
Population Dynamics of a Two Phages–One Host Infection System Using Escherichia coli Strain ECOR57 and Phages vB_EcoP_SU10 and vB_EcoD_SU57


Abstract
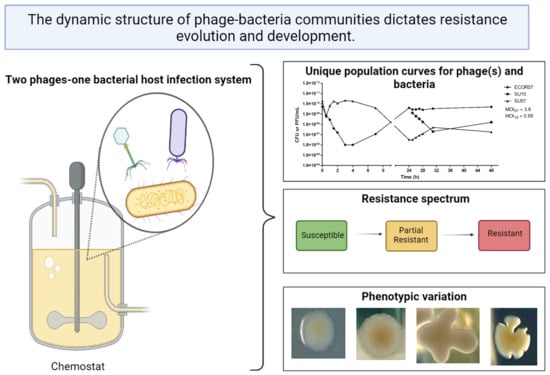
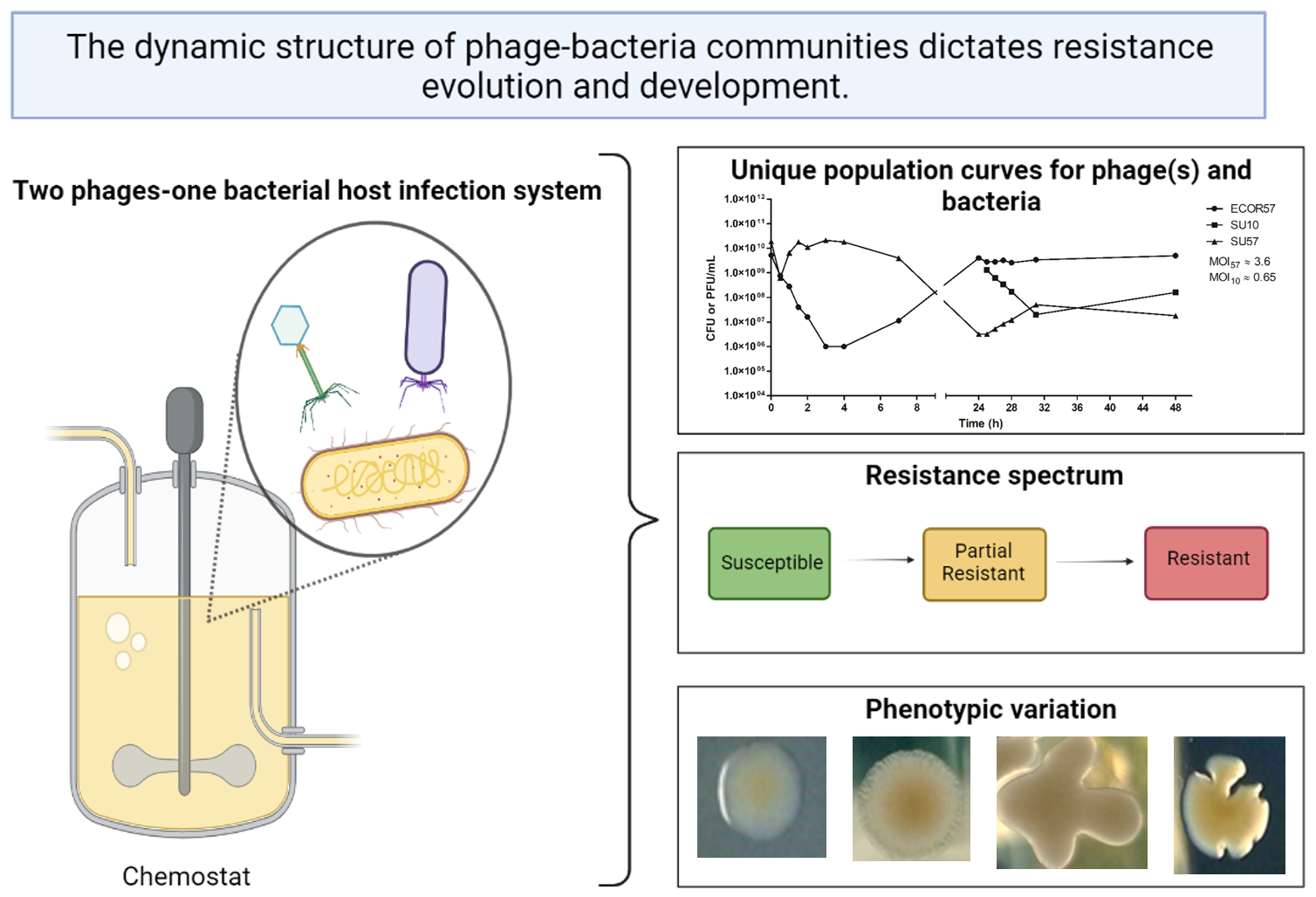
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
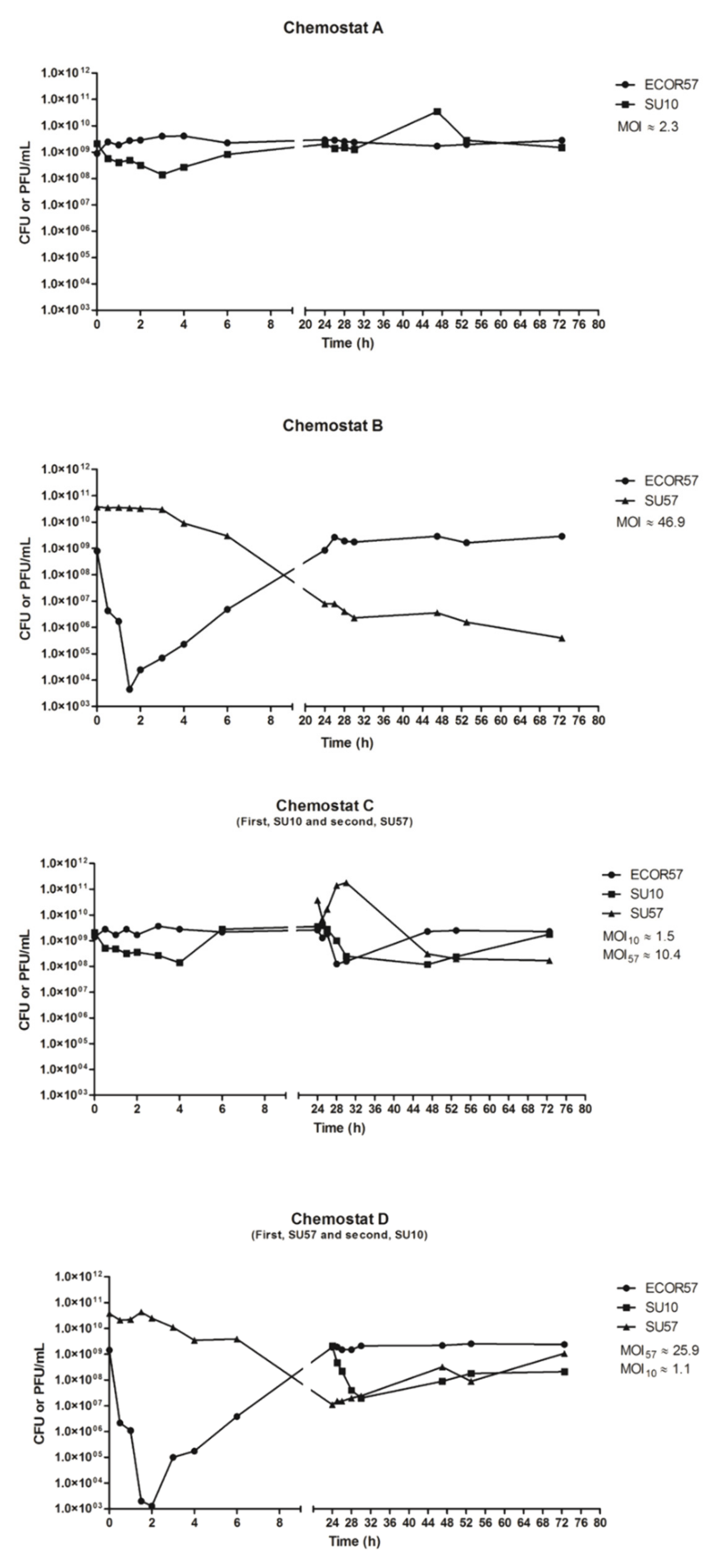
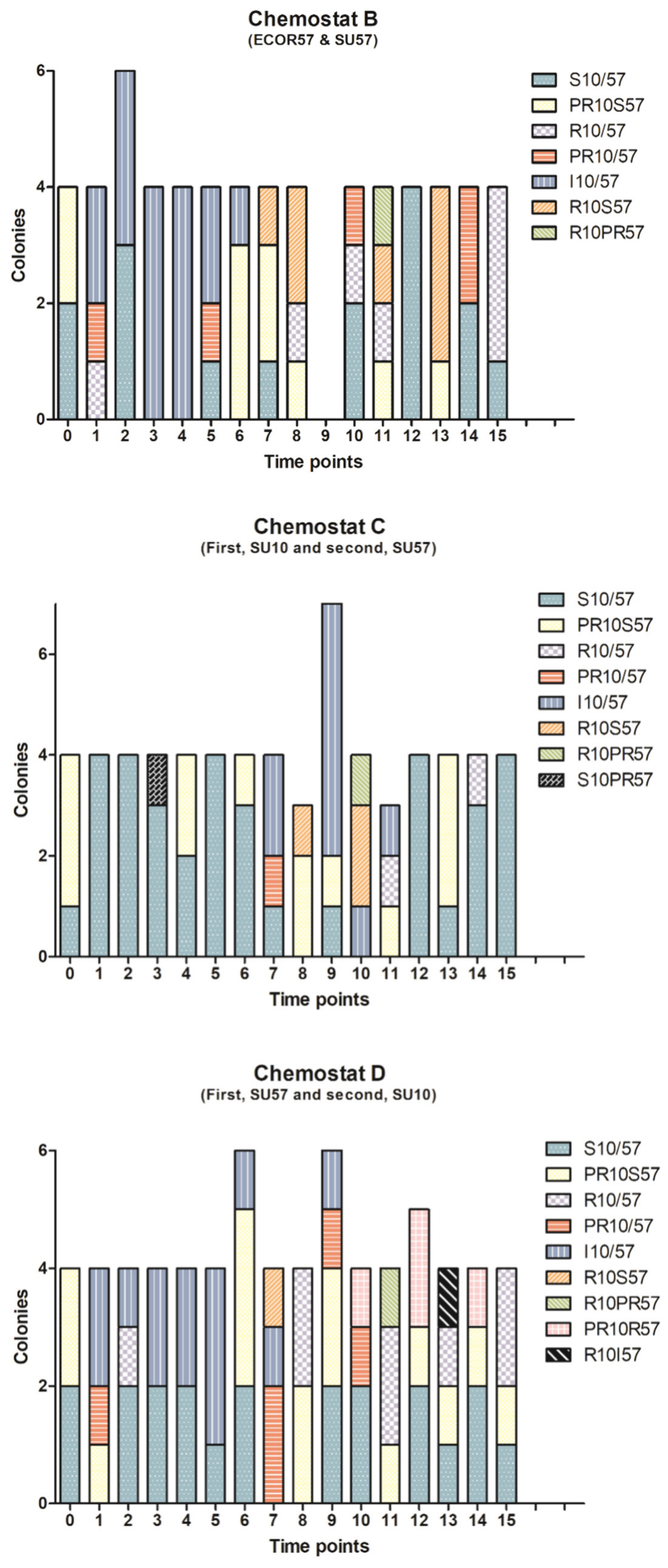
2.1. Population Dynamics of a Two Phages–One Host System Using SU10/SU57 and ECOR57
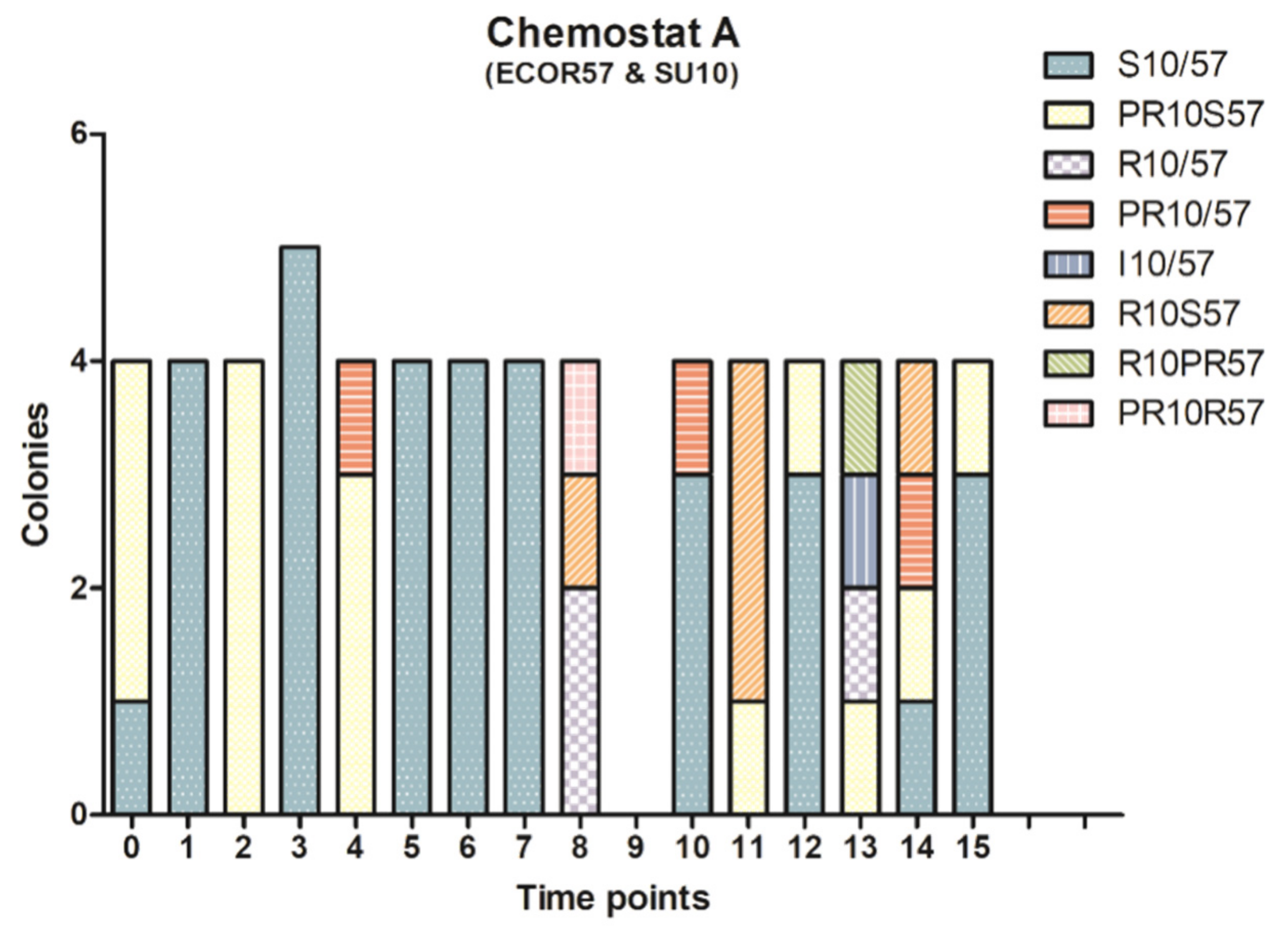
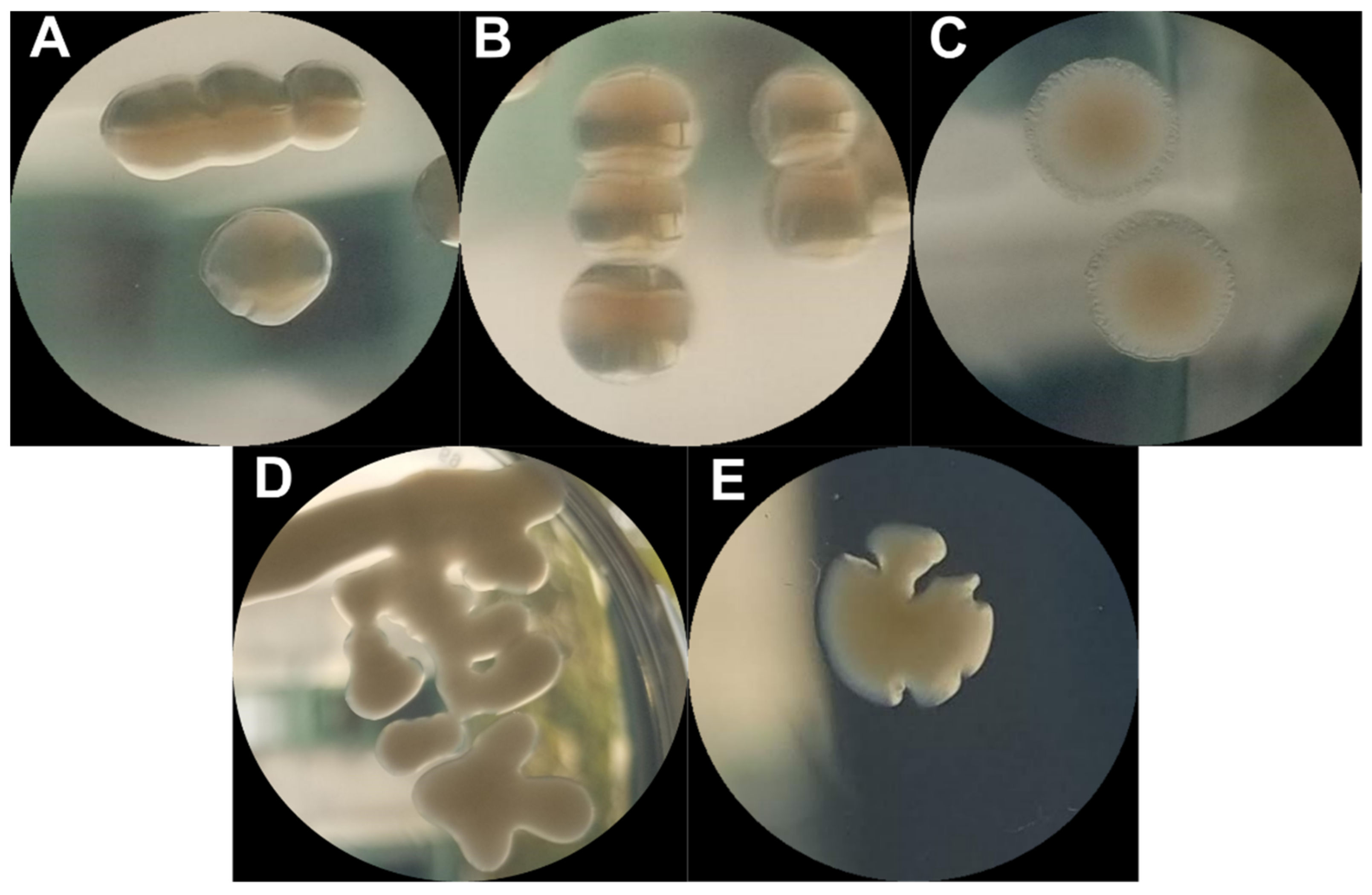
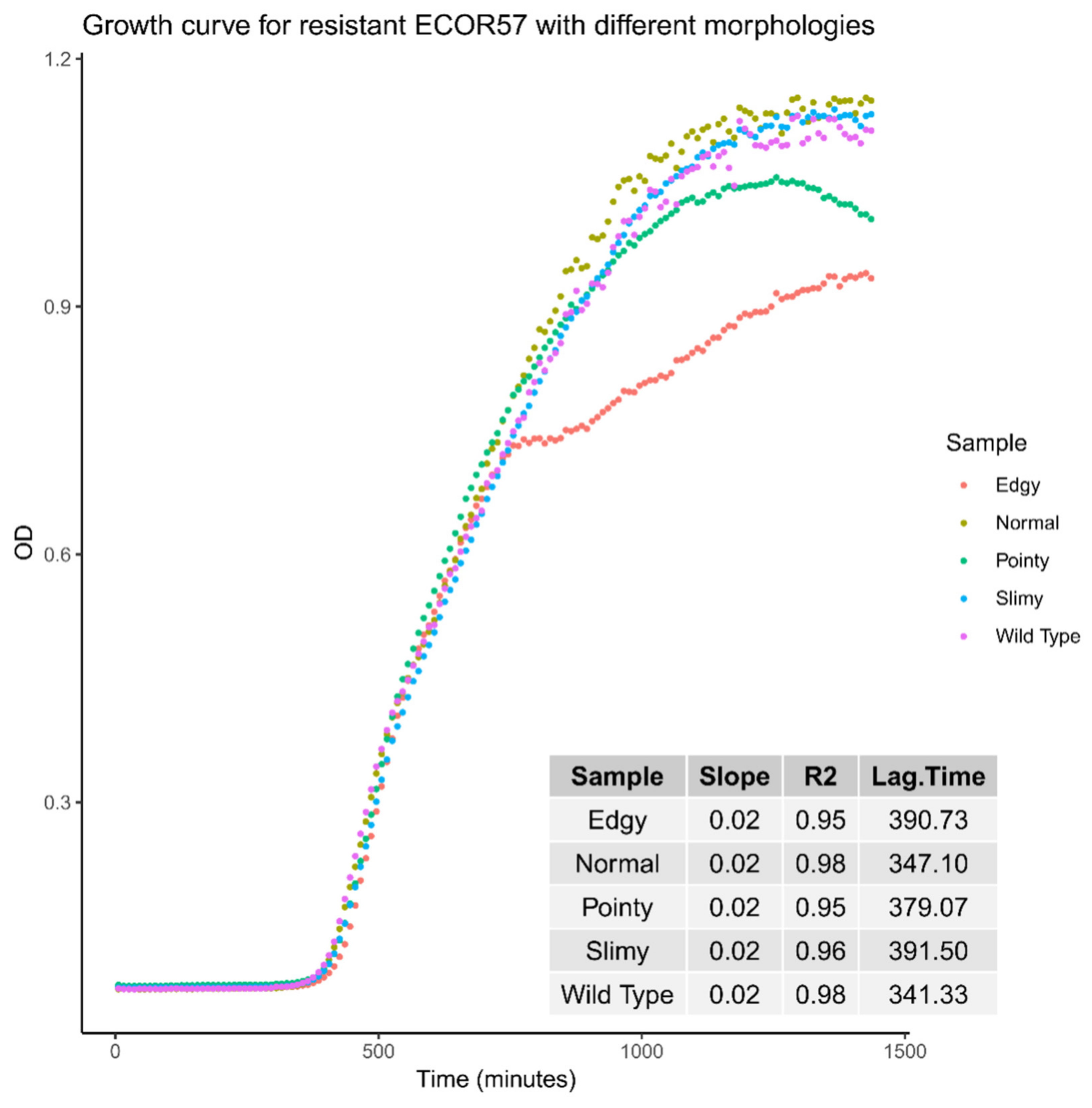
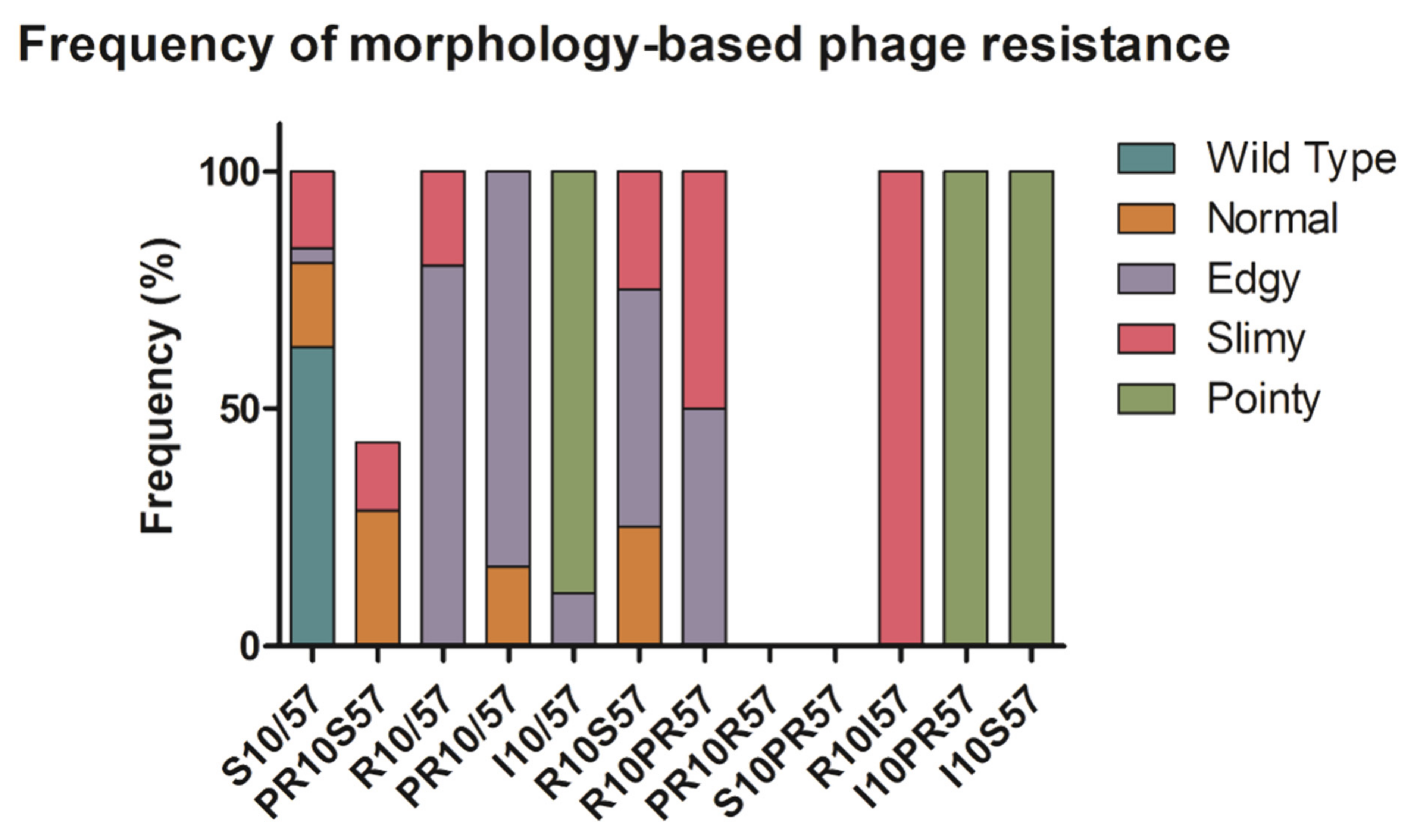
2.2. Phenotypic Diversity among ECOR57 Bacteria following Exposure to Two Phages
3. Discussion
3.1. Population Dynamics and Resistance Patterns of a Two Phages–One Host System
3.2. Phenotypic Diversity within a Two Phages–One Host System
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Phage Propagation, Purification, and Quantification
4.3. Chemostat Setup
4.4. Determination of Adsorption Rate Constant
4.5. Efficiency of Plating (EOP) and Frequency of Phage Resistance Development
4.6. DNA Extraction, Sequencing, Bioinformatics, and Genomic Analyses
4.7. Bacterial Growth Analyses
4.8. Luria–Delbrück Fluctuation Tests
4.9. Antibiotic Resistance Testing
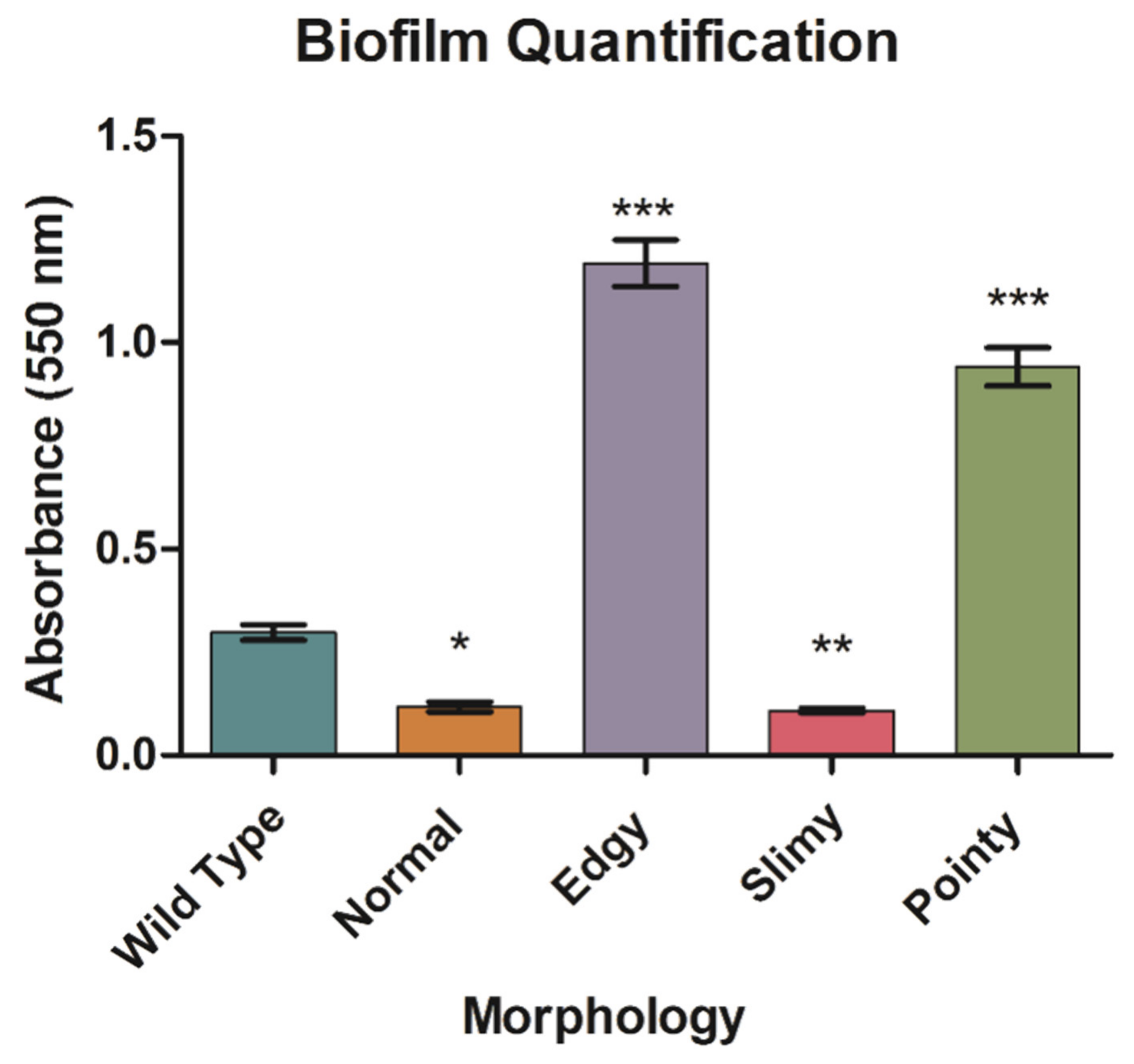
4.10. Biofilm Formation and Quantification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the Gut Microbiota in Disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Sutton, T.D.S.; Hill, C. Gut Bacteriophage: Current Understanding and Challenges. Front. Endocrinol. 2019, 10, 784. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Zablocki, O.; Howell, A.; Bolduc, B.; Sullivan, M.B. The Human Gut Virome Database. bioRxiv 2019, 2019, 655910. [Google Scholar] [CrossRef]
- Von Wintersdorff, C.J.H.; Penders, J.; van Niekerk, J.M.; Mills, N.D.; Majumder, S.; van Alphen, L.B.; Savelkoul, P.H.M.; Wolffs, P.F.G. Dissemination of Antimicrobial Resistance in Microbial Ecosystems through Horizontal Gene Transfer. Front. Microbiol. 2016, 7, 173. [Google Scholar] [CrossRef] [Green Version]
- Hoyles, L.; McCartney, A.L.; Neve, H.; Gibson, G.R.; Sanderson, J.D.; Heller, K.J.; van Sinderen, D. Characterization of Virus-like Particles Associated with the Human Faecal and Caecal Microbiota. Res. Microbiol. 2014, 165, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Chiang, Y.N.; Penadés, J.R.; Chen, J. Genetic Transduction by Phages and Chromosomal Islands: The New and Noncanonical. PLoS Pathog. 2019, 15, e1007878. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, D.; Baldridge, M.T.; Handley, S.A. Phages and Human Health: More Than Idle Hitchhikers. Viruses 2019, 11, 587. [Google Scholar] [CrossRef] [Green Version]
- Kasman, L.M.; Porter, L.D. Bacteriophages. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Maurice, C.F. Considering the Other Half of the Gut Microbiome: Bacteriophages. mSystems 2019, 4, e00102–e00119. [Google Scholar] [CrossRef] [Green Version]
- Casjens, S.; Palmer, N.; Vugt, R.V.; Huang, W.M.; Stevenson, B.; Rosa, P.; Lathigra, R.; Sutton, G.; Peterson, J.; Dodson, R.J.; et al. A Bacterial Genome in Flux: The Twelve Linear and Nine Circular Extrachromosomal DNAs in an Infectious Isolate of the Lyme Disease Spirochete Borrelia Burgdorferi. Mol. Microbiol. 2000, 35, 490–516. [Google Scholar] [CrossRef]
- Casjens, S. Prophages and Bacterial Genomics: What Have We Learned so Far? Mol. Microbiol. 2003, 49, 277–300. [Google Scholar] [CrossRef]
- Brüssow, H.; Canchaya, C.; Hardt, W.-D. Phages and the Evolution of Bacterial Pathogens: From Genomic Rearrangements to Lysogenic Conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [Green Version]
- Doss, J.; Culbertson, K.; Hahn, D.; Camacho, J.; Barekzi, N. A Review of Phage Therapy against Bacterial Pathogens of Aquatic and Terrestrial Organisms. Viruses 2017, 9, 50. [Google Scholar] [CrossRef] [Green Version]
- Cieplak, T.; Soffer, N.; Sulakvelidze, A.; Nielsen, D.S. A Bacteriophage Cocktail Targeting Escherichia Coli Reduces E. Coli in Simulated Gut Conditions, While Preserving a Non-Targeted Representative Commensal Normal Microbiota. Gut Microbes 2018, 9, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Makky, S.; Dawoud, A.; Safwat, A.; Abdelsattar, A.S.; Rezk, N.; El-Shibiny, A. The Bacteriophage Decides Own Tracks: When They Are with or against the Bacteria. Curr. Res. Microb. Sci. 2021, 2, 100050. [Google Scholar] [CrossRef]
- Cisek, A.A.; Dąbrowska, I.; Gregorczyk, K.P.; Wyżewski, Z. Phage Therapy in Bacterial Infections Treatment: One Hundred Years After the Discovery of Bacteriophages. Curr. Microbiol. 2017, 74, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Young, R. Phage Lysis: Three Steps, Three Choices, One Outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef]
- Bull, J.J.; Vegge, C.S.; Schmerer, M.; Chaudhry, W.N.; Levin, B.R. Phenotypic Resistance and the Dynamics of Bacterial Escape from Phage Control. PLoS ONE 2014, 9, e94690. [Google Scholar] [CrossRef]
- Rostøl, J.T.; Marraffini, L. (Ph)Ighting Phages—How Bacteria Resist Their Parasites. Cell Host Microbe 2019, 25, 184–194. [Google Scholar] [CrossRef] [Green Version]
- Schwechheimer, C.; Kuehn, M.J. Outer-Membrane Vesicles from Gram-Negative Bacteria: Biogenesis and Functions. Nat. Rev. Microbiol. 2015, 13, 605–619. [Google Scholar] [CrossRef] [Green Version]
- Harvey, H.; Bondy-Denomy, J.; Marquis, H.; Sztanko, K.M.; Davidson, A.R.; Burrows, L.L. Pseudomonas Aeruginosa Defends against Phages through Type IV Pilus Glycosylation. Nat. Microbiol. 2018, 3, 47–52. [Google Scholar] [CrossRef]
- Pedruzzi, I.; Rosenbusch, J.P.; Locher, K.P. Inactivation in Vitro of the Escherichia Coli Outer Membrane Protein FhuA by a Phage T5-Encoded Lipoprotein. FEMS Microbiol. Lett. 1998, 168, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Clément, J.M.; Lepouce, E.; Marchal, C.; Hofnung, M. Genetic Study of a Membrane Protein: DNA Sequence Alterations Due to 17 LamB Point Mutations Affecting Adsorption of Phage Lambda. EMBO J. 1983, 2, 77–80. [Google Scholar] [CrossRef]
- Eriksen, R.S.; Svenningsen, S.L.; Sneppen, K.; Mitarai, N. A Growing Microcolony Can Survive and Support Persistent Propagation of Virulent Phages. Proc. Natl. Acad. Sci. USA 2018, 115, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Vidakovic, L.; Singh, P.K.; Hartmann, R.; Nadell, C.D.; Drescher, K. Dynamic Biofilm Architecture Confers Individual and Collective Mechanisms of Viral Protection. Nat. Microbiol. 2018, 3, 26–31. [Google Scholar] [CrossRef]
- Zheng, Y.; Roberts, R.J.; Kasif, S. Identification of Genes with Fast-Evolving Regions in Microbial Genomes. Nucleic Acids Res. 2004, 32, 6347–6357. [Google Scholar] [CrossRef] [Green Version]
- Wright, R.C.T.; Friman, V.-P.; Smith, M.C.M.; Brockhurst, M.A. Resistance Evolution against Phage Combinations Depends on the Timing and Order of Exposure. mBio 2019, 10, e01652–e01719. [Google Scholar] [CrossRef] [Green Version]
- Gurney, J.; Aldakak, L.; Betts, A.; Gougat-Barbera, C.; Poisot, T.; Kaltz, O.; Hochberg, M.E. Network Structure and Local Adaptation in Co-Evolving Bacteria-Phage Interactions. Mol. Ecol. 2017, 26, 1764–1777. [Google Scholar] [CrossRef]
- Zeng, Z.; Salmond, G.P.C. Bacteriophage Host Range Evolution through Engineered Enrichment Bias, Exploiting Heterologous Surface Receptor Expression. Environ. Microbiol. 2020, 22, 5207–5221. [Google Scholar] [CrossRef]
- Wright, R.C.T.; Friman, V.-P.; Smith, M.C.M.; Brockhurst, M.A. Cross-Resistance Is Modular in Bacteria–Phage Interactions. PLoS Biol. 2018, 16, e2006057. [Google Scholar] [CrossRef]
- Nilsson, A.S. Pharmacological Limitations of Phage Therapy. Upsala J. Med. Sci. 2019, 124, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.R.; Vos, D.D.; Friman, V.-P.; Pirnay, J.-P.; Buckling, A. Effects of Sequential and Simultaneous Applications of Bacteriophages on Populations of Pseudomonas Aeruginosa In Vitro and in Wax Moth Larvae. Appl. Environ. Microbiol. 2012, 78, 5646–5652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, B.K.; Abedon, S.T. Phage Therapy Pharmacology Phage Cocktails. Adv. Appl. Microbiol. 2012, 78, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Koonjan, S.; Seijsing, F.; Cooper, C.J.; Nilsson, A.S. Infection Kinetics and Phylogenetic Analysis of VB_EcoD_SU57, a Virulent T1-Like Drexlerviridae Coliphage. Front. Microbiol. 2020, 11, 2439. [Google Scholar] [CrossRef]
- Abedon, S.T.; Herschler, T.D.; Stopar, D. Bacteriophage Latent-Period Evolution as a Response to Resource Availability. Appl. Environ. Microbiol. 2001, 67, 4233–4241. [Google Scholar] [CrossRef] [Green Version]
- Abedon, S.T. Phage Therapy Dosing: The Problem(s) with Multiplicity of Infection (MOI). Bacteriophage 2016, 6, e1220348. [Google Scholar] [CrossRef] [Green Version]
- Stern, A.; Sorek, R. The Phage-Host Arms Race: Shaping the Evolution of Microbes. Bioessays 2011, 33, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Weitz, J.S.; Hartman, H.; Levin, S.A. Coevolutionary Arms Races between Bacteria and Bacteriophage. Proc. Natl. Acad. Sci. USA 2005, 102, 9535–9540. [Google Scholar] [CrossRef] [Green Version]
- Abdelsattar, A.S.; Dawooud, A.; Rezk, N.; Makky, S.; Safwat, A.; Richards, P.J.; El-Shibiny, A. How to Train Your Phage: The Recent Efforts in Phage Training. Biologics 2021, 1, 70–88. [Google Scholar] [CrossRef]
- Borin, J.M.; Avrani, S.; Barrick, J.E.; Petrie, K.L.; Meyer, J.R. Coevolutionary Phage Training Leads to Greater Bacterial Suppression and Delays the Evolution of Phage Resistance. Proc. Natl. Acad. Sci. USA 2021, 118, 118. [Google Scholar] [CrossRef]
- Schmerer, M.; Molineux, I.J.; Bull, J.J. Synergy as a Rationale for Phage Therapy Using Phage Cocktails. PeerJ 2014, 2, e590. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, A.S. Phage Therapy—Constraints and Possibilities. Ups. J. Med. Sci. 2014, 119, 192–198. [Google Scholar] [CrossRef]
- Ochman, H.; Selander, R.K. Standard Reference Strains of Escherichia Coli from Natural Populations. J. Bacteriol. 1984, 157, 690–693. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, M.K.; Eriksson, H.; Kasuga, K.; Haggård-Ljungquist, E.; Nilsson, A.S. Genomic, Proteomic, Morphological, and Phylogenetic Analyses of VB_EcoP_SU10, a Podoviridae Phage with C3 Morphology. PLoS ONE 2014, 9, e116294. [Google Scholar] [CrossRef] [Green Version]
- Lang, G.I. Measuring Mutation Rates Using the Luria-Delbrück Fluctuation Assay. In Genome Instability: Methods and Protocols; Methods in Molecular Biology; Muzi-Falconi, M., Brown, G.W., Eds.; Springer: New York, NY, USA, 2018; pp. 21–31. ISBN 978-1-4939-7306-4. [Google Scholar]
- Bohannan, B.J.M.; Kerr, B.; Jessup, C.M.; Hughes, J.B.; Sandvik, G. Trade-Offs and Coexistence in Microbial Microcosms. Antonie Van Leeuwenhoek 2002, 81, 107–115. [Google Scholar] [CrossRef]
- Scanlan, P.D.; Buckling, A. Co-Evolution with Lytic Phage Selects for the Mucoid Phenotype of Pseudomonas Fluorescens SBW25. ISME J. 2012, 6, 1148–1158. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, M.K.K.; Nilsson, A.S. Isolation of Phages for Phage Therapy: A Comparison of Spot Tests and Efficiency of Plating Analyses for Determination of Host Range and Efficacy. PLoS ONE 2015, 10, e0127606. [Google Scholar] [CrossRef] [Green Version]
- Kutter, E.; de Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S.T. Phage Therapy in Clinical Practice: Treatment of Human Infections. Curr. Pharm. Biotechnol. 2010, 11, 69–86. [Google Scholar] [CrossRef]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage Treatment of Human Infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef] [Green Version]
- Abedon, S.T. Thinking about Microcolonies as Phage Targets. Bacteriophage 2012, 2, 200–204. [Google Scholar] [CrossRef] [Green Version]
- Abedon, S.T. Bacteriophage Secondary Infection. Virol. Sin. 2015, 30, 3–10. [Google Scholar] [CrossRef]
- Loc-Carrillo, C.; Abedon, S.T. Pros and Cons of Phage Therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Abedon, S.T.; Thomas-Abedon, C. Phage Therapy Pharmacology. Curr. Pharm. Biotechnol. 2010, 11, 28–47. [Google Scholar] [CrossRef]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage Resistance Mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Silva, J.B.; Storms, Z.; Sauvageau, D. Host Receptors for Bacteriophage Adsorption. FEMS Microbiol. Lett. 2016, 363, fnw002. [Google Scholar] [CrossRef] [Green Version]
- Windels, E.M.; Michiels, J.E.; van den Bergh, B.; Fauvart, M.; Michiels, J. Antibiotics: Combatting Tolerance to Stop Resistance. mBio 2019, 10, e02095–e02119. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, W.N.; Pleška, M.; Shah, N.N.; Weiss, H.; McCall, I.C.; Meyer, J.R.; Gupta, A.; Guet, C.C.; Levin, B.R. Leaky Resistance and the Conditions for the Existence of Lytic Bacteriophage. PLoS Biol. 2018, 16, e2005971. [Google Scholar] [CrossRef]
- Boldogh, I.; Albrecht, T.; Porter, D.D. Persistent Viral Infections. In Medical Microbiology; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996; ISBN 978-0-9631172-1-2. [Google Scholar]
- Tan, D.; Zhang, Y.; Cheng, M.; Le, S.; Gu, J.; Bao, J.; Qin, J.; Guo, X.; Zhu, T. Characterization of Klebsiella Pneumoniae ST11 Isolates and Their Interactions with Lytic Phages. Viruses 2019, 11, 1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laanto, E.; Bamford, J.K.H.; Laakso, J.; Sundberg, L.-R. Phage-Driven Loss of Virulence in a Fish Pathogenic Bacterium. PLoS ONE 2012, 7, e53157. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, W.; Lee, E.; Worthy, A.; Weiss, Z.; Grabowicz, M.; Vega, N.; Levin, B. Mucoidy, a General Mechanism for Maintaining Lytic Phage in Populations of Bacteria. FEMS Microbiol. Ecol. 2020, 96, 96. [Google Scholar] [CrossRef] [PubMed]
- Koskella, B.; Lin, D.M.; Buckling, A.; Thompson, J.N. The Costs of Evolving Resistance in Heterogeneous Parasite Environments. Proc. R. Soc. B Biol. Sci. 2012, 279, 1896–1903. [Google Scholar] [CrossRef]
- Wisniewski-Dyé, F.; Vial, L. Phase and Antigenic Variation Mediated by Genome Modifications. Antonie van Leeuwenhoek 2008, 94, 493–515. [Google Scholar] [CrossRef]
- Van der Woude, M.W.; Bäumler, A.J. Phase and Antigenic Variation in Bacteria. Clin. Microbiol. Rev. 2004, 17, 581–611. [Google Scholar] [CrossRef] [Green Version]
- Hasman, H.; Schembri, M.A.; Klemm, P. Antigen 43 and Type 1 Fimbriae Determine Colony Morphology of Escherichia Coli K-12. J. Bacteriol. 2000, 182, 1089–1095. [Google Scholar] [CrossRef] [Green Version]
- Danese, P.N.; Pratt, L.A.; Dove, S.L.; Kolter, R. The Outer Membrane Protein, Antigen 43, Mediates Cell-to-Cell Interactions within Escherichia Coli Biofilms. Mol. Microbiol. 2000, 37, 424–432. [Google Scholar] [CrossRef]
- Zöllner, R.; Oldewurtel, E.R.; Kouzel, N.; Maier, B. Phase and Antigenic Variation Govern Competition Dynamics through Positioning in Bacterial Colonies. Sci. Rep. 2017, 7, 12151. [Google Scholar] [CrossRef] [Green Version]
- Oldewurtel, E.R.; Kouzel, N.; Dewenter, L.; Henseler, K.; Maier, B. Differential Interaction Forces Govern Bacterial Sorting in Early Biofilms. eLife 2015, 4, e10811. [Google Scholar] [CrossRef]
- Flemming, H.-C.; Wingender, J. The Biofilm Matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Lembre, P.; Lorentz, C.; Martino, P.D. Exopolysaccharides of the Biofilm Matrix: A Complex Biophysical World; IntechOpen: London, UK, 2012; ISBN 978-953-51-0819-1. [Google Scholar]
- Simmons, E.L.; Bond, M.C.; Koskella, B.; Drescher, K.; Bucci, V.; Nadell, C.D. Biofilm Structure Promotes Coexistence of Phage-Resistant and Phage-Susceptible Bacteria. mSystems 2020, 5, e00877-19. [Google Scholar] [CrossRef]
- Cimdins, A.; Simm, R.; Li, F.; Lüthje, P.; Thorell, K.; Sjöling, Å.; Brauner, A.; Römling, U. Alterations of C-di-GMP Turnover Proteins Modulate Semi-constitutive Rdar Biofilm Formation in Commensal and Uropathogenic Escherichia Coli. Microbiologyopen 2017, 6, e00508. [Google Scholar] [CrossRef]
- Römling, U. Characterization of the Rdar Morphotype, a Multicellular Behaviour in Enterobacteriaceae. Cell. Mol. Life Sci. CMLS 2005, 62, 1234–1246. [Google Scholar] [CrossRef] [PubMed]
- Grantcharova, N.; Peters, V.; Monteiro, C.; Zakikhany, K.; Römling, U. Bistable Expression of CsgD in Biofilm Development of Salmonella Enterica Serovar Typhimurium. J. Bacteriol. 2010, 192, 456–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Römling, U.; Bian, Z.; Hammar, M.; Sierralta, W.D.; Normark, S. Curli Fibers Are Highly Conserved between Salmonella Typhimurium and Escherichia Coli with Respect to Operon Structure and Regulation. J. Bacteriol. 1998, 180, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Hammar, M.; Bian, Z.; Normark, S. Nucleator-Dependent Intercellular Assembly of Adhesive Curli Organelles in Escherichia Coli. Proc. Natl. Acad. Sci. USA 1996, 93, 6562–6566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zogaj, X.; Nimtz, M.; Rohde, M.; Bokranz, W.; Römling, U. The Multicellular Morphotypes of Salmonella Typhimurium and Escherichia Coli Produce Cellulose as the Second Component of the Extracellular Matrix. Mol. Microbiol. 2001, 39, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Römling, U.; Galperin, M.Y.; Gomelsky, M. Cyclic Di-GMP: The First 25 Years of a Universal Bacterial Second Messenger. Microbiol. Mol. Biol. Rev. 2013, 77, 1–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambrook, J.; Russell, D.W. Precipitation of Bacteriophage λ Particles from Large-Scale Lysates. Cold Spring Harb. Protoc. 2006, 2006, 39. [Google Scholar] [CrossRef]
- Kropinski, A.M.; Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of Bacteriophages by Double Agar Overlay Plaque Assay. In Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization and Interactions; Methods in Molecular BiologyTM; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 69–76. ISBN 978-1-60327-164-6. [Google Scholar]
- Gratia, A. The Numerical Relation between Lysogenic Bacteria and the Phage Particles which they carry. Ann. Inst. Pasteur. 1936, 57, 652–676. [Google Scholar]
- Cornejo, O.E.; Rozen, D.E.; May, R.M.; Levin, B.R. Oscillations in Continuous Culture Populations of Streptococcus Pneumoniae: Population Dynamics and the Evolution of Clonal Suicide. Proc. R. Soc. B Boil. Sci. 2009, 276, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Naghili, H.; Tajik, H.; Mardani, K.; Razavi Rouhani, S.M.; Ehsani, A.; Zare, P. Validation of Drop Plate Technique for Bacterial Enumeration by Parametric and Nonparametric Tests. Veter. Res. Forum Int. Q. J. 2013, 4, 179–183. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Jandu, N.; Ho, N.K.L.; Donato, K.A.; Karmali, M.A.; Mascarenhas, M.; Duffy, S.P.; Tailor, C.; Sherman, P.M. Enterohemorrhagic Escherichia Coli O157: H7 Gene Expression Profiling in Response to Growth in the Presence of Host Epithelia. PLoS ONE 2009, 4, e4889. [Google Scholar] [CrossRef]
- Reid, S.D.; Herbelin, C.J.; Bumbaugh, A.C.; Selander, R.K.; Whittam, T.S. Parallel Evolution of Virulence in Pathogenic Escherichia Coli. Nature 2000, 406, 64–67. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence with Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Herigstad, B.; Hamilton, M.; Heersink, J. How to Optimize the Drop Plate Method for Enumerating Bacteria. J. Microbiol. Methods 2001, 44, 121–129. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Carpenter, D. Find Growth Rates. 2012. Available online: https://github.com/decarpen/growth-curves/blob/master/find_gr.R (accessed on 15 December 2021).
- O′Toole, G.A. Microtiter Dish Biofilm Formation Assay. J. Vis. Exp. 2011, 47, e2437. [Google Scholar] [CrossRef]
- Walker, D.; Roach, D.R.; Debarbieux, L. Phage Therapy: Awakening a Sleeping Giant. Emerg. Top. Life Sci. 2017, 1, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, B.R.; Bull, J.J. Population and Evolutionary Dynamics of Phage Therapy. Nat. Rev. Microbiol. 2004, 2, 166–173. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koonjan, S.; Cardoso Palacios, C.; Nilsson, A.S. Population Dynamics of a Two Phages–One Host Infection System Using Escherichia coli Strain ECOR57 and Phages vB_EcoP_SU10 and vB_EcoD_SU57. Pharmaceuticals 2022, 15, 268. https://doi.org/10.3390/ph15030268
Koonjan S, Cardoso Palacios C, Nilsson AS. Population Dynamics of a Two Phages–One Host Infection System Using Escherichia coli Strain ECOR57 and Phages vB_EcoP_SU10 and vB_EcoD_SU57. Pharmaceuticals. 2022; 15(3):268. https://doi.org/10.3390/ph15030268
Chicago/Turabian StyleKoonjan, Shazeeda, Carlos Cardoso Palacios, and Anders S. Nilsson. 2022. "Population Dynamics of a Two Phages–One Host Infection System Using Escherichia coli Strain ECOR57 and Phages vB_EcoP_SU10 and vB_EcoD_SU57" Pharmaceuticals 15, no. 3: 268. https://doi.org/10.3390/ph15030268
APA StyleKoonjan, S., Cardoso Palacios, C., & Nilsson, A. S. (2022). Population Dynamics of a Two Phages–One Host Infection System Using Escherichia coli Strain ECOR57 and Phages vB_EcoP_SU10 and vB_EcoD_SU57. Pharmaceuticals, 15(3), 268. https://doi.org/10.3390/ph15030268