
Repurposing Antifungals for Host-Directed Antiviral Therapy?


Abstract
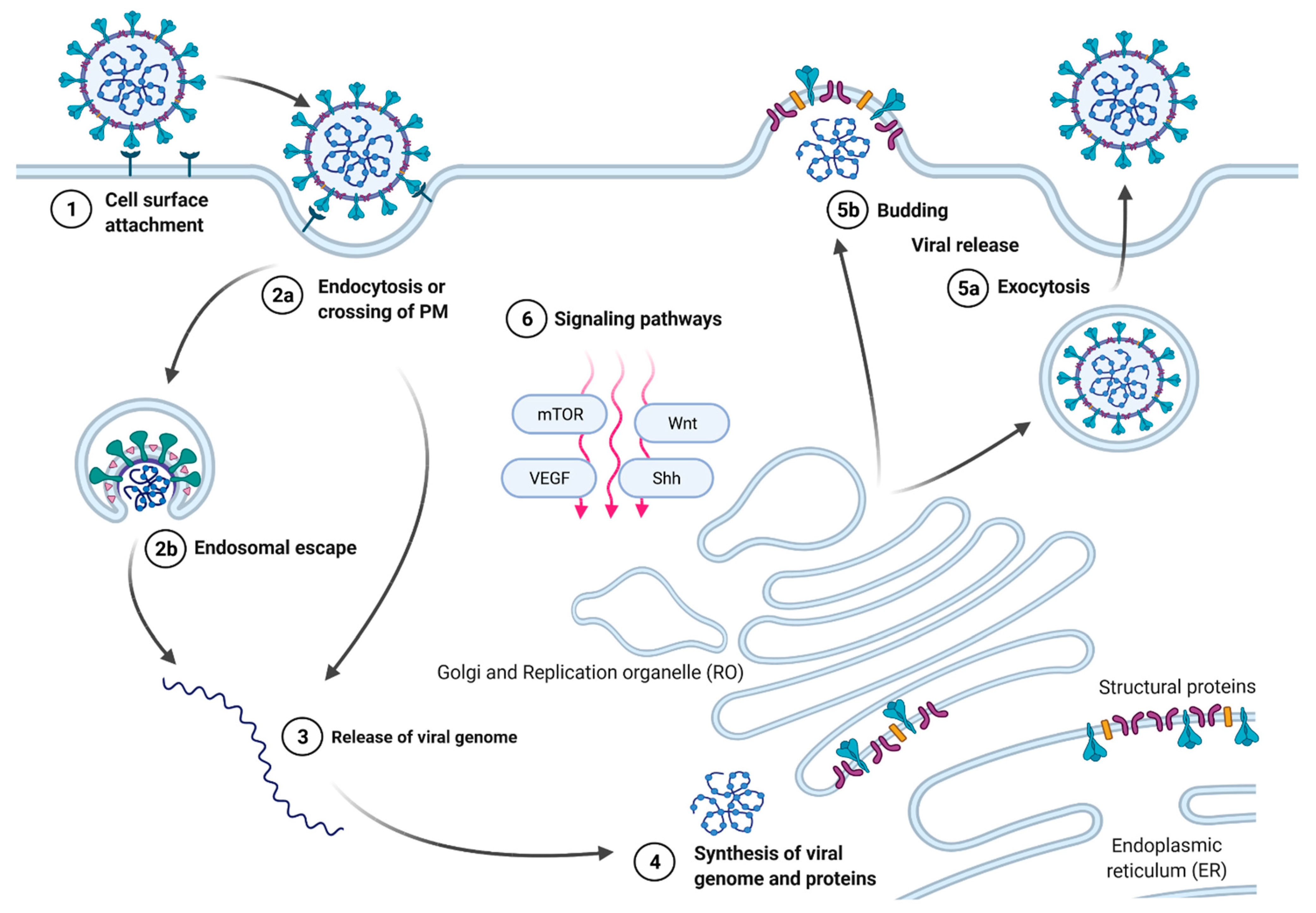
:1. Introduction
2. Antifungal Drugs
2.1. Polyenes—Disruptors of Fungal Membrane
2.2. Flucytosine—A Selective Inhibitor of Fungal Nuclear Acid Synthesis
2.3. Echinocandins—Noncompetitive Inhibitors of (1,3)-β-D-Glucan Synthase
2.4. Azoles—Inhibitors of Ergosterol Biosynthesis
2.4.1. Itraconazole Directly Interacts with the Endolysosomal Cholesterol Transporter NPC1
2.4.2. Itraconazole Interferes with OSBP and OSBP-Related Proteins (ORP) Functionality
2.4.3. Targeting mTOR Signaling via Itraconazole
2.4.4. Itraconazole, a Modulator of Hedgehog Signaling
2.4.5. Itraconazole and Its Inhibitory Effect on Wnt Signaling
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cascella, M.; Rajnik, M.; Cuomo, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation and Treatment Coronavirus (COVID-19); StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Kumar, S.; Çalışkan, D.M.; Janowski, J.; Faist, A.; Conrad, B.C.G.; Lange, J.; Ludwig, S.; Brunotte, L. Beyond Vaccines: Clinical Status of Prospective COVID-19 Therapeutics. Front. Immunol. 2021, 12, 752227. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Anand, U.; Jakhmola, S.; Indari, O.; Jha, H.C.; Chen, Z.S.; Tripathi, V.; Pérez de la Lastra, J.M. Potential Therapeutic Targets and Vaccine Development for SARS-CoV-2/COVID-19 Pandemic Management: A Review on the Recent Update. Front. Immunol. 2021, 12, 658519. [Google Scholar] [CrossRef]
- Martin, D.P.; Weaver, S.; Tegally, H.; San, J.E.; Shank, S.D.; Wilkinson, E.; Lucaci, A.G.; Giandhari, J.; Naidoo, S.; Pillay, Y.; et al. The emergence and ongoing convergent evolution of the SARS-CoV-2 N501Y lineages. Cell 2021, 184, 5189–5200.e7. [Google Scholar] [CrossRef]
- de Souza, U.J.B.; Dos Santos, R.N.; Campos, F.S.; Lourenço, K.L.; da Fonseca, F.G.; Spilki, F.R. High rate of mutational events in sars-cov-2 genomes across brazilian geographical regions, february 2020 to june 2021. Viruses 2021, 13, 1806. [Google Scholar] [CrossRef]
- Hurt, A.C.; Holien, J.K.; Parker, M.W.; Barr, I.G. Oseltamivir resistance and the H274Y neuraminidase mutation in seasonal, pandemic and highly pathogenic influenza viruses. Drugs 2009, 69, 2523–2531. [Google Scholar] [CrossRef]
- Trebbien, R.; Pedersen, S.S.; Vorborg, K.; Franck, K.T.; Fischer, T.K. Development of oseltamivir and zanamivir resistance in influenza A(H1N1)pdm09 virus, Denmark, 2014. Eurosurveillance 2017, 22, 30445. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H.E.; Dorhoi, A.; Hotchkiss, R.S.; Bartenschlager, R. Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discov. 2018, 17, 35–56. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Carolus, H.; Pierson, S.; Lagrou, K.; Van Dijck, P. Amphotericin b and other polyenes—Discovery, clinical use, mode of action and drug resistance. J. Fungi 2020, 6, 321. [Google Scholar] [CrossRef]
- Kim, H.; Kim, S.J.; Park, S.N.; Oh, J.W. Antiviral effect of amphotericin B on Japanese encephalitis virus replication. J. Microbiol. Biotechnol. 2004, 14, 121–127. [Google Scholar]
- Shiota, H.; Jones, B.R.; Schaffner, C.P. Anti-Herpes Simplex Virus (HSV) Effect of Amphotericin B Methyl Ester In Vivo. In Parasites, Fungi, and Viruses; Springer US: Boston, MA, USA, 1976; pp. 339–346. [Google Scholar]
- Konopka, K.; Guo, L.S.S.; Düzgüneş, N. Anti-HIV activity of amphotericin B-cholesteryl sulfate colloidal dispersion in vitro. Antiviral Res. 1999, 42, 197–209. [Google Scholar] [CrossRef]
- Jordan, G.W.; Humphreys, S.; Zee, Y.C. Effect of amphotericin B methyl ester on vesicular stomatitis virus morphology. Antimicrob. Agents Chemother. 1978, 13, 340–341. [Google Scholar] [CrossRef] [Green Version]
- Padda, I.S.; Parmar, M. Flucytosine; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Hüttel, W. Echinocandins: Structural diversity, biosynthesis, and development of antimycotics. Appl. Microbiol. Biotechnol. 2021, 105, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-J.; Liu, F.-C.; Yeh, C.-T.; Yang, C.M.; Lin, C.-C.; Lin, T.-Y.; Hsieh, P.-S.; Hu, M.-K.; Gong, Z.; Lu, J.-W. Micafungin is a novel anti-viral agent of chikungunya virus through multiple mechanisms. Antiviral Res. 2018, 159, 134–142. [Google Scholar] [CrossRef]
- Kim, C.; Kang, H.; Kim, D.E.; Song, J.H.; Choi, M.; Kang, M.; Lee, K.; Kim, H.S.; Shin, J.S.; Jeong, H.; et al. Antiviral activity of micafungin against enterovirus 71. Virol. J. 2016, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-C.; Lu, J.-W.; Yeh, C.-T.; Lin, T.-Y.; Liu, F.-C.; Ho, Y.-J. Micafungin Inhibits Dengue Virus Infection through the Disruption of Virus Binding, Entry, and Stability. Pharmaceuticals 2021, 14, 338. [Google Scholar] [CrossRef]
- Vergoten, G.; Bailly, C. In silico analysis of echinocandins binding to the main proteases of coronaviruses PEDV (3CLpro) and SARS-CoV-2 (Mpro). Silico Pharmacol. 2021, 9, 1–10. [Google Scholar] [CrossRef]
- Shafiei, M.; Peyton, L.; Hashemzadeh, M.; Foroumadi, A. History of the development of antifungal azoles: A review on structures, SAR, and mechanism of action. Bioorg. Chem. 2020, 104, 104240. [Google Scholar] [CrossRef]
- Schloer, S.; Brunotte, L.; Mecate-Zambrano, A.; Zheng, S.; Tang, J.; Ludwig, S.; Rescher, U. Drug synergy of combinatory treatment with remdesivir and the repurposed drugs fluoxetine and itraconazole effectively impairs SARS-CoV-2 infection in vitro. Br. J. Pharmacol. 2021, 178, 2339–2350. [Google Scholar] [CrossRef]
- Van Damme, E.; De Meyer, S.; Bojkova, D.; Ciesek, S.; Cinatl, J.; De Jonghe, S.; Jochmans, D.; Leyssen, P.; Buyck, C.; Neyts, J.; et al. In vitro activity of itraconazole against SARS-CoV-2. J. Med. Virol. 2021, 93, 4454–4460. [Google Scholar] [CrossRef] [PubMed]
- Schloer, S.; Goretzko, J.; Kühnl, A.; Brunotte, L.; Ludwig, S.; Rescher, U. The clinically licensed antifungal drug itraconazole inhibits influenza virus in vitro and in vivo. Emerg. Microbes Infect. 2019, 8, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flemming, A. Antivirals: Achilles heel of Ebola viral entry. Nat. Rev. Drug Discov. 2011, 10, 731. [Google Scholar] [CrossRef]
- Head, S.A.; Shi, W.Q.; Yang, E.J.; Nacev, B.A.; Hong, S.Y.; Pasunooti, K.K.; Li, R.J.; Shim, J.S.; Liu, J.O. Simultaneous targeting of NPC1 and VDAC1 by itraconazole leads to synergistic inhibition of MTOR signaling and angiogenesis. ACS Chem. Biol. 2017, 12, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Kummer, S.; Lander, A.; Goretzko, J.; Kirchoff, N.; Rescher, U.; Schloer, S. Pharmacologically induced endolysosomal cholesterol imbalance through clinically licensed drugs itraconazole and fluoxetine impairs Ebola virus infection in vitro. Emerg. Microbes Infect. 2022, 11, 195–207. [Google Scholar] [CrossRef]
- Rhoden, E.; Ng, T.F.F.; Campagnoli, R.; Nix, W.A.; Konopka-Anstadt, J.; Selvarangan, R.; Briesach, L.; Oberste, M.S.; Weldon, W.C. Antifungal triazole posaconazole targets an early stage of the parechovirus A3 life cycle. Antimicrob. Agents Chemother. 2020, 64, e02372-19. [Google Scholar] [CrossRef] [Green Version]
- Meutiawati, F.; Bezemer, B.; Strating, J.R.P.M.; Overheul, G.J.; Žusinaite, E.; van Kuppeveld, F.J.M.; van Cleef, K.W.R.; van Rij, R.P. Posaconazole inhibits dengue virus replication by targeting oxysterol-binding protein. Antiviral Res. 2018, 157, 68–79. [Google Scholar] [CrossRef]
- Strating, J.R.P.M.; van der Linden, L.; Albulescu, L.; Bigay, J.; Arita, M.; Delang, L.; Leyssen, P.; van der Schaar, H.M.; Lanke, K.H.W.; Thibaut, H.J.; et al. Itraconazole Inhibits Enterovirus Replication by Targeting the Oxysterol-Binding Protein. Cell Rep. 2015, 10, 600–615. [Google Scholar] [CrossRef] [Green Version]
- Mercorelli, B.; Luganini, A.; Celegato, M.; Palù, G.; Gribaudo, G.; Lepesheva, G.I.; Loregian, A. The clinically approved antifungal drug posaconazole inhibits human cytomegalovirus replication. Antimicrob. Agents Chemother. 2020, 64, e00056-20. [Google Scholar] [CrossRef]
- Mesa-Arango, A.C.; Scorzoni, L.; Zaragoza, O. It only takes one to do many jobs: Amphotericin B as antifungal and immunomodulatory drug. Front. Microbiol. 2012, 3, 286. [Google Scholar] [CrossRef] [Green Version]
- Malewicz, B.; Momsen, M.; Jenkin, H.M.; Borowski, E. Potentiation of antiviral activity of acyclovir by polyene macrolide antibiotics. Antimicrob. Agents Chemother. 1984, 25, 772–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marak, B.N.; Dowarah, J.; Khiangte, L.; Singh, V.P. Step toward repurposing drug discovery for COVID-19 therapeutics through in silico approach. Drug Dev. Res. 2021, 82, 374–392. [Google Scholar] [CrossRef] [PubMed]
- Di Mambro, T.; Guerriero, I.; Aurisicchio, L.; Magnani, M.; Marra, E. The yin and yang of current antifungal therapeutic strategies: How can we harness our natural defenses? Front. Pharmacol. 2019, 10, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona, E.M.; Limper, A.H. Overview of Treatment Approaches for Fungal Infections. Clin. Chest Med. 2017, 38, 393–402. [Google Scholar] [CrossRef]
- Gintjee, T.J.; Donnelley, M.A.; Thompson, G.R. Aspiring Antifungals: Review of Current Antifungal Pipeline Developments. J. Fungi 2020, 6, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Daele, R.; Spriet, I.; Wauters, J.; Maertens, J.; Mercier, T.; Van Hecke, S.; Brüggemann, R. Antifungal drugs: What brings the future? Med. Mycol. 2019, 57, S328–S343. [Google Scholar] [CrossRef] [Green Version]
- Sheehan, D.J.; Hitchcock, C.A.; Sibley, C.M. Current and emerging azole antifungal agents. Clin. Microbiol. Rev. 1999, 12, 40–79. [Google Scholar] [CrossRef] [Green Version]
- Francois, I.; Cammue, B.; Borgers, M.; Ausma, J.; Dispersyn, G.; Thevissen, K. Azoles: Mode of Antifungal Action and Resistance Development. Effect of Miconazole on Endogenous Reactive Oxygen Species Production in Candida albicans. Antiinfect. Agents Med. Chem. 2006, 5, 3–13. [Google Scholar] [CrossRef]
- Lass-Flörl, C. Triazole antifungal agents in invasive fungal infections: A comparative review. Drugs 2011, 71, 2405–2419. [Google Scholar] [CrossRef]
- Kelly, S.L.; Lamb, D.C.; Baldwin, B.C.; Corran, A.J.; Kelly, D.E. Characterization of Saccharomyces cerevisiae CYP61, Sterol Δ22-Desaturase, and Inhibition by Azole Antifungal Agents. J. Biol. Chem. 1997, 272, 9986–9988. [Google Scholar] [CrossRef] [Green Version]
- Minnebruggen, G.V.; Francois, I.E.J.A.; Cammue, B.P.A.; Thevissen, K.; Vroome, V.; Borgers, M.; Shroot, B. A General Overview on Past, Present and Future Antimycotics. Open Mycol. J. 2010, 4, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Warrilow, A.G.; Parker, J.E.; Kelly, D.E.; Kelly, S.L. Azole Affinity of Sterol 14-Demethylase (CYP51) Enzymes from Candida albicans and Homo sapiens. Antimicrob. Agents Chemother. 2013, 57, 1352–1360. [Google Scholar] [CrossRef] [Green Version]
- Munayyer, H.K.; Mann, P.A.; Chau, A.S.; Yarosh-Tomaine, T.; Greene, J.R.; Hare, R.S.; Heimark, L.; Palermo, R.E.; Loebenberg, D.; McNicholas, P.M. Posaconazole is a potent inhibitor of sterol 14α-demethylation in yeasts and molds. Antimicrob. Agents Chemother. 2004, 48, 3690–3696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinh, M.N.; Lu, F.; Li, X.; Das, A.; Liang, Q.; De Brabander, J.K.; Brown, M.S.; Goldstein, J.L.; Chang, T.-Y.; Maxfield, F.R. Triazoles inhibit cholesterol export from lysosomes by binding to NPC1. Proc. Natl. Acad. Sci. USA 2017, 114, 89–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanier, M.; Millat, G. Niemann-Pick disease type C. Clin. Genet. 2003, 64, 269–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, S.R. NPC intracellular cholesterol transporter 1 (NPC1)-mediated cholesterol export from lysosomes. J. Biol. Chem. 2019, 294, 1706–1709. [Google Scholar] [CrossRef] [Green Version]
- Herbert, A.S.; Davidson, C.; Kuehne, A.I.; Bakken, R.; Braigen, S.Z.; Gunn, K.E.; Whelan, S.P.; Brummelkamp, T.R.; Twenhafel, N.A.; Chandran, K.; et al. Niemann-pick C1 is essential for ebolavirus replication and pathogenesis in vivo. MBio 2015, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Côté, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar] [CrossRef]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.H.; Obernosterer, G.; Raaben, M.; Herbert, A.S.; Deffieu, M.S.; Krishnan, A.; Ndungo, E.; Sandesara, R.G.; Carette, J.E.; Kuehne, A.I.; et al. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 2012, 31, 1947–1960. [Google Scholar] [CrossRef] [Green Version]
- Coleman, E.M.; Walker, T.N.; Hildreth, J.E.K. Loss of Niemann Pick type C proteins 1 and 2 greatly enhances HIV infectivity and is associated with accumulation of HIV Gag and cholesterol in late endosomes/lysosomes. Virol. J. 2012, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Leao, I.C.; Coleman, E.M.; Broughton, R.S.; Hildreth, J.E.K. Deficiency of Niemann-Pick Type C-1 Protein Impairs Release of Human Immunodeficiency Virus Type 1 and Results in Gag Accumulation in Late Endosomal/Lysosomal Compartments. J. Virol. 2009, 83, 7982–7995. [Google Scholar] [CrossRef] [Green Version]
- van ′t Wout, A.B.; Swain, J.V.; Schindler, M.; Rao, U.; Pathmajeyan, M.S.; Mullins, J.I.; Kirchhoff, F. Nef Induces Multiple Genes Involved in Cholesterol Synthesis and Uptake in Human Immunodeficiency Virus Type 1-Infected T Cells. J. Virol. 2005, 79, 10053–10058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, F.; Liang, Q.; Abi-Mosleh, L.; Das, A.; de Brabander, J.K.; Goldstein, J.L.; Brown, M.S. Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. Elife 2015, 4, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liscum, L.; Faust, J.R. The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-β-[2-(diethylamino)ethoxy]androst-5-en-17-one. J. Biol. Chem. 1989, 264, 11796–11806. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Cenedella, R.J.; Jacob, R.; Borchman, D.; Tang, D.; Neely, A.R.; Samadi, A.; Mason, R.P.; Sexton, P. Direct perturbation of lens membrane structure may contribute to cataracts caused by U18666A, an oxidosqualene cyclase inhibitor. J. Lipid Res. 2004, 45, 1232–1241. [Google Scholar] [CrossRef] [Green Version]
- Liesenborghs, L.; Spriet, I.; Jochmans, D.; Belmans, A.; Gyselinck, I.; Teuwen, L.-A.; ter Horst, S.; Dreesen, E.; Geukens, T.; Engelen, M.M.; et al. Itraconazole for COVID-19: Preclinical studies and a proof-of-concept randomized clinical trial. EBioMedicine 2021, 66, 103288. [Google Scholar] [CrossRef]
- Ridgway, N.D.; Dawson, P.A.; Ho, Y.K.; Brown, M.S.; Goldstein, J.L. Translocation of oxysterol binding protein to Golgi apparatus triggered by ligand binding. J. Cell Biol. 1992, 116, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Mesmin, B.; Bigay, J.; Moser von Filseck, J.; Lacas-Gervais, S.; Drin, G.; Antonny, B. A Four-Step Cycle Driven by PI(4)P Hydrolysis Directs Sterol/PI(4)P Exchange by the ER-Golgi Tether OSBP. Cell 2013, 155, 830–843. [Google Scholar] [CrossRef] [Green Version]
- Weber-Boyvat, M.; Zhong, W.; Yan, D.; Olkkonen, V.M. Oxysterol-binding proteins: Functions in cell regulation beyond lipid metabolism. Biochem. Pharmacol. 2013, 86, 89–95. [Google Scholar] [CrossRef]
- Raychaudhuri, S.; Prinz, W.A. The diverse functions of oxysterol-binding proteins. Annu. Rev. Cell Dev. Biol. 2010, 26, 157–177. [Google Scholar] [CrossRef] [Green Version]
- Hsu, N.Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arita, M.; Kojima, H.; Nagano, T.; Okabe, T.; Wakita, T.; Shimizu, H. Phosphatidylinositol 4-Kinase III Beta Is a Target of Enviroxime-Like Compounds for Antipoliovirus Activity. J. Virol. 2011, 85, 2364–2372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arita, M. Phosphatidylinositol-4 kinase III beta and oxysterol-binding protein accumulate unesterified cholesterol on poliovirus-induced membrane structure. Microbiol. Immunol. 2014, 58, 239–256. [Google Scholar] [CrossRef]
- Roulin, P.S.; Lötzerich, M.; Torta, F.; Tanner, L.B.; Van Kuppeveld, F.J.M.; Wenk, M.R.; Greber, U.F. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe 2014, 16, 677–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; Van Der Welle, R.E.N.; Mydock-McGrane, L.; Jiang, X.; Van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.Y.; Davis, O.B.; Shin, H.R.; Zhang, J.; Berdan, C.A.; Jiang, X.; Counihan, J.L.; Ory, D.S.; Nomura, D.K.; Zoncu, R. ER–lysosome contacts enable cholesterol sensing by mTORC1 and drive aberrant growth signalling in Niemann–Pick type C. Nat. Cell Biol. 2019, 21, 1206–1218. [Google Scholar] [CrossRef]
- Karam, B.S.; Morris, R.S.; Bramante, C.T.; Puskarich, M.; Zolfaghari, E.J.; Lotfi-Emran, S.; Ingraham, N.E.; Charles, A.; Odde, D.J.; Tignanelli, C.J. mTOR inhibition in COVID-19: A commentary and review of efficacy in RNA viruses. J. Med. Virol. 2021, 93, 1843–1846. [Google Scholar] [CrossRef]
- Shin, Y.K.; Liu, Q.; Tikoo, S.K.; Babiuk, L.A.; Zhou, Y. Effect of the phosphatidylinositol 3-kinase/Akt pathway on influenza A virus propagation. J. Gen. Virol. 2007, 88, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Jackson, D.; Chen, Y.H.; Lamb, R.A.; Randall, R.E. Influenza A virus NS1 protein binds p85β and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shives, K.D.; Beatman, E.L.; Chamanian, M.; O’Brien, C.; Hobson-Peters, J.; Beckham, J.D. West Nile Virus-Induced Activation of Mammalian Target of Rapamycin Complex 1 Supports Viral Growth and Viral Protein Expression. J. Virol. 2014, 88, 9458–9471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, S.K.; Shrivastava, S.; Meyer, K.; Ray, R.B.; Ray, R. Hepatitis C Virus Activates the mTOR/S6K1 Signaling Pathway in Inhibiting IRS-1 Function for Insulin Resistance. J. Virol. 2012, 86, 6315–6322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joubert, P.E.; Stapleford, K.; Guivel-Benhassine, F.; Vignuzzi, M.; Schwartz, O.; Albert, M.L. Inhibition of mTORC1 Enhances the Translation of Chikungunya Proteins via the Activation of the MnK/eIF4E Pathway. PLoS Pathog. 2015, 11, e1005091. [Google Scholar] [CrossRef]
- Das, I.; Basantray, I.; Mamidi, P.; Nayak, T.K.; Pratheek, B.M.; Chattopadhyay, S.; Chattopadhyay, S. Heat shock protein 90 positively regulates Chikungunya virus replication by stabilizing viral non-structural protein nsP2 during infection. PLoS One 2014, 9, e100531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; He, X.; Zhu, G.; Tu, H.; Liu, Z.; Li, W.; Han, S.; Yin, J.; Peng, B.; Liu, W. Coxsackievirus A16 elicits incomplete autophagy involving the mTOR and ERK pathways. PLoS One 2015, 10, e0122109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuss-Duerkop, S.K.; Wang, J.; Mena, I.; White, K.; Metreveli, G.; Sakthivel, R.; Mata, M.A.; Muñoz-Moreno, R.; Chen, X.; Krammer, F.; et al. Influenza Virus Differentially Activates mTORC1 and mTORC2 Signaling to Maximize Late Stage Replication. PLoS Pathog. 2017, 13, e1006635. [Google Scholar] [CrossRef] [PubMed]
- Bakar, F.A.; Ng, L.F.P. Nonstructural proteins of alphavirus—potential targets for drug development. Viruses 2018, 10, 71. [Google Scholar] [CrossRef] [Green Version]
- Mohankumar, V.; Dhanushkodi, N.R.; Raju, R. Sindbis virus replication, is insensitive to rapamycin and torin1, and suppresses Akt/mTOR pathway late during infection in HEK cells. Biochem. Biophys. Res. Commun. 2011, 406, 262–267. [Google Scholar] [CrossRef] [Green Version]
- Joubert, P.E.; Werneke, S.W.; de la Calle, C.; Guivel-Benhassine, F.; Giodini, A.; Peduto, L.; Levine, B.; Schwartz, O.; Lenschow, D.J.; Albert, M.L. Chikungunya virus-induced autophagy delays caspase-dependent cell death. J. Exp. Med. 2012, 209, 1029–1047. [Google Scholar] [CrossRef]
- Thaa, B.; Biasiotto, R.; Eng, K.; Neuvonen, M.; Götte, B.; Rheinemann, L.; Mutso, M.; Utt, A.; Varghese, F.; Balistreri, G.; et al. Differential Phosphatidylinositol-3-Kinase-Akt-mTOR Activation by Semliki Forest and Chikungunya Viruses Is Dependent on nsP3 and Connected to Replication Complex Internalization. J. Virol. 2015, 89, 11420–11437. [Google Scholar] [CrossRef] [Green Version]
- Klaitong, P.; Smith, D.R. Roles of non-structural protein 4a in flavivirus infection. Viruses 2021, 13, 2077. [Google Scholar] [CrossRef]
- Lahon, A.; Arya, R.P.; Banerjea, A.C. Dengue Virus Dysregulates Master Transcription Factors and PI3K/AKT/mTOR Signaling Pathway in Megakaryocytes. Front. Cell. Infect. Microbiol. 2021, 11, 715208. [Google Scholar] [CrossRef]
- Albentosa-González, L.; de Oya, N.J.; Arias, A.; Clemente-Casares, P.; Martin-Acebes, M.Á.; Saiz, J.C.; Sabariegos, R.; Mas, A. Akt kinase intervenes in flavivirus replication by interacting with viral protein ns5. Viruses 2021, 13, 896. [Google Scholar] [CrossRef]
- Urbanowski, M.D.; Hobman, T.C. The West Nile Virus Capsid Protein Blocks Apoptosis through a Phosphatidylinositol 3-Kinase-Dependent Mechanism. J. Virol. 2013, 87, 872–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-J.; Liao, C.-L.; Lin, Y.-L. Flavivirus Activates Phosphatidylinositol 3-Kinase Signaling To Block Caspase-Dependent Apoptotic Cell Death at the Early Stage of Virus Infection. J. Virol. 2005, 79, 8388–8399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, A.; Panda, S.; Kudmulwar, D.; Chhatbar, S.P.; Nayak, S.C.; Krishnan, H.H. Hepatitis C virus NS5A binds to the mRNA cap-binding eukaryotic translation initiation 4F (elF4F) complex and up-regulates host translation initiation machinery through elF4E-binding protein 1 inactivation. J. Biol. Chem. 2012, 287, 5042–5058. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Nakao, H.; Tan, S.-L.; Polyak, S.J.; Neddermann, P.; Vijaysri, S.; Jacobs, B.L.; Katze, M.G. Subversion of Cell Signaling Pathways by Hepatitis C Virus Nonstructural 5A Protein via Interaction with Grb2 and P85 Phosphatidylinositol 3-Kinase. J. Virol. 2002, 76, 9207–9217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Street, A.; Macdonald, A.; Crowder, K.; Harris, M. The Hepatitis C Virus NS5A Protein Activates a Phosphoinositide 3-Kinase-dependent Survival Signaling Cascade. J. Biol. Chem. 2004, 279, 12232–12241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Sage, V.; Cinti, A.; Amorim, R.; Mouland, A.J. Adapting the stress response: Viral subversion of the mTOR signaling pathway. Viruses 2016, 8, 152. [Google Scholar] [CrossRef] [Green Version]
- Mannová, P.; Beretta, L. Activation of the N-Ras–PI3K–Akt-mTOR Pathway by Hepatitis C Virus: Control of Cell Survival and Viral Replication. J. Virol. 2005, 79, 8742–8749. [Google Scholar] [CrossRef] [Green Version]
- Clippinger, A.J.; Maguire, T.G.; Alwine, J.C. Human Cytomegalovirus Infection Maintains mTOR Activity and Its Perinuclear Localization during Amino Acid Deprivation. J. Virol. 2011, 85, 9369–9376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudchodkar, S.B.; Del Prete, G.Q.; Maguire, T.G.; Alwine, J.C. AMPK-Mediated Inhibition of mTOR Kinase Is Circumvented during Immediate-Early Times of Human Cytomegalovirus Infection. J. Virol. 2007, 81, 3649–3651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Alwine, J.C. Human Cytomegalovirus Major Immediate-Early Proteins and Simian Virus 40 Large T Antigen Can Inhibit Apoptosis through Activation of the Phosphatidylinositide 3′-OH Kinase Pathway and the Cellular Kinase Akt. J. Virol. 2002, 76, 3731–3738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchet, F.P.; Moris, A.; Nikolic, D.S.; Lehmann, M.; Cardinaud, S.; Stalder, R.; Garcia, E.; Dinkins, C.; Leuba, F.; Wu, L.; et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 2010, 32, 654–669. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Rawat, P.; Bruckman, R.S.; Spector, S.A. Human Immunodeficiency Virus Type 1 Nef Inhibits Autophagy through Transcription Factor EB Sequestration. PLoS Pathog. 2015, 11, e1005018. [Google Scholar] [CrossRef] [Green Version]
- Cinti, A.; Le Sage, V.; Milev, M.P.; Valiente-Echeverría, F.; Crossie, C.; Miron, M.-J.; Panté, N.; Olivier, M.; Mouland, A.J. HIV-1 enhances mTORC1 activity and repositions lysosomes to the periphery by co-opting Rag GTPases. Sci. Rep. 2017, 7, 5515. [Google Scholar] [CrossRef] [Green Version]
- Heredia, A.; Le, N.; Gartenhaus, R.B.; Sausville, E.; Medina-Moreno, S.; Zapata, J.C.; Davis, C.; Gallo, R.C.; Redfield, R.R. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9412–9417. [Google Scholar] [CrossRef] [Green Version]
- Donia, M.; McCubrey, J.A.; Bendtzen, K.; Nicoletti, F. Potential use of rapamycin in HIV infection. Br. J. Clin. Pharmacol. 2010, 70, 784–793. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Jiang, X.; Liu, D.; Fan, Z.; Hu, X.; Yan, J.; Wang, M.; Gao, G.F. Autophagy is involved in influenza A virus replication. Autophagy 2009, 5, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Zhirnov, O.P.; Konakova, T.E.; Garten, W.; Klenk, H.-D. Caspase-Dependent N-Terminal Cleavage of Influenza Virus Nucleocapsid Protein in Infected Cells. J. Virol. 1999, 73, 10158–10163. [Google Scholar] [CrossRef] [Green Version]
- Tsubamoto, H.; Inoue, K.; Sakata, K.; Ueda, T.; Takeyama, R.; Shibahara, H.; Sonoda, T. Itraconazole inhibits AKT/mTOR signaling and proliferation in endometrial cancer cells. Anticancer Res. 2017, 37, 515–520. [Google Scholar] [CrossRef] [Green Version]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Cork, G.K.; Thompson, J.; Slawson, C. Real Talk: The Inter-play Between the mTOR, AMPK, and Hexosamine Biosynthetic Pathways in Cell Signaling. Front. Endocrinol. (Lausanne) 2018, 9, 522. [Google Scholar] [CrossRef] [PubMed]
- Nacev, B.A.; Grassi, P.; Dell, A.; Haslam, S.M.; Liu, J.O. The antifungal drug itraconazole inhibits Vascular Endothelial Growth Factor Receptor 2 (VEGFR2) glycosylation, trafficking, and signaling in endothelial cells. J. Biol. Chem. 2011, 286, 44045–44056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrancken, K.; Paeshuyse, J.; Liekens, S. Angiogenic activity of hepatitis B and C viruses. Antivir. Chem. Chemother. 2012, 22, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; He, L.; Zhang, E.; Shi, J.; Zhang, Q.; Le, A.D.; Zhou, K.; Tang, X. Overexpression of human papillomavirus (HPV) type 16 oncoproteins promotes angiogenesis via enhancing HIF-1α and VEGF expression in non-small cell lung cancer cells. Cancer Lett. 2011, 311, 160–170. [Google Scholar] [CrossRef]
- Hassan, M.; Selimovic, D.; El-Khattouti, A.; Soell, M.; Ghozlan, H.; Haikel, Y.; Abdelkader, O.; Megahed, M. Hepatitis C virus-mediated angiogenesis: Molecular mechanisms and therapeutic strategies. World J. Gastroenterol. 2014, 20, 15467–15475. [Google Scholar] [CrossRef]
- Smelkinson, M.G.; Guichard, A.; Teijaro, J.R.; Malur, M.; Loureiro, M.E.; Jain, P.; Ganesan, S.; Zúñiga, E.I.; Krug, R.M.; Oldstone, M.B.; et al. Influenza NS1 directly modulates Hedgehog signaling during infection. PLoS Pathog. 2017, 13, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.W. Targeting the sonic hedgehog signaling pathway: Review of smoothened and GLI inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, T.D.A.; Witek, R.P.; Syn, W.K.; Choi, S.S.; Bradrick, S.; Karaca, G.F.; Agboola, K.M.; Jung, Y.; Omenetti, A.; Moylan, C.A.; et al. Viral factors induce Hedgehog pathway activation in humans with viral hepatitis, cirrhosis, and hepatocellular carcinoma. Lab. Investig. 2010, 90, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Zompetta, C.; Vescarelli, E.; Rizzello, C.; Cardi, A.; Valia, S.; Antonelli, G.; Marchese, C.; Torrisi, M.R.; Faggioni, A.; et al. HCV derived from sera of HCVinfected patients induces profibrotic effects in human primary fibroblasts by activating GLI2. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.Y.; Cho, H.K.; Hong, S.P.; Cheong, J.H. Hepatitis B virus X protein stimulates the Hedgehog-Gli activation through protein stabilization and nuclear localization of Gli1 in liver cancer cells. Cancer Lett. 2011, 309, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a Commonly Used Antifungal that Inhibits Hedgehog Pathway Activity and Cancer Growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef] [Green Version]
- Dirix, L. Discovery and exploitation of novel targets by approved drugs. J. Clin. Oncol. 2014, 32, 720–721. [Google Scholar] [CrossRef]
- Deng, H.; Huang, L.; Liao, Z.; Liu, M.; Li, Q.; Xu, R. Itraconazole inhibits the Hedgehog signaling pathway thereby inducing autophagy-mediated apoptosis of colon cancer cells. Cell Death Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [Green Version]
- van Zuylen, W.J.; Rawlinson, W.D.; Ford, C.E. The Wnt pathway: A key network in cell signalling dysregulated by viruses. Rev. Med. Virol. 2016, 26, 340–355. [Google Scholar] [CrossRef]
- Shackelford, J.; Maier, C.; Pagano, J.S. Epstein-Barr virus activates β-catenin in type III latently infected B lymphocyte lines: Association with deubiquitinating enzymes. Proc. Natl. Acad. Sci. USA 2003, 100, 15572–15576. [Google Scholar] [CrossRef] [Green Version]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.; Kim, H.S.; Lim, S.O.; Yu, D.Y.; Jung, G. Hepatitis B viral X protein interacts with tumor suppressor adenomatous polyposis coli to activate Wnt/β-catenin signaling. Cancer Lett. 2011, 300, 162–172. [Google Scholar] [CrossRef]
- Xie, Q.; Chen, L.; Shan, X.; Shan, X.; Tang, J.; Zhou, F.; Chen, Q.; Quan, H.; Nie, D.; Zhang, W.; et al. Epigenetic silencing of SFRP1 and SFRP5 by hepatitis B virus X protein enhances hepatoma cell tumorigenicity through Wnt signaling pathway. Int. J. Cancer 2014, 135, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Umer, M.; Qureshi, S.A.; Hashmi, Z.Y.; Raza, A.; Ahmad, J.; Rahman, M.; Iqbal, M. Promoter hypermethylation of Wnt pathway inhibitors in hepatitis C virus - Induced multistep hepatocarcinogenesis. Virol. J. 2014, 11, 117. [Google Scholar] [CrossRef] [Green Version]
- Popova, S.A.; Buczacki, S.J.A. Itraconazole perturbs colorectal cancer dormancy through SUFU-mediated WNT inhibition. Mol. Cell. Oncol. 2018, 5, e1494950. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Liu, M.; Wang, Q.; Shen, Y.; Mei, H.; Li, D.; Liu, W. Itraconazole exerts its anti-melanoma effect by suppressing Hedgehog, Wnt, and PI3K/mTOR signaling pathways. Oncotarget 2017, 8, 28510–28525. [Google Scholar] [CrossRef] [Green Version]
- Shyr, Z.A.; Cheng, Y.S.; Lo, D.C.; Zheng, W. Drug combination therapy for emerging viral diseases. Drug Discov. Today 2021, 26, 2367–2376. [Google Scholar] [CrossRef]
- Govorkova, E.A.; Webster, R.G. Combination chemotherapy for influenza. Viruses 2010, 2, 1510–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, J.D.; Piefer, A.J. Emerging drug targets for antiretroviral therapy. Drugs 2005, 65, 1747–1766. [Google Scholar] [CrossRef]
- Schloer, S.; Goretzko, J.; Pleschka, S.; Ludwig, S.; Rescher, U. Combinatory Treatment with Oseltamivir and Itraconazole Targeting Both Virus and Host Factors in Influenza A Virus Infection. Viruses 2020, 12, 703. [Google Scholar] [CrossRef]
- Domínguez-Gil Hurlé, A.; Sánchez Navarro, A.; García Sánchez, M.J. Therapeutic drug monitoring of itraconazole and the relevance of pharmacokinetic interactions. Clin. Microbiol. Infect. 2006, 12, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Prentice, A.G.; Glasmacher, A. Making sense of itraconazole pharmacokinetics. J. Antimicrob. Chemother. 2005, 56, i17–i22. [Google Scholar] [CrossRef] [Green Version]
- Jaruratanasirikul, S.; Sriwiriyajan, S. Effect of omeprazole on the pharmacokinetics of itraconazole. Eur. J. Clin. Pharmacol. 1998, 54, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Lestner, J.; Hope, W.W. Itraconazole: An update on pharmacology and clinical use for treatment of invasive and allergic fungal infections. Expert Opin. Drug Metab. Toxicol. 2013, 9, 911–926. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.K.; Park, S.J.; Shim, E.J.; Mun, J.H.; Kim, E.Y.; Shin, J.G.; Shon, J.H. Increased oral bioavailability of itraconazole and its active metabolite, 7-hydroxyitraconazole, when coadministered with a vitamin C beverage in healthy participants. J. Clin. Pharmacol. 2011, 51, 444–451. [Google Scholar] [CrossRef]
- Vena, A.; Muñoz, P.; Mateos, M.; Guinea, J.; Galar, A.; Pea, F.; Alvarez-Uria, A.; Escribano, P.; Bouza, E. Therapeutic Drug Monitoring of Antifungal Drugs: Another Tool to Improve Patient Outcome? Infect. Dis. Ther. 2020, 9, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Tverdek, F.P.; Kofteridis, D.; Kontoyiannis, D.P. Antifungal agents and liver toxicity: A complex interaction. Expert Rev. Anti. Infect. Ther. 2016, 14, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Ashbee, H.R.; Barnes, R.A.; Johnson, E.M.; Richardson, M.D.; Gorton, R.; Hope, W.W. Therapeutic drug monitoring (TDM) of antifungal agents: Guidelines from the british society for medical mycology. J. Antimicrob. Chemother. 2014, 69, 1162–1176. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Antifungal Drug Family | Mechanism of Action | Clinical Use as Antifungals | Antiviral Potential |
|---|---|---|---|
| Polyenes | Bind sterol components and form pores, resulting in a compromised fungal plasma membrane [11]. | Aspergillosis, cryptococcosis, candidiasis, zygomycosis, fusariosis, coccidioidomycosis, paracoccidioidomycosis, histoplasmosis, blastomycosis, mucormycosis, penicilliosis, and phaeohyphomycosis [11]. | Japanese encephalitis virus [12], herpes simplex virus (HSV) [13], human immunodeficiency virus (HIV) [14], rubella virus [12], vesicular stomatitis virus (VSV) [15] |
| Flucytosine | Interferes with fungal nucleic acid synthesis [16]. | Candidiasis, and cryptococcosis [16]. | Not known |
| Echinocandins | Inhibit the fungal enzyme β1,3-glucan synthase, leading to incomplete fungal cell wall formation [17]. | Aspergillosis, and candidiasis [17]. | Chikungunya virus (CHIKV) [18], enteroviruses [19], dengue virus [20], SARS-CoV-2 [21], Sindbis virus (SINV) and Semliki Forest virus (SFV) [18] |
| Azoles | Primarily inhibit the fungal sterol biosynthesis, leading to compromised fungal membranes [22]. | Aspergillosis, candidiasis, and cryptococcosis [22]. | SARS-CoV-2 [23,24], influenza virus [25], Ebola virus [26,27,28], Parechovirus A3 [29], dengue virus [30], enteroviruses [31], human cytomegalovirus [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schloer, S.; Goretzko, J.; Rescher, U. Repurposing Antifungals for Host-Directed Antiviral Therapy? Pharmaceuticals 2022, 15, 212. https://doi.org/10.3390/ph15020212
Schloer S, Goretzko J, Rescher U. Repurposing Antifungals for Host-Directed Antiviral Therapy? Pharmaceuticals. 2022; 15(2):212. https://doi.org/10.3390/ph15020212
Chicago/Turabian StyleSchloer, Sebastian, Jonas Goretzko, and Ursula Rescher. 2022. "Repurposing Antifungals for Host-Directed Antiviral Therapy?" Pharmaceuticals 15, no. 2: 212. https://doi.org/10.3390/ph15020212
APA StyleSchloer, S., Goretzko, J., & Rescher, U. (2022). Repurposing Antifungals for Host-Directed Antiviral Therapy? Pharmaceuticals, 15(2), 212. https://doi.org/10.3390/ph15020212