| Rank | Compound (CID) | Structure | Molecular Weight | LogP | #Rotatable Bonds | #Acceptors | #Donors | Surface Area |
|---|---|---|---|---|---|---|---|---|
| 1 | 129696793 |  | 384.387 | 4.7673 | 4 | 5 | 2 | 165.349 |
| 2 | 10813589 |  | 454.478 | 5.246 | 6 | 6 | 0 | 195.555 |
| 3 | 10991656 |  | 348.354 | 4.3729 | 1 | 5 | 0 | 148.966 |
| 4 | 10524567 |  | 392.495 | 5.6003 | 1 | 4 | 0 | 171.717 |
| 5 | 10575055 |  | 439.423 | 4.991 | 5 | 6 | 2 | 186.688 |
| Property | Model Name | Predicted Value | Unit | ||||
|---|---|---|---|---|---|---|---|
| 129696793 | 10813589 | 10991656 | 10524567 | 10575055 | |||
| Absorption | Water solubility | −4.456 | −5.914 | −4.984 | −5.161 | −3.903 | Numeric (log mol/L) |
| Caco2 permeability | 1.089 | 1.073 | 1.004 | 1.116 | 0.549 | Numeric (log Papp in 10−6 cm/s) | |
| Intestinal absorption (human) | 90.869 | 98.737 | 95.566 | 96.435 | 88.343 | Numeric (% Absorbed) | |
| Skin Permeability | −2.735 | −2.731 | −2.589 | −2.729 | −2.734 | Numeric (log Kp) | |
| P-glycoprotein substrate | Yes | No | Yes | No | Yes | Categorical (Yes/No) | |
| P-glycoprotein I inhibitor | Yes | Yes | Yes | Yes | No | Categorical (Yes/No) | |
| P-glycoprotein II inhibitor | Yes | Yes | Yes | Yes | Yes | Categorical (Yes/No) | |
| Distribution | VDss (human) | −0.696 | −0.157 | 0.121 | 0.363 | −0.432 | Numeric (log L/kg) |
| Fraction unbound (human) | 0.039 | 0.223 | 0.195 | 0.03 | 0.208 | Numeric (Fu) | |
| BBB permeability | −0.371 | −0.749 | 0.358 | −0.005 | −0.956 | Numeric (log BB) | |
| CNS permeability | −1.883 | −1.903 | −1.595 | −1.608 | −2.889 | Numeric (log PS) | |
| Metabolism | CYP2D6 substrate | No | No | No | No | No | Categorical (Yes/No) |
| CYP3A4 substrate | Yes | Yes | Yes | Yes | Yes | Categorical (Yes/No) | |
| CYP1A2 inhibitor | Yes | No | Yes | No | No | Categorical (Yes/No) | |
| CYP2C19 inhibitor | Yes | Yes | Yes | Yes | No | Categorical (Yes/No) | |
| CYP2C9 inhibitor | Yes | Yes | Yes | Yes | Yes | Categorical (Yes/No) | |
| CYP2D6 inhibitor | No | No | No | No | No | Categorical (Yes/No) | |
| CYP3A4 inhibitor | Yes | Yes | Yes | Yes | No | Categorical (Yes/No) | |
| Excretion | Total Clearance | 0.184 | 0.81 | 0.345 | 0.087 | 0.551 | Numeric (log mL/min/kg) |
| Renal OCT2 substrate | No | No | No | No | No | Categorical (Yes/No) | |
| Toxicity | AMES toxicity | Yes | Yes | No | No | No | Categorical (Yes/No) |
| Max. tolerated dose (human) | 0.204 | 0.64 | −0.242 | −0.067 | 0.742 | Numeric (log mg/kg/day) | |
| hERG I inhibitor | No | No | No | No | No | Categorical (Yes/No) | |
| hERG II inhibitor | Yes | Yes | No | No | Yes | Categorical (Yes/No) | |
| Oral Rat Acute Toxicity (LD50) | 2.767 | 2.734 | 2.086 | 3.018 | 2.656 | Numeric (mol/kg) | |
| Oral Rat Chronic Toxicity (LOAEL) | 0.914 | 0.805 | 1.269 | 1.713 | 0.755 | Numeric (log mg/kg_bw/day) | |
| Hepatotoxicity | Yes | No | No | No | Yes | Categorical (Yes/No) | |
| Skin Sensitization | No | No | No | No | No | Categorical (Yes/No) | |
| T.Pyriformis toxicity | 0.29 | 0.287 | 0.555 | 0.491 | 0.285 | Numeric (log ug/L) | |
| Minnow toxicity | 0.09 | −2.72 | −0.483 | −0.22 | −1.62 | Numeric (log mM) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AlZahrani, W.M.; AlGhamdi, S.A.; Zughaibi, T.A.; Rehan, M. Exploring the Natural Compounds in Flavonoids for Their Potential Inhibition of Cancer Therapeutic Target MEK1 Using Computational Methods. Pharmaceuticals 2022, 15, 195. https://doi.org/10.3390/ph15020195
AlZahrani WM, AlGhamdi SA, Zughaibi TA, Rehan M. Exploring the Natural Compounds in Flavonoids for Their Potential Inhibition of Cancer Therapeutic Target MEK1 Using Computational Methods. Pharmaceuticals. 2022; 15(2):195. https://doi.org/10.3390/ph15020195
Chicago/Turabian StyleAlZahrani, Wejdan M., Shareefa A. AlGhamdi, Torki A. Zughaibi, and Mohd Rehan. 2022. "Exploring the Natural Compounds in Flavonoids for Their Potential Inhibition of Cancer Therapeutic Target MEK1 Using Computational Methods" Pharmaceuticals 15, no. 2: 195. https://doi.org/10.3390/ph15020195
APA StyleAlZahrani, W. M., AlGhamdi, S. A., Zughaibi, T. A., & Rehan, M. (2022). Exploring the Natural Compounds in Flavonoids for Their Potential Inhibition of Cancer Therapeutic Target MEK1 Using Computational Methods. Pharmaceuticals, 15(2), 195. https://doi.org/10.3390/ph15020195