
A Lead-Based Fragment Library Screening of the Glycosyltransferase WaaG from Escherichia coli

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
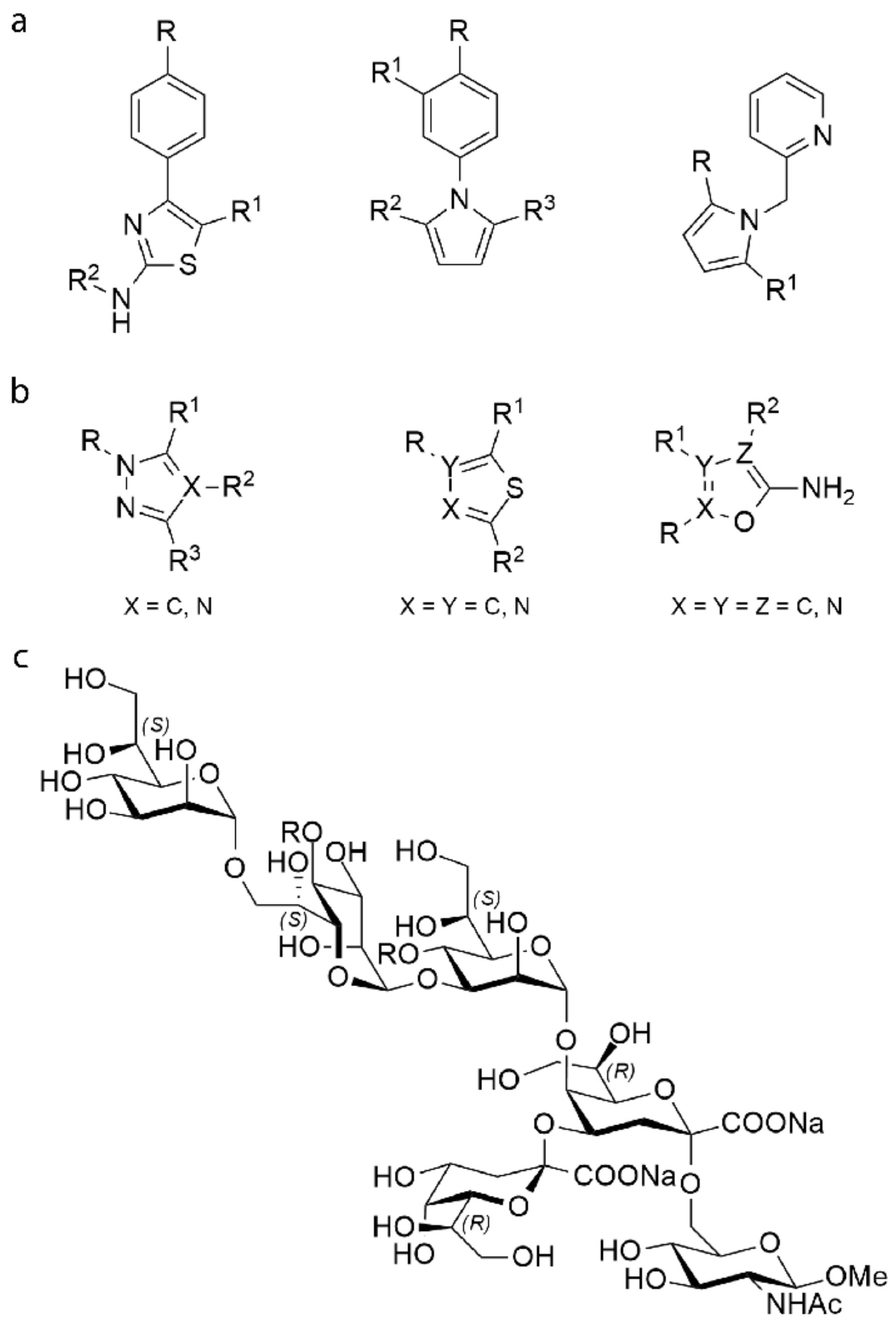
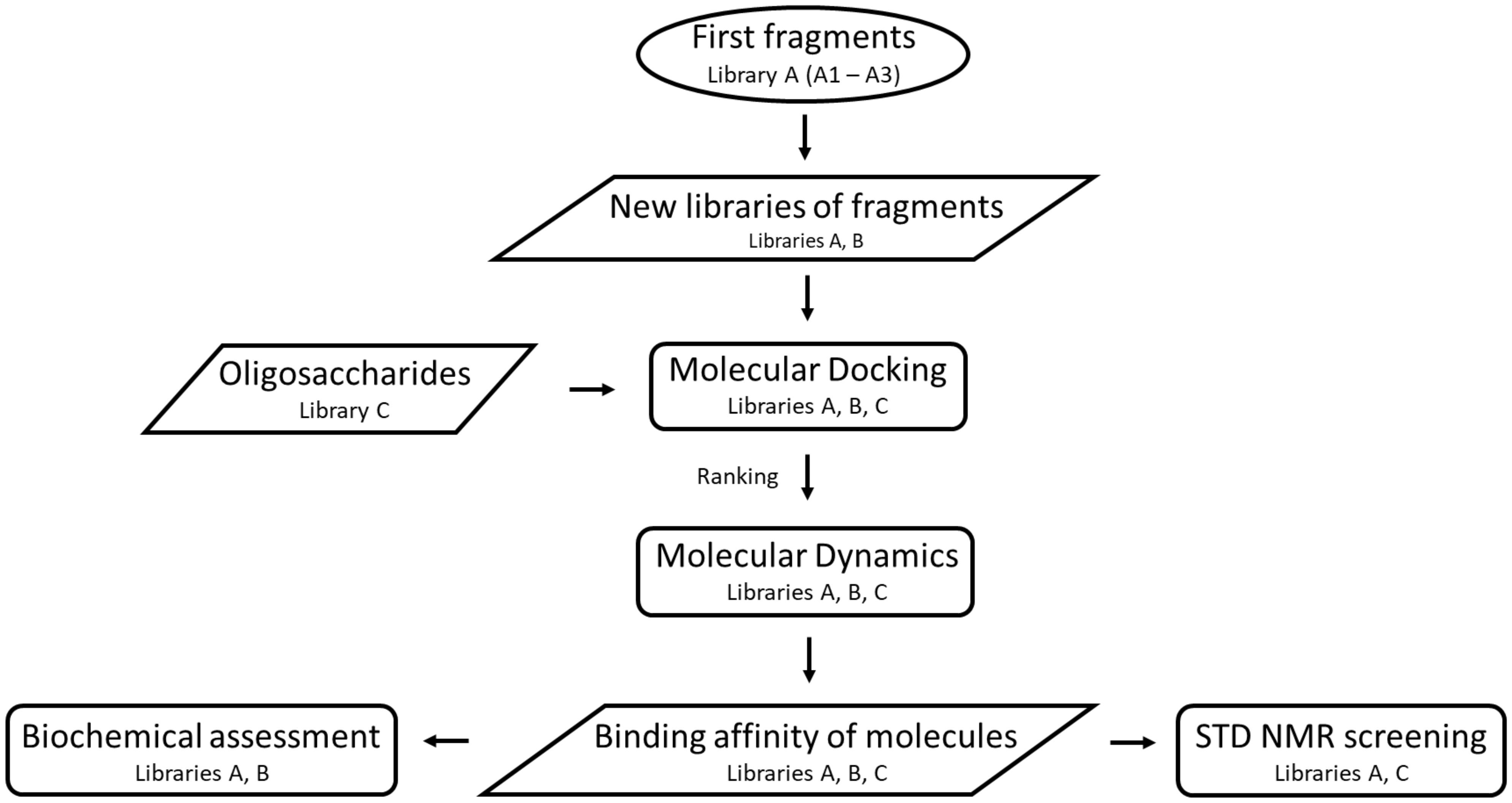
2.1. Construction of Libraries
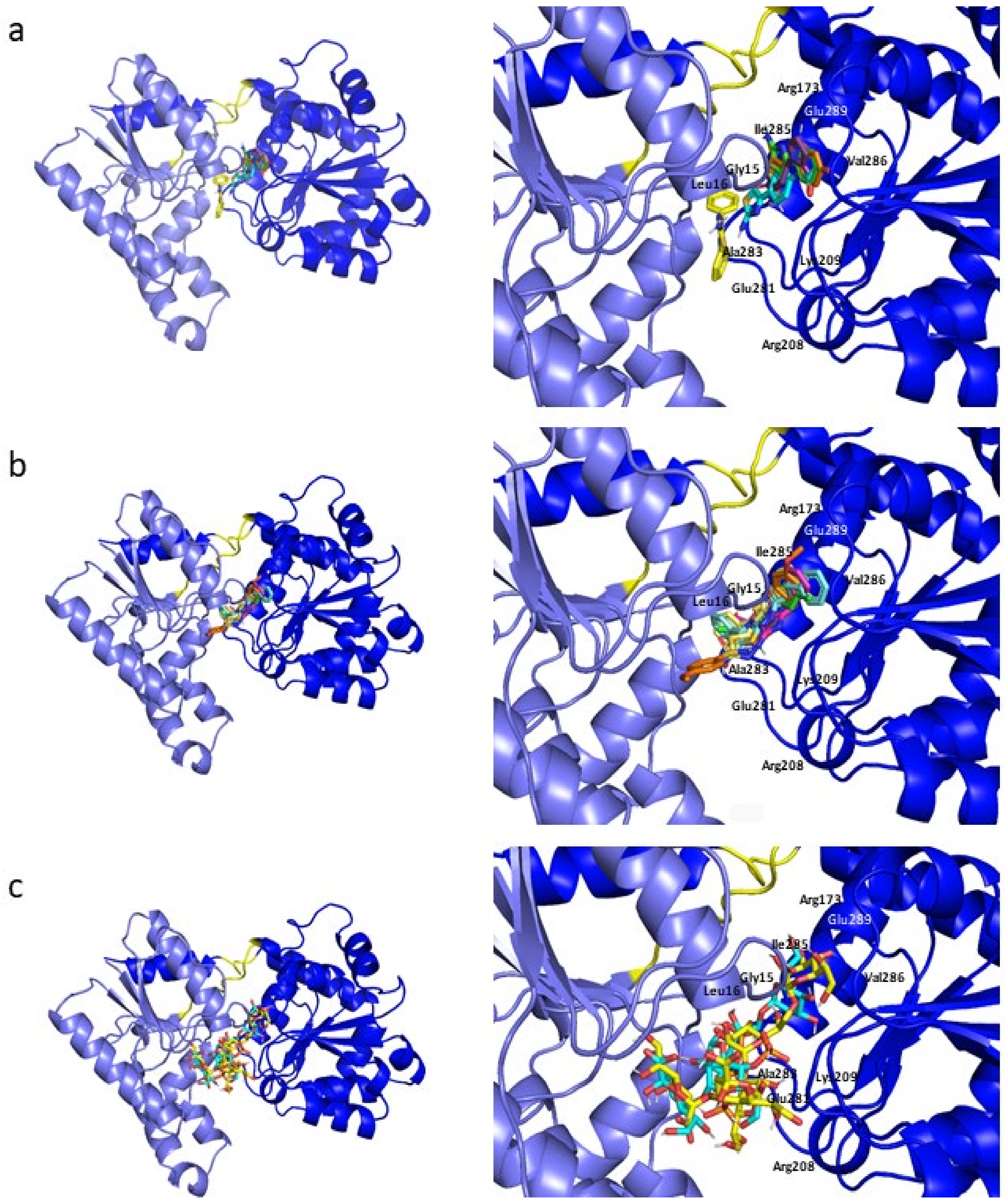
2.2. Molecular Docking
2.3. Molecular Dynamics Simulations
2.4. Binding Free Energy (BFE) Calculations
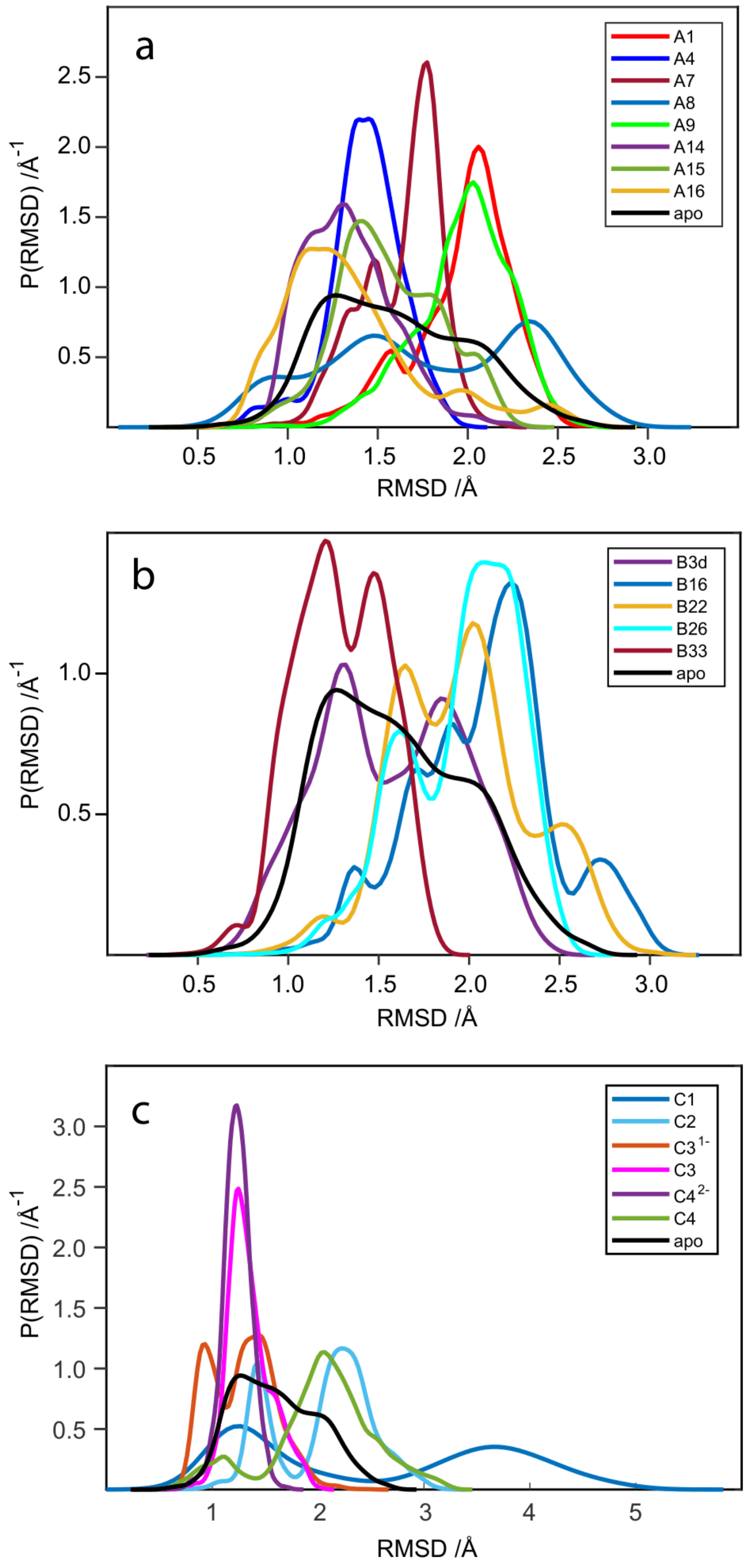
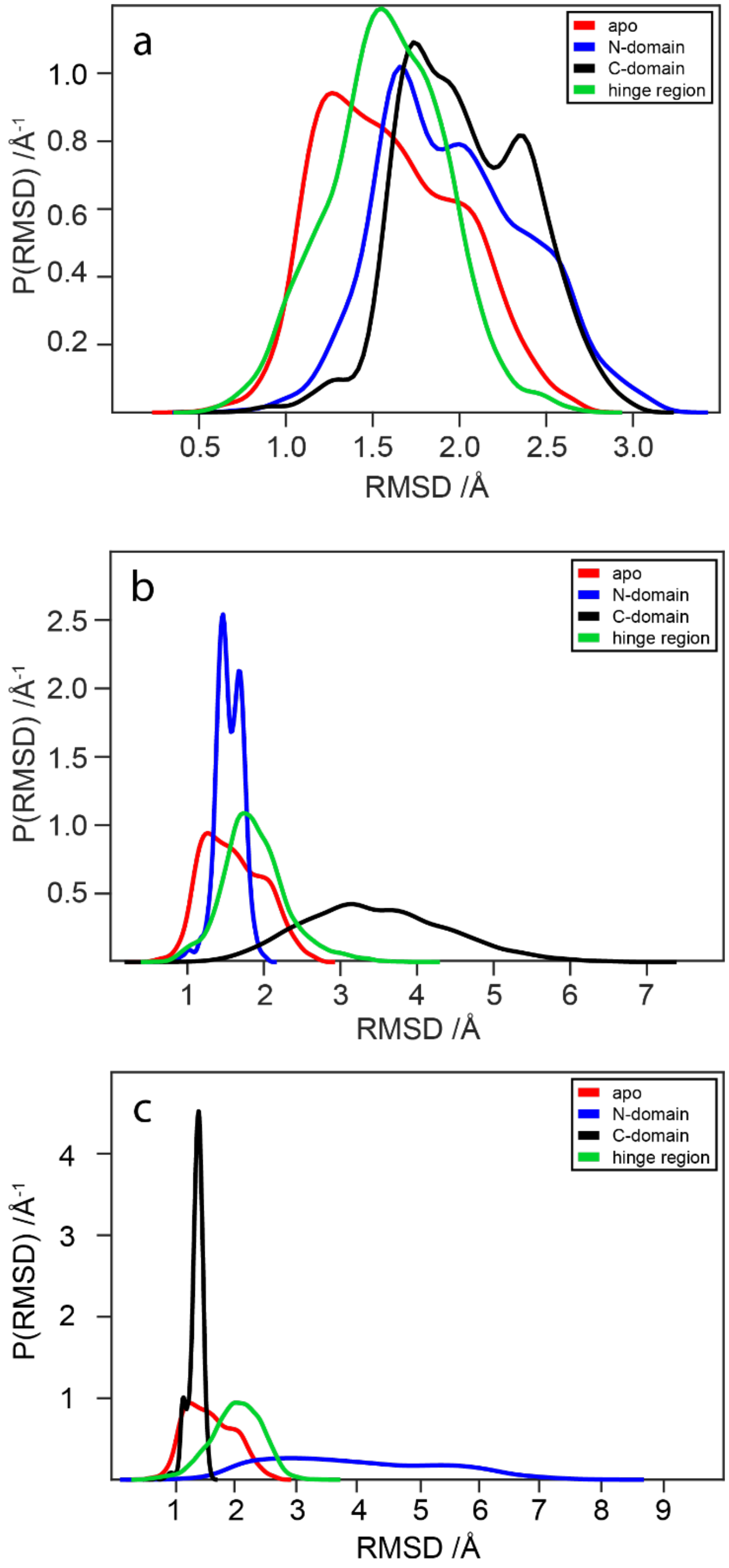
2.5. RMSD Analysis of WaaG Backbone
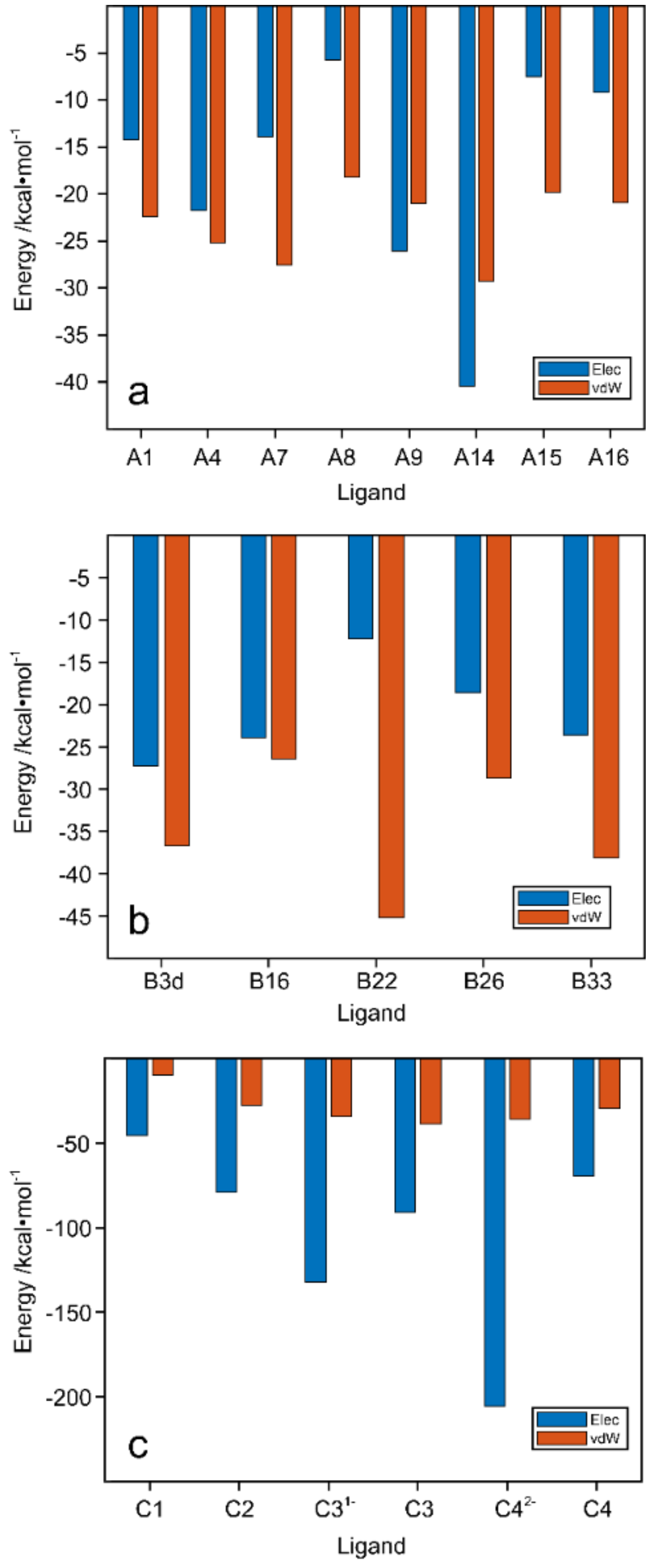
2.6. Electrostatic and van der Waals Contributions to WaaG/Ligand Binding
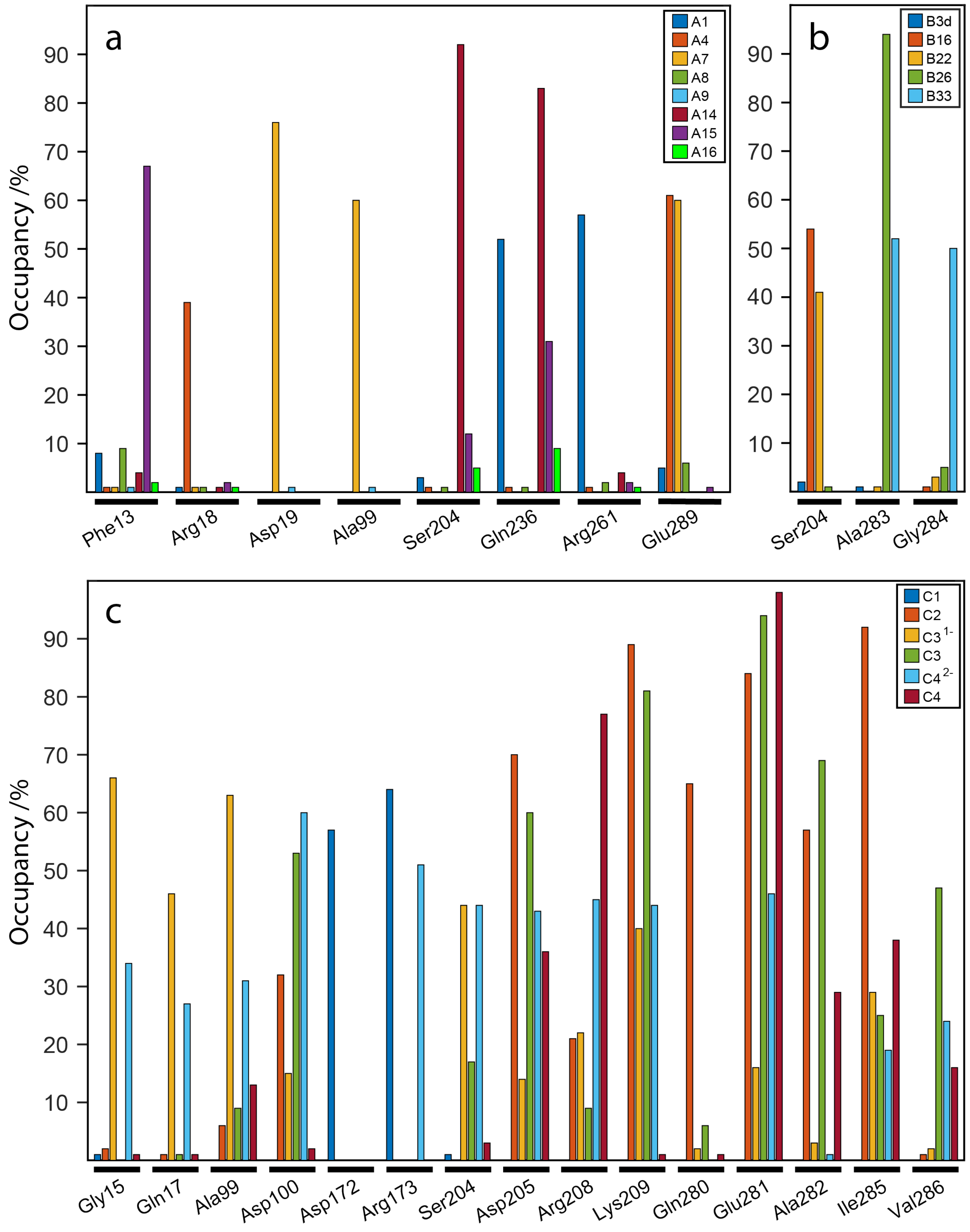
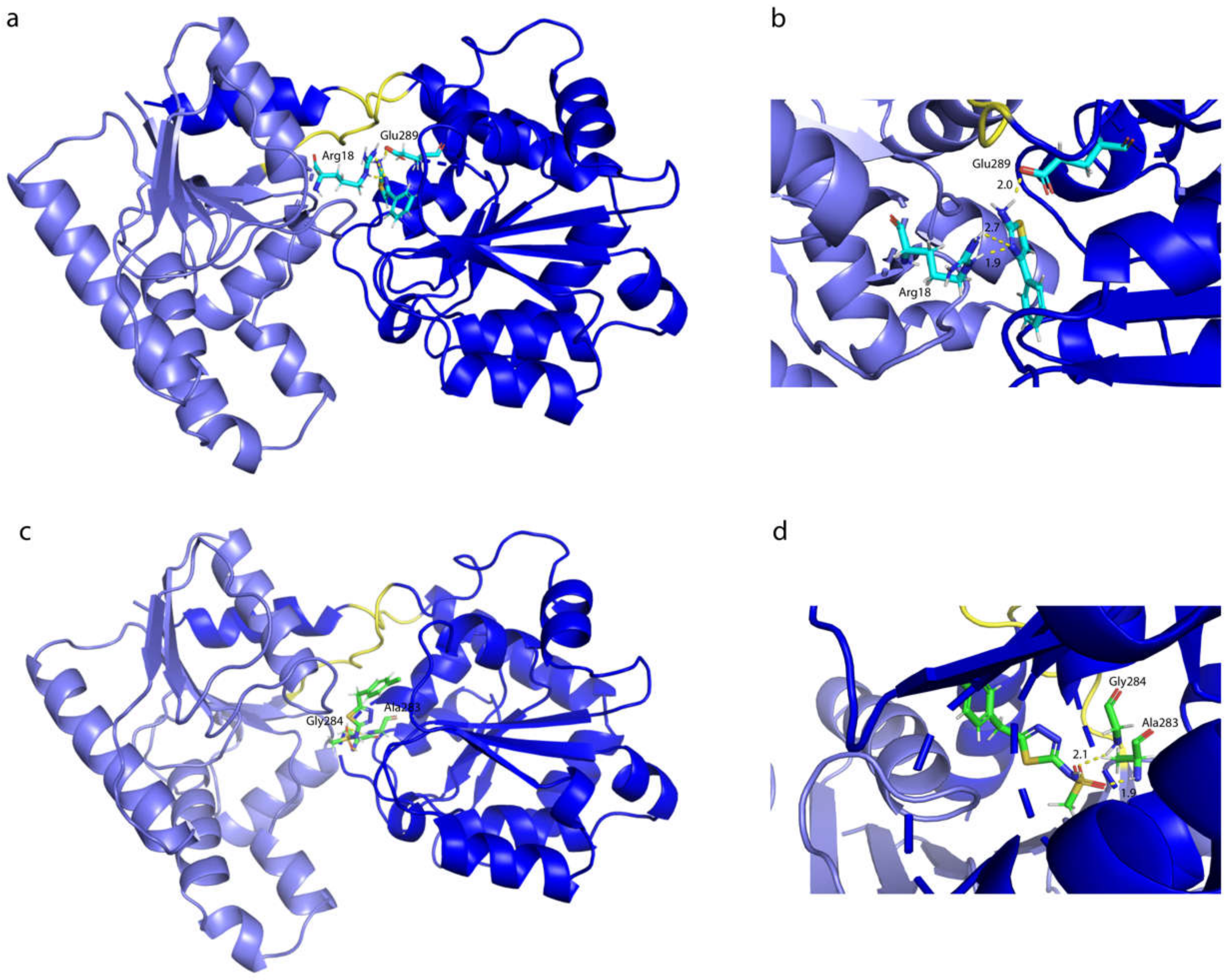
2.7. Influence of the H-Bonds for WaaG/Ligand Binding Affinity
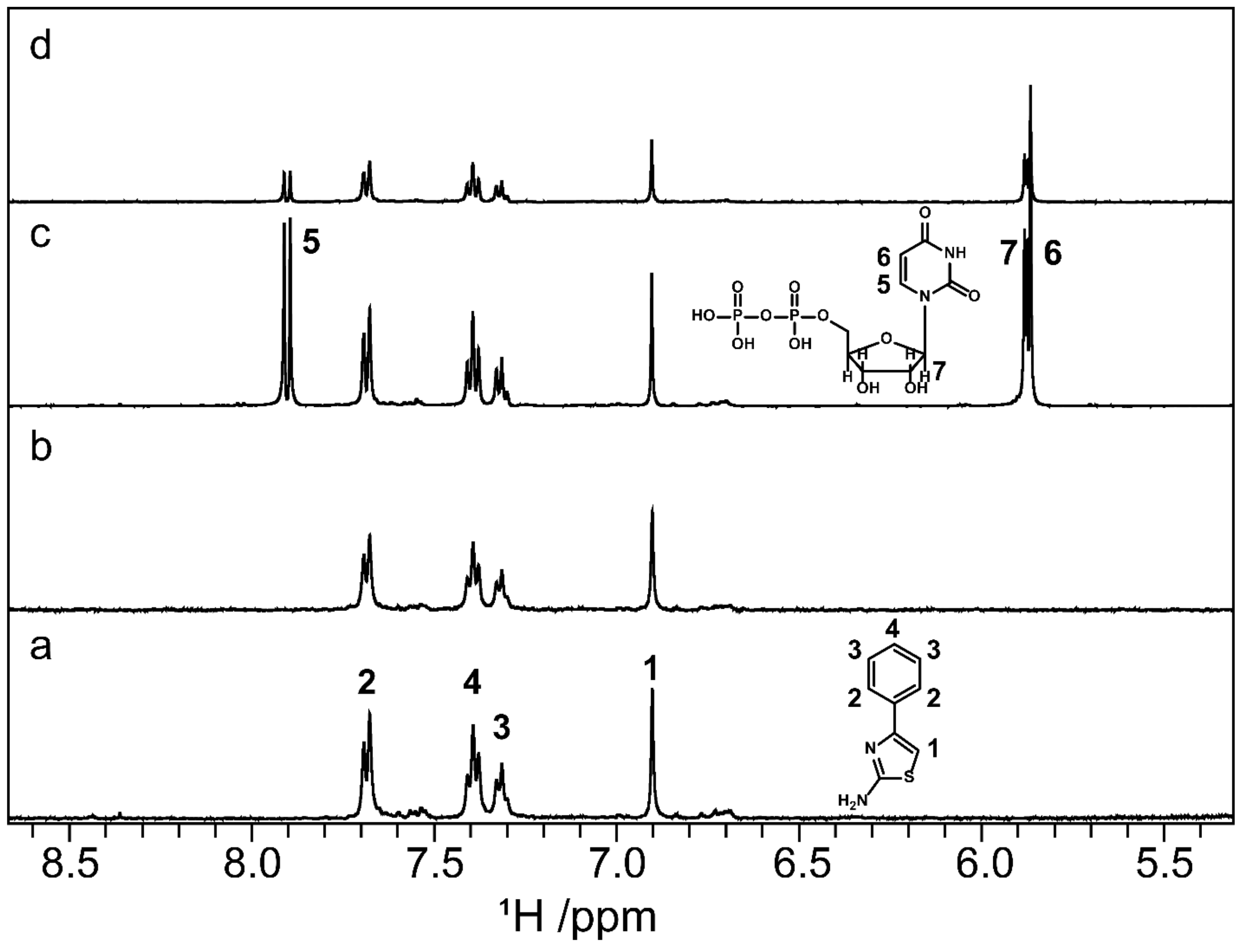
2.8. Screening by NMR Spectroscopy
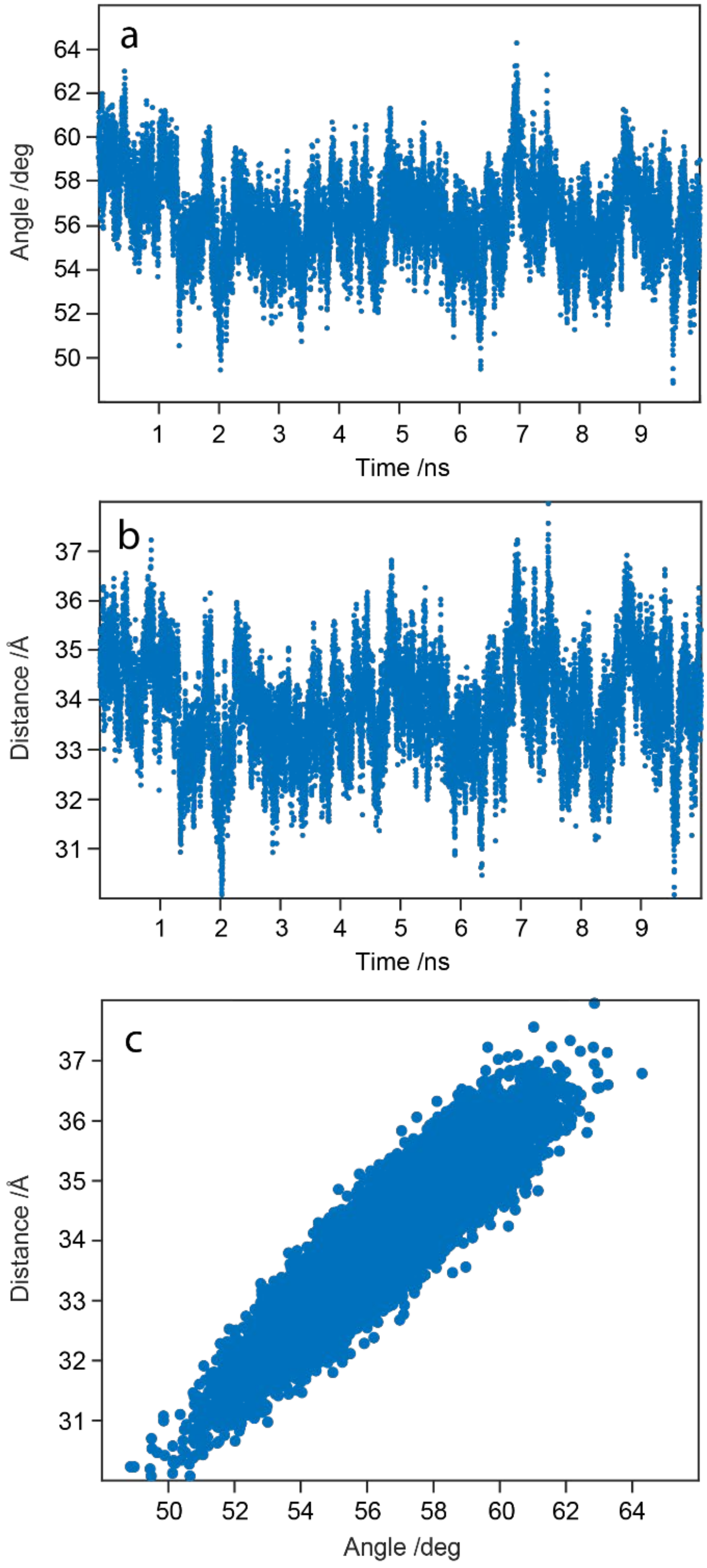
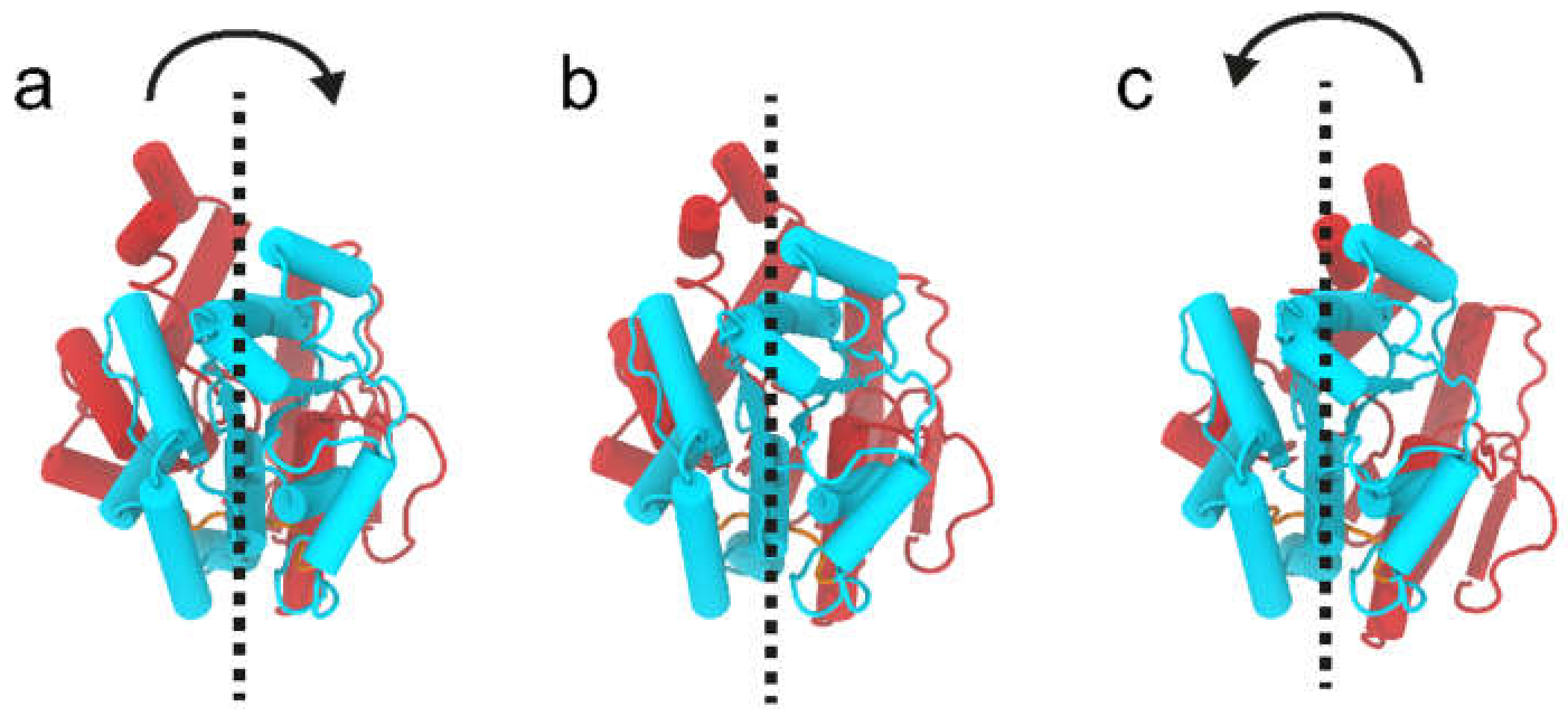
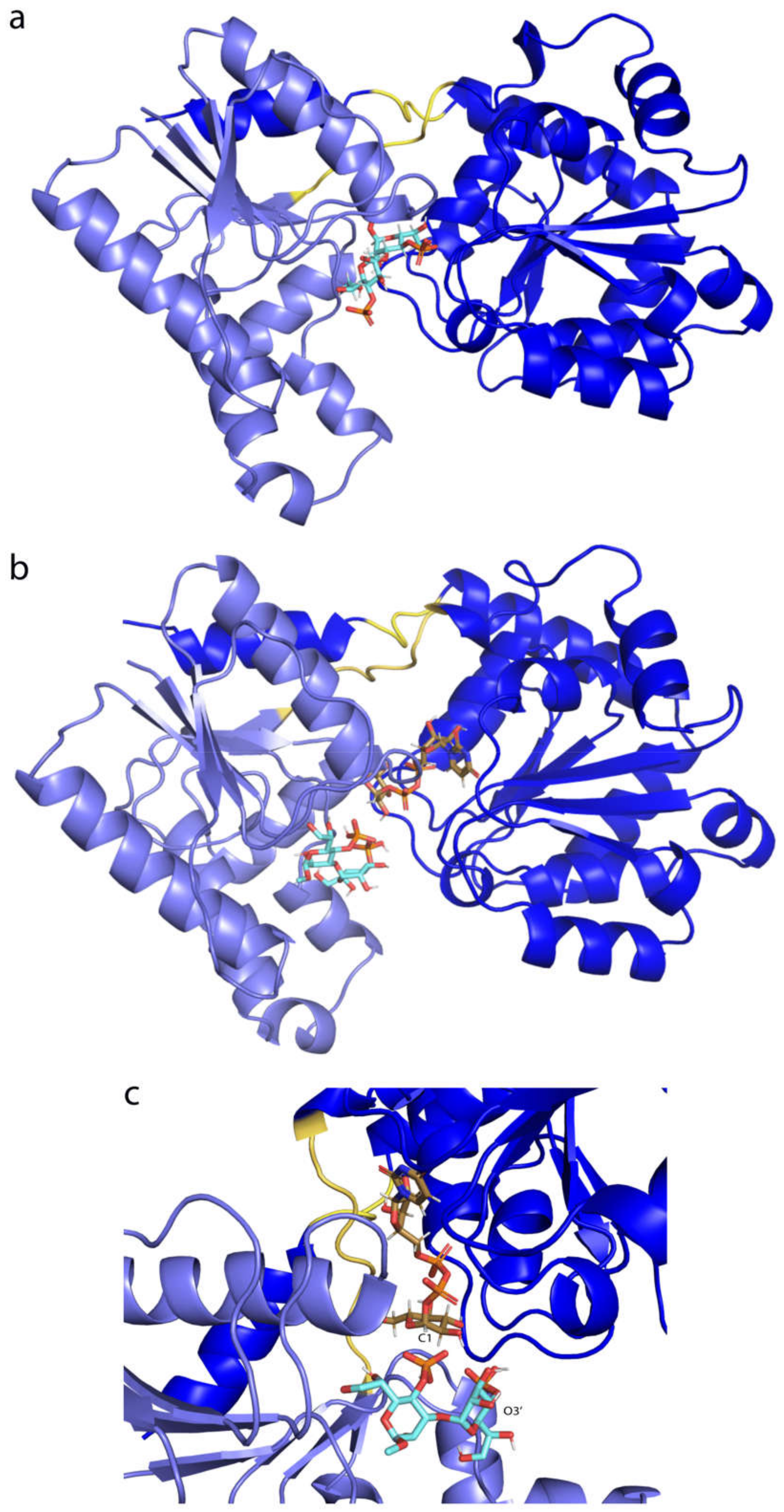
2.9. WaaG Has a Twisting-like Dynamics Involving N- and C-Domains
2.10. In Vitro WaaG Inhibition Assay
3. Discussion
4. Materials and Methods
4.1. General Experimental Methods
4.1.1. 2-Acetamido-5-methyl-4-phenyl-1,3-thiazol (A11)
4.1.2. 2-Formamido-5-methyl-4-phenyl-1,3-thiazol (A12)
4.1.3. 2-Amino-3,5-dimethyl-4-phenyl-1,3-thiazolium acetate (A13)
4.1.4. Methyl l-glycero-α-d-manno-heptopyranosyl-(1→3)-l-glycero-α-d-manno-heptopyranoside (C2)
4.2. Molecular Docking
4.3. Molecular Dynamics Simulations
4.4. Binding Free Energy Calculation
4.5. RMSD Analysis
4.6. MD Post-Processing: Interactions, Angle, Distance and Spearman Correlation
4.7. Saturation Transfer Difference NMR Spectroscopy
4.8. Sample Preparation for STD NMR Spectroscopy
4.9. Biochemical In Vitro Assessment/In Vitro WaaG Activity Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Croxen, M.A.; Finlay, B.B. Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 2010, 8, 26–38. [Google Scholar] [CrossRef]
- World Health Organization. Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug Resistant Bacterial Infections, Including Tuberculosis. 2017. Available online: https://apps.who.int/iris/handle/10665/311820 (accessed on 14 January 2022).
- Levin, S. The crisis in antibiotic resistance. Infect. Dis. Clin. Pract. 1993, 2, 53. [Google Scholar] [CrossRef]
- World Health Organization. Outbreaks of E. coli O104:H4 Infection. 2011. Available online: https://www.euro.who.int/en/countries/germany/outbreaks-of-e.-coli-o104h4-infection (accessed on 14 January 2022).
- World Health Organization. Antimicrobial Resistance. 2020. Available online: https://www.who.int/health-topics/antimicrobial-resistance (accessed on 14 January 2022).
- Bernal, P.; Molina-Santiago, C.; Daddaoua, A.; Llamas, M.A. Antibiotic adjuvants: Identification and clinical use. Microb. Biotechnol. 2013, 6, 445–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ejim, L.; Farha, M.A.; Falconer, S.B.; Wildenhain, J.; Coombes, B.K.; Tyers, M.; Brown, E.D.; Wright, G.D. Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nat. Chem. Biol. 2011, 7, 348–350. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.L.; Rossi, L.; de Pascale, G.; Wright, G.D. A forward chemical screen identifies antibiotic adjuvants in Escherichia coli. ACS Chem. Biol. 2012, 7, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
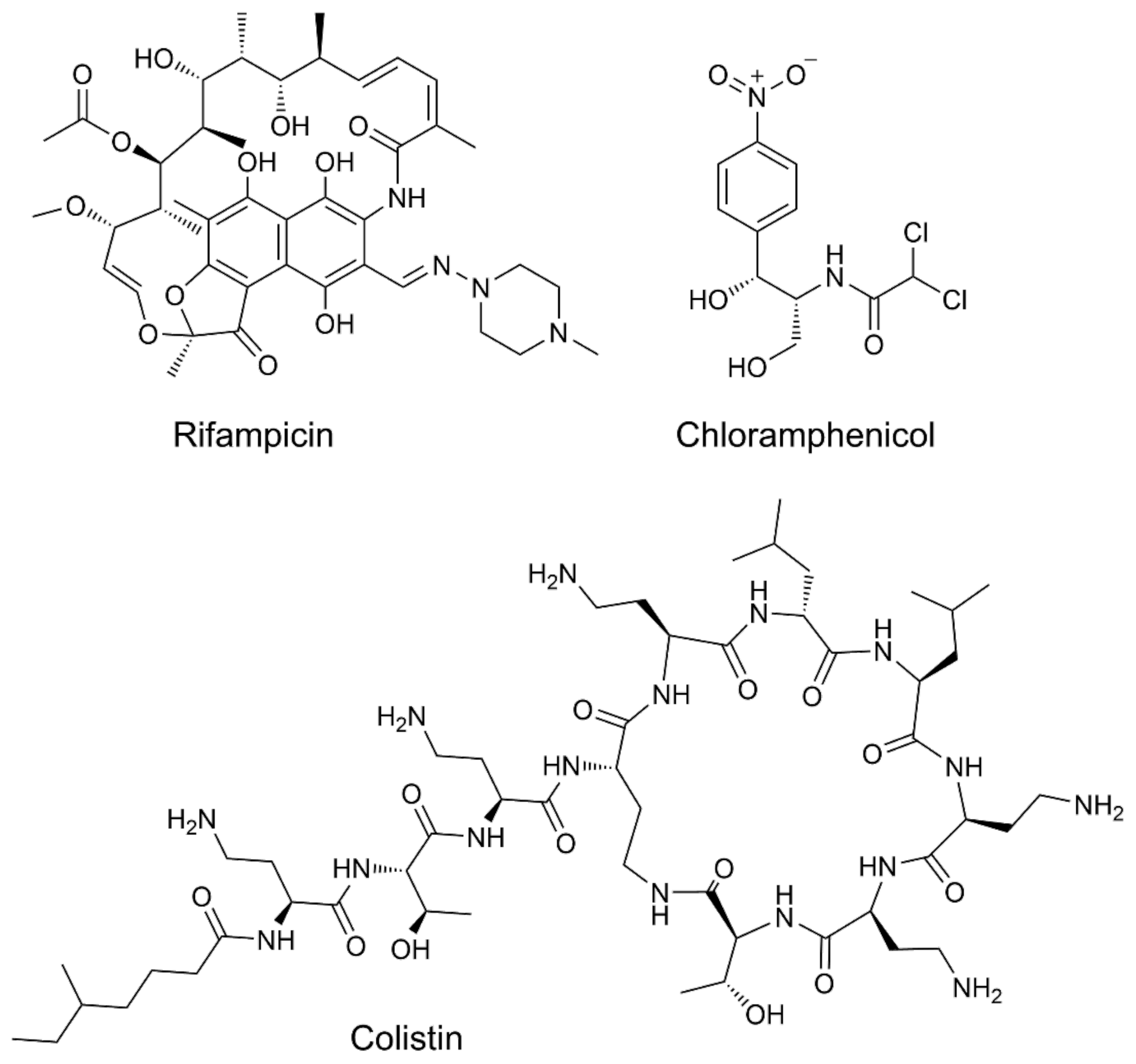
- Harris, T.L.; Worthington, R.J.; Hittle, L.E.; Zurawski, D.V.; Ernst, R.K.; Melander, C. Small molecule downregulation of PmrAB reverses lipid a modification and breaks colistin resistance. ACS Chem. Biol. 2014, 9, 122–127. [Google Scholar] [CrossRef]
- Worthington, R.J.; Melander, C. Combination approaches to combat multidrug-resistant bacteria. Trends Biotechnol. 2013, 31, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Delcour, A.H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta-Proteins Proteom. 2009, 1794, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Pagès, J.M.; James, C.E.; Winterhalter, M. The porin and the permeating antibiotic: A selective diffusion barrier in Gram-negative bacteria. Nat. Rev. Microbiol. 2008, 6, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Cox, G.; Wright, G.D. Intrinsic antibiotic resistance: Mechanisms, origins, challenges and solutions. Int. J. Med. Microbiol. 2013, 303, 287–292. [Google Scholar] [CrossRef]
- Bolla, J.M.; Alibert-Franco, S.; Handzlik, J.; Chevalier, J.; Mahamoud, A.; Boyer, G.; Kieć-Kononowicz, K.; Pags, J.M. Strategies for bypassing the membrane barrier in multidrug resistant Gram-negative bacteria. FEBS Lett. 2011, 585, 1682–1690. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Molecular basis of bacteria outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, N.; Wu, T.; Kahne, D.; Silhavy, T.J. Probing the barrier function of the outer membrane with chemical conditionality. ACS Chem. Biol. 2006, 1, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Raetz, C.R.H.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
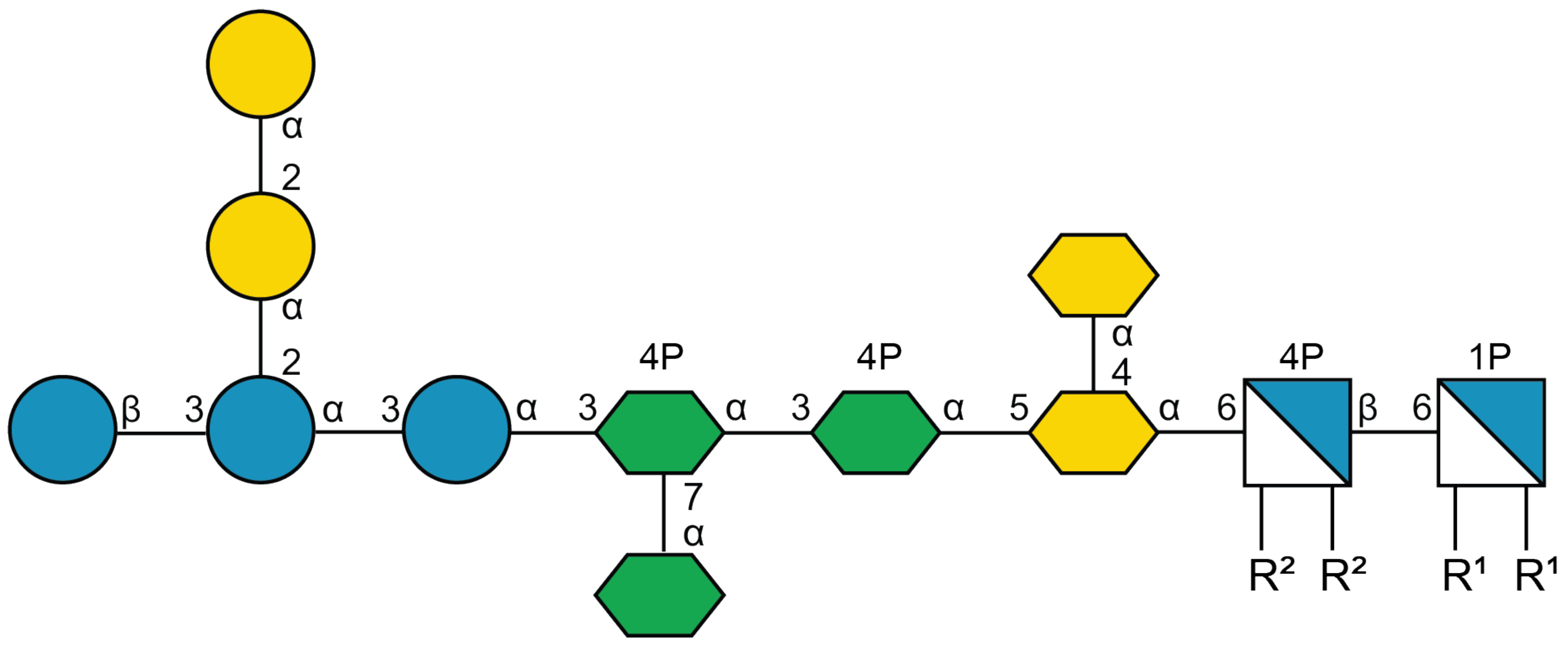
- Yethon, J.A.; Vinogradov, E.; Perry, M.B.; Whitfield, C. Mutation of the lipopolysaccharide core glycosyltransferase encoded by waaG destabilizes the outer membrane of Escherichia coli by interfering with core phosphorylation. J. Bacteriol. 2000, 182, 5620–5623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.; Tran, L.; Becket, E.; Lee, K.; Chinn, L.; Park, E.; Tran, K.; Miller, J.H. Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: Generating an antibiotic bar code. Antimicrob. Agents Chemother. 2010, 54, 1393–1403. [Google Scholar] [CrossRef] [Green Version]
- Vinogradov, E.V.; van der Drift, K.; Thomas-Oates, J.E.; Meshkov, S.; Brade, H.; Holst, O. The structures of the carbohydrate backbones of the lipopolysaccharides from Escherichia coli rough mutants F470 (R1 core type) and F576 (R2 core type). Eur. J. Biochem. 1999, 261, 629–639. [Google Scholar] [CrossRef]
- Neelamegham, S.; Aoki-Kinoshita, K.; Bolton, E.; Frank, M.; Lisacek, F.; Lütteke, T.; O’Boyle, N.; Packer, N.H.; Stanley, P.; Toukach, P.; et al. Updates to the Symbol Nomenclature for Glycans guidelines. Glycobiology 2019, 29, 620–624. [Google Scholar] [CrossRef]
- Landström, J.; Persson, K.; Rademacher, C.; Lundborg, M.; Wakarchuk, W.; Peters, T.; Widmalm, G. Small molecules containing hetero-bicyclic ring systems compete with UDP-Glc for binding to WaaG glycosyltransferase. Glycoconj. J. 2012, 29, 491–502. [Google Scholar] [CrossRef]
- Muheim, C.; Bakali, A.; Engström, O.; Wieslander, Å.; Daley, D.O.; Widmalm, G. Identification of a fragment-based scaffold that inhibits the glycosyltransferase WaaG from Escherichia coli. Antibiotics 2016, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Wagstaff, J.L.; Taylor, S.L.; Howard, M.J. Recent developments and applications of saturation transfer difference nuclear magnetic resonance (STD NMR) spectroscopy. Mol. Biosyst. 2013, 9, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.J. Fragment Screening by NMR. In Protein-Ligand Interactions Methods and Applications; Daviter, T., Johnson, C.M., McLaughlin, S.H., Williams, M.A., Eds.; Methods in Molecular Biology; Springer Nature: Cham, Switzerland, 2021; pp. 247–270. [Google Scholar]
- Meyer, B.; Peters, T. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew. Chem.-Int. Ed. 2003, 42, 864–890. [Google Scholar] [CrossRef] [PubMed]
- Walpole, S.; Monaco, S.; Nepravishta, R.; Angulo, J. STD NMR as a Technique for Ligand Screening and Structural Studies. Methods Enzymol. 2019, 615, 423–451. [Google Scholar] [CrossRef]
- Price, A.J.; Howard, S.; Cons, B.D. Fragment-based drug discovery and its application to challenging drug targets. Essays Biochem. 2017, 61, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Xie, X.Q.S. Computational Fragment-Based Drug Design: Current Trends, Strategies, and Applications. AAPS J. 2018, 20, 59. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Xu, Z. Using LeDock as a docking tool for computational drug design. IOP Conf. Ser. Earth Environ. Sci. 2019, 218, 012143. [Google Scholar] [CrossRef]
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef] [Green Version]
- Liebeschuetz, J.W.; Cole, J.C.; Korb, O. Pose prediction and virtual screening performance of GOLD scoring functions in a standardized test. J. Comput. Aided. Mol. Des. 2012, 26, 737–748. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Lundborg, M.; Ali, E.; Widmalm, G. An in silico virtual screening study for the design of norovirus inhibitors: Fragment-based molecular docking and binding free energy calculations. Carbohydr. Res. 2013, 378, 133–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
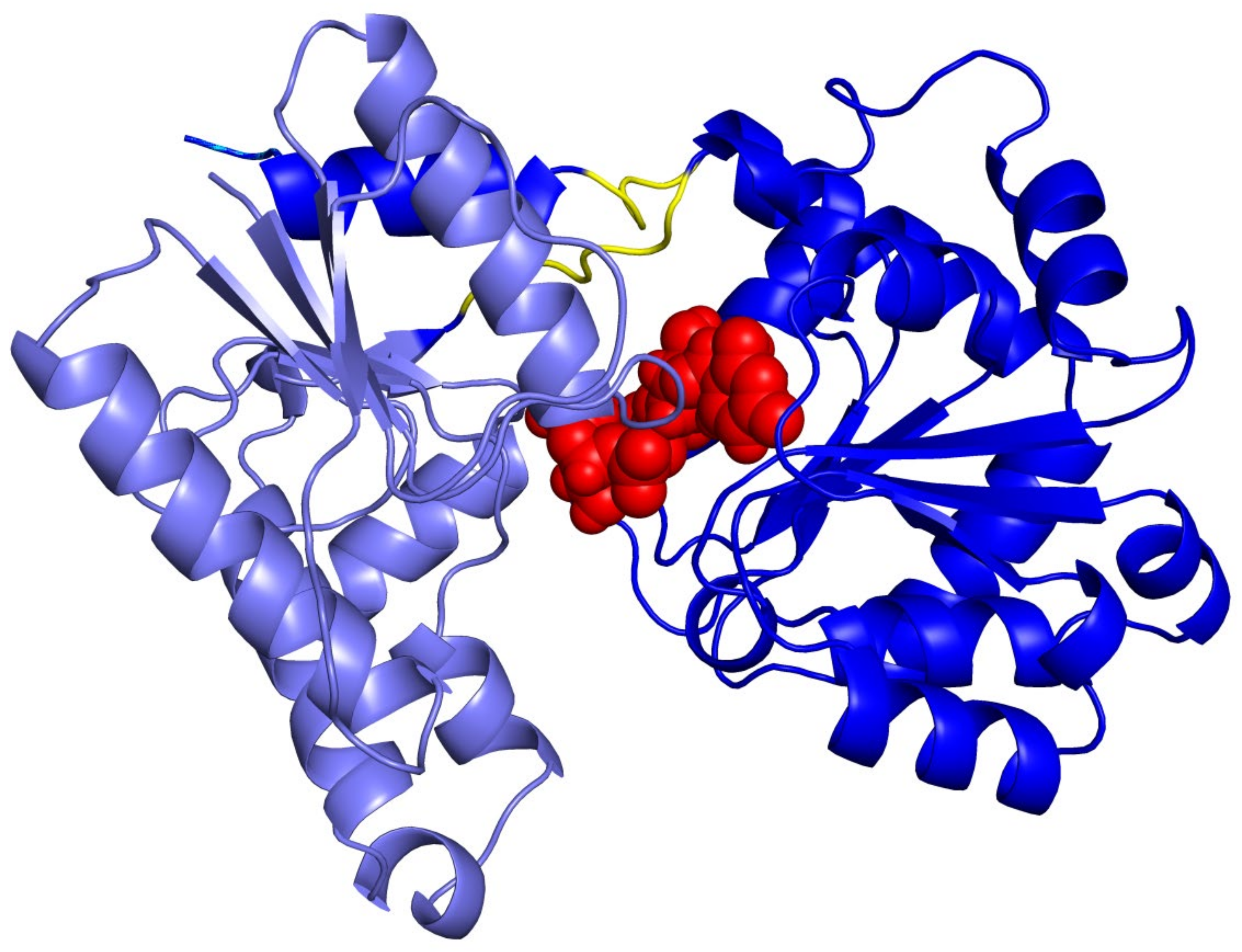
- Martinez-Fleites, C.; Proctor, M.; Roberts, S.; Bolam, D.N.; Gilbert, H.J.; Davies, G.J. Insights into the Synthesis of Lipopolysaccharide and Antibiotics through the Structures of Two Retaining Glycosyltransferases from Family GT4. Chem. Biol. 2006, 13, 1143–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Åqvist, J.; Medina, C.; Samuelsson, J.E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. Des. Sel. 1994, 7, 385–391. [Google Scholar] [CrossRef]
- Rifai, E.A.; van Dijk, M.; Geerke, D.P. Recent Developments in Linear Interaction Energy Based Binding Free Energy Calculations. Front. Mol. Biosci. 2020, 7, 114. [Google Scholar] [CrossRef]
- Jiang, L.; Sun, Q.; Li, L.; Lu, F.; Liu, F. Molecular Insights into the Inhibitory Effect of GV971 Components Derived from Marine Acidic Oligosaccharides against the Conformational Transition of Aβ42 Monomers. ACS Chem. Neurosci. 2021, 12, 3772–3784. [Google Scholar] [CrossRef] [PubMed]
- Landström, J.; Bergström, M.; Hamark, C.; Ohlson, S.; Widmalm, G. Combining weak affinity chromatography, NMR spectroscopy and molecular simulations in carbohydrate-lysozyme interaction studies. Org. Biomol. Chem. 2012, 10, 3019–3032. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem.-Int. Ed. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Laatikainen, R.; Niemitz, M.; Weber, U.; Sundeun, J.; Hassinen, T.; Vepsäläinen, J. General strategies for total-lineshape-type spectral analysis of NMR spectra using integral-transform iterator. J. Magn. Reson.-Ser. A 1996, 120, 1–10. [Google Scholar] [CrossRef]
- Rönnols, J.; Pendrill, R.; Fontana, C.; Hamark, C.; D’Ortoli, T.A.; Engström, O.; Ståhle, J.; Zaccheus, M.V.; Säwén, E.; Hahn, L.E.; et al. Complete 1H and 13C NMR chemical shift assignments of mono- to tetrasaccharides as basis for NMR chemical shift predictions of oligosaccharides using the computer program CASPER. Carbohydr. Res. 2013, 380, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Jansson, P.; Widmalm, G. NMR Studies of some 1,2- and 1,3-Linked Disaccharides. J. Chem. Soc. 1992, 2, 1085–1090. [Google Scholar] [CrossRef]
- Stanetty, C.; Walter, M.; Kosma, P. Convergent Synthesis of 4-O-Phosphorylated L-glycero-D-manno-Heptosyl Lipopolysaccharide Core Oligosaccharides Based on Regioselective Cleavage of a 6,7-O-Tetraisopropyldisiloxane-1,3-diyl Protecting Group. J. Org. Chem. 2014, 79, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Method 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-based Graphical User Interface for CHARMM. J. Chem. Theory Comput. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Cheng, X.; Islam, S.M.; Huang, L.; Rui, H.; Zhu, A.; Lee, H.S.; Qi, Y.; Han, W.; Vanommeslaeghe, K.; et al. CHARMM-GUI PDB Manipulator for Advanced Modeling and Simulations of Proteins Containing Non-standard Residues. Adv. Protein Chem. Struct. Biol. 2014, 96, 235–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Lee, J.; Jo, S.; Brooks, C.L., III; Lee, S.; Im, W. CHARMM-GUI Ligand Reader and Modeler for CHARMM Force Field Generation of Small Molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Song, K.C.; Desaire, H.; MacKerell, J.A.D.; Im, W. Glycan Reader: Automated Sugar Identification and Simulation Preparation for Carbohydrates and Glycoproteins. J. Comput. Chem. 2011, 32, 3135–3141. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-J.; Lee, J.; Patel, D.S.; Ma, H.; Lee, H.S.; Jo, S.; Im, W. Glycan Reader is Improved to Recognize Most Sugar Types and Chemical Modifications in the Protein Data Bank. Bioinformatics 2017, 33, 3051–3057. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Lee, J.; Qi, Y.; Kern, N.R.; Lee, H.S.; Jo, S.; Joung, I.; Joo, K.; Lee, J.; Im, W. CHARMM-GUI Glycan Modeler for Modeling and Simulation of Carbohydrates and Glycoconjugates. Glycobiology 2019, 29, 320–331. [Google Scholar] [CrossRef]
- Durell, S.R.; Brooks, B.R.; Ben-Naim, A. Solvent-induced forces between two hydrophilic groups. J. Phys. Chem. 1994, 98, 2198–2202. [Google Scholar] [CrossRef]
- Liu, H.; Hou, T. CaFE: A tool for binding affinity prediction using end-point free energy methods. Bioinformatics 2016, 32, 2216–2218. [Google Scholar] [CrossRef] [Green Version]
- Lecca, P.; Laurenzi, I.; Jordan, F. Deterministic chemical kinetics. Determ. Versus Stoch. Model. Biochem. Syst. Biol. 2013, 1–34. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LIGAND | ΔGbind | Calc KD (mM) | LIGAND | ΔGbind | Calc KD (mM) | LIGAND | ΔGbind | Calc KD (mM) |
|---|---|---|---|---|---|---|---|---|
| A7 | −5.65 | 0.11 | B33 | −6.29 | 0.04 | C42− | −9.14 | 0.0004 |
| A4 | −4.58 | 0.62 | B16 | −3.89 | 1.89 | C4 | −8.70 | 0.0008 |
| A9 | −3.34 | 4.55 | B22 | −3.18 | 5.97 | C2 | −5.97 | 0.07 |
| A15 | −1.73 | 61.10 | B3d | −2.19 | 29.09 | C3 | −4.21 | 1.13 |
| A14 | −0.81 | 269.26 | B26 | −1.03 | 190.48 | C1 | −4.20 | 1.15 |
| A1 | −0.64 | 354.70 | C31− | −0.72 | 313.08 | |||
| A8 | >0 | > 103 | ||||||
| A16 | >0 | > 103 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riu, F.; Ruda, A.; Engström, O.; Muheim, C.; Mobarak, H.; Ståhle, J.; Kosma, P.; Carta, A.; Daley, D.O.; Widmalm, G. A Lead-Based Fragment Library Screening of the Glycosyltransferase WaaG from Escherichia coli. Pharmaceuticals 2022, 15, 209. https://doi.org/10.3390/ph15020209
Riu F, Ruda A, Engström O, Muheim C, Mobarak H, Ståhle J, Kosma P, Carta A, Daley DO, Widmalm G. A Lead-Based Fragment Library Screening of the Glycosyltransferase WaaG from Escherichia coli. Pharmaceuticals. 2022; 15(2):209. https://doi.org/10.3390/ph15020209
Chicago/Turabian StyleRiu, Federico, Alessandro Ruda, Olof Engström, Claudio Muheim, Hani Mobarak, Jonas Ståhle, Paul Kosma, Antonio Carta, Daniel O. Daley, and Göran Widmalm. 2022. "A Lead-Based Fragment Library Screening of the Glycosyltransferase WaaG from Escherichia coli" Pharmaceuticals 15, no. 2: 209. https://doi.org/10.3390/ph15020209
APA StyleRiu, F., Ruda, A., Engström, O., Muheim, C., Mobarak, H., Ståhle, J., Kosma, P., Carta, A., Daley, D. O., & Widmalm, G. (2022). A Lead-Based Fragment Library Screening of the Glycosyltransferase WaaG from Escherichia coli. Pharmaceuticals, 15(2), 209. https://doi.org/10.3390/ph15020209