
Small Molecule Induced FLT3 Degradation


Abstract
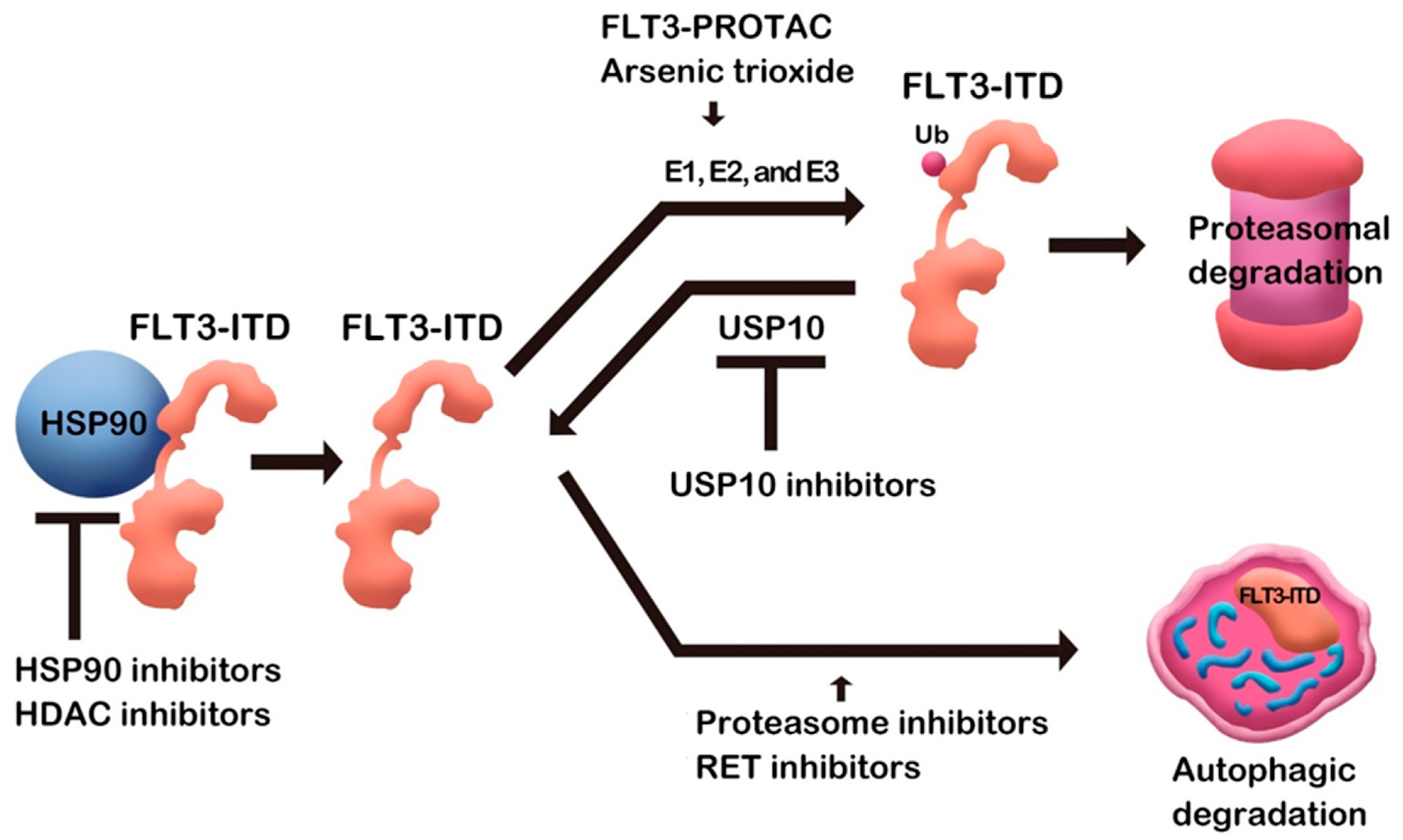
:1. Introduction
2. HSP90-Mediated FLT3 Degradation
2.1. Hsp90 Inhibitors
2.2. Green Tea Catechins
2.3. FLT3 as Hsp90 Client Protein
3. Proteasome Inhibitor
4. Arsenic Trioxide
5. HDAC Inhibitors
6. RET Inhibitors
7. FLT3 PROTAC
8. Ubiquitin-Proteasome System for FLT3
9. FLT3 Phosphorylation Status and Degradation
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dale, B.; Cheng, M.; Park, K.-S.; Kaniskan, H.; Xiong, Y.; Jin, J. Advancing targeted protein degradation for cancer therapy. Nat. Cancer 2021, 21, 638–654. [Google Scholar] [CrossRef] [PubMed]
- Newell, L.F.; Cook, R.J. Advances in acute myeloid leukemia. BMJ 2021, 375, n2026. [Google Scholar] [CrossRef] [PubMed]
- DiPiro, J.T. Pharmacotherapy: A Pathophysiologic Approach, 10th ed.; McGraw-Hill Medical: New York, NY, USA, 2016. [Google Scholar]
- Stirewalt, D.L.; Radich, J.P. The role of FLT3 in haematopoietic malignancies. Nat. Cancer 2003, 3, 650–665. [Google Scholar] [CrossRef]
- Nakao, M.; Yokota, S.; Iwai, T.; Kaneko, H.; Horiike, S.; Kashima, K.; Sonoda, Y.; Fujimoto, T.; Misawa, S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996, 10, 1911–1918. [Google Scholar]
- Wang, W.; Wang, X.-Q.; Xu, X.-P.; Lin, G.-W. Prevalence and prognostic significance of FLT3 gene mutations in patients with acute leukaemia: Analysis of patients from the shanghai leukaemia co-operative group. J. Int. Med. Res. 2010, 38, 432–442. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Kiyoi, H.; Nakano, Y.; Suzuki, R.; Kodera, Y.; Miyawaki, S.; Asou, N.; Kuriyama, K.; Yagasaki, F.; Shimazaki, C.; et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001, 97, 2434–2439. [Google Scholar] [CrossRef] [Green Version]
- Levis, M. Midostaurin approved for FLT3-mutated AML. Blood 2017, 129, 3403–3406. [Google Scholar] [CrossRef]
- Pulte, E.D.; Norsworthy, K.J.; Wang, Y.; Xu, Q.; Qosa, H.; Gudi, R.; Przepiorka, D.; Fu, W.; Okusanya, O.O.; Goldberg, K.B.; et al. FDA approval summary: Gilteritinib for relapsed or refractory acute myeloid leukemia with a FLT3 mutation. Clin. Cancer Res. 2021, 27, 3515–3521. [Google Scholar] [CrossRef]
- Lovly, C.; Shaw, A.T. Molecular Pathways: Resistance to Kinase Inhibitors and Implications for Therapeutic Strategies. Clin. Cancer Res. 2014, 20, 2249–2256. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, E.P.; Sattler, M.B.; Ray, A.K.; Griffin, J.D. Drug resistance in mutant FLT3-positive AML. Oncogene 2010, 29, 5120–5134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyoi, H.; Kawashima, N.; Ishikawa, Y. FLT3 mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Sci. 2019, 111, 312–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, V.E.; Smith, C.C. FLT3 mutations in acute myeloid leukemia: Key concepts and emerging controversies. Front. Oncol. 2020, 10, 612880. [Google Scholar] [CrossRef] [PubMed]
- Fröhling, S.; Scholl, C.; Levine, R.L.; Loriaux, M.; Boggon, T.J.; Bernard, O.; Berger, R.; Döhner, H.; Döhner, K.; Ebert, B.L.; et al. Identification of driver and passenger mutations of FLT3 by high-throughput DNA sequence analysis and functional assessment of candidate alleles. Cancer Cell 2007, 12, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.C.; Zhang, C.; Lin, K.C.; Lasater, E.A.; Zhang, Y.; Massi, E.; Damon, L.E.; Pendleton, M.; Bashir, A.; Sebra, R.; et al. Characterizing and overriding the structural mechanism of the quizartinib-resistant flt3 “Gatekeeper” F691L mutation with PLX3397. Cancer Discov. 2015, 5, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Scholl, S.; Fleischmann, M.; Schnetzke, U.; Heidel, F.H. Molecular mechanisms of resistance to FLT3 inhibitors in acute myeloid Leukemia: Ongoing challenges and future treatments. Cells 2020, 9, 2493. [Google Scholar] [CrossRef]
- Yao, Q.; Nishiuchi, R.; Li, Q.; Kumar, A.R.; Hudson, W.A.; Kersey, J.H. FLT3 expressing leukemias are selectively sensitive to inhibitors of the molecular chaperone heat shock protein 90 through destabilization of signal transduction-associated kinases. Clin. Cancer Res. 2003, 9, 4483–4493. [Google Scholar]
- Ly, B.T.K.; Chi, H.T.; Yamagishi, M.; Kano, Y.; Hara, Y.; Nakano, K.; Sato, Y.; Watanabe, T. Inhibition of FLT3 expression by green tea catechins in FLT3 Mutated-AML cells. PLoS ONE 2013, 8, e66378. [Google Scholar] [CrossRef] [Green Version]
- Minami, Y.; Kiyoi, H.; Yamamoto, Y.; Ueda, R.; Saito, H.; Naoe, T. Selective apoptosis of tandemly duplicated FLT3-transformed leukemia cells by Hsp90 inhibitors. Leukemia 2002, 16, 1535–1540. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Nishiuchi, R.; Kitamura, T.; Kersey, J.H. Human leukemias with mutated FLT3 kinase are synergistically sensitive to FLT3 and Hsp90 inhibitors: The key role of the STAT5 signal transduction pathway. Leukemia 2005, 19, 1605–1612. [Google Scholar] [CrossRef]
- Yao, Q.; Weigel, B.; Kersey, J. Synergism between etoposide and 17-AAG in leukemia cells: Critical roles for Hsp90, FLT3, Topoisomerase II, Chk1, and Rad51. Clin. Cancer Res. 2007, 13, 1591–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Shaer, L.; Walsby, E.; Gilkes, A.; Tonks, A.; Walsh, V.; Mills, K.; Burnett, A.; Rowntree, C. Heat shock protein 90 inhibition is cytotoxic to primary AML cells expressing mutant FLT3 and results in altered downstream signalling. Br. J. Haematol. 2008, 141, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Oshikawa, G.; Nagao, T.; Wu, N.; Kurosu, T.; Miura, O. c-Cbl and Cbl-b ligases mediate 17-allylaminodemethoxygeldanamycin-induced degradation of autophosphorylated FLT3 kinase with internal tandem duplication through the ubiquitin proteasome pathway. J. Biol. Chem. 2011, 286, 30263–30273. [Google Scholar] [CrossRef] [Green Version]
- Ly, B.T.K.; Chi, H.T. ETV6/FLT3 fusion is a novel client protein of Hsp90. Oncol. Res. 2018, 26, 1201–1205. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Kancha, R.K.; Duyster, J. Targeting Oncoprotein Stability Overcomes Drug Resistance Caused by FLT3 Kinase Domain Mutations. PLoS ONE 2014, 9, e97116. [Google Scholar] [CrossRef] [Green Version]
- Workman, P.; Burrows, F.; Neckers, L.; Rosen, N. Drugging the cancer chaperone HSP90: Combinatorial therapeutic exploitation of oncogene addiction and tumor Stress. Ann. N. Y. Acad. Sci. 2007, 1113, 202–216. [Google Scholar] [CrossRef]
- Larrue, C.; Saland, E.; Boutzen, H.; Vergez, F.; David, M.; Joffre, C.; Hospital, M.-A.; Tamburini, J.; Delabesse, E.; Manenti, S.; et al. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood 2016, 127, 882–892. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.-N.; Tang, Y.-L.; Zhang, Y.-C.; Zhang, Z.-H.; Liu, X.-J.; Ke, Z.-Y.; Li, Y.; Tan, H.-Z.; Huang, L.-B.; Luo, X.-Q. Arsenic trioxide and all-trans-retinoic acid selectively exert synergistic cytotoxicity against FLT3-ITD AML cells via co-inhibition of FLT3 signaling pathways. Leuk. Lymphoma 2017, 58, 2426–2438. [Google Scholar] [CrossRef]
- Liang, C.; Peng, C.-J.; Wang, L.-N.; Li, Y.; Zheng, L.-M.; Fan, Z.; Huang, D.-P.; Tang, W.-Y.; Zhang, X.-L.; Huang, L.-B.; et al. Arsenic trioxide and all-trans retinoic acid suppress the expression of FLT3-ITD. Leuk. Lymphoma 2020, 61, 2692–2699. [Google Scholar] [CrossRef]
- Nagai, K.; Hou, L.; Li, L.; Nguyen, B.; Seale, T.; Shirley, C.; Ma, H.; Levis, M.; Ghiaur, G.; Duffield, A.; et al. Combination of ATO with FLT3 TKIs eliminates FLT3/ITD+ leukemia cells through reduced expression of FLT3. Oncotarget 2018, 9, 32885–32899. [Google Scholar] [CrossRef] [Green Version]
- Slingerland, M.; Guchelaar, H.J.; Gelderblom, H. Histone deacetylase inhibitors: An overview of the clinical studies in solid tumors. Anticancer Drugs 2014, 25, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Bali, P.; George, P.; Cohen, P.; Tao, J.; Guo, F.; Sigua, C.; Vishvanath, A.; Scuto, A.; Annavarapu, S.; Fiskus, W.; et al. Superior activity of the combination of histone deacetylase inhibitor LAQ824 and the FLT-3 Kinase inhibitor PKC412 against human acute myelogenous leukemia cells with mutant FLT-3. Clin. Cancer Res. 2004, 10, 4991–4997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, P.; Bali, P.; Annavarapu, S.; Scuto, A.; Fiskus, W.; Guo, F.; Sigua, C.; Sondarva, G.; Moscinski, L.; Atadja, P.; et al. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood 2005, 105, 1768–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudat, S.; Pfaus, A.; Cheng, Y.Y.; Holtmann, J.; Ellegast, J.; Bühler, C.; Di Marcantonio, D.; Martinez, E.; Göllner, S.; Wickenhauser, C.; et al. RET-Mediated autophagy suppression as targetable co-dependence in acute myeloid leukemia. Leukemia 2018, 32, 2189–2202. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-T.; Dobrovolsky, D.; Paulk, J.; Yang, G.; Weisberg, E.L.; Doctor, Z.M.; Buckley, D.L.; Cho, J.-H.; Ko, E.; Jang, J.; et al. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem. Biol. 2018, 25, 88–99.e6. [Google Scholar] [CrossRef] [Green Version]
- Burslem, G.M.; Song, J.; Chen, X.; Hines, J.; Crews, C.M. Enhancing antiproliferative activity and selectivity of a FLT-3 Inhibitor by proteolysis targeting chimera conversion. J. Am. Chem. Soc. 2018, 140, 16428–16432. [Google Scholar] [CrossRef]
- Chen, Y.; Yuan, X.; Tang, M.; Shi, M.; Yang, T.; Liu, K.; Deng, D.; Chen, L. Degrading FLT3-ITD protein by proteolysis targeting chimera (PROTAC). Bioorg. Chem. 2022, 119, 105508. [Google Scholar] [CrossRef]
- Donovan, K.A.; Ferguson, F.M.; Bushman, J.W.; Eleuteri, N.A.; Bhunia, D.; Ryu, S.; Tan, L.; Shi, K.; Yue, H.; Liu, X.; et al. Mapping the Degradable Kinome Provides a Resource for Expedited Degrader Development. Cell 2020, 183, 1714–1731.e10. [Google Scholar] [CrossRef]
- Nalepa, G.; Rolfe, M.; Harper, J. Drug discovery in the ubiquitin–proteasome system. Nat. Rev. Drug Discov. 2006, 5, 596–613. [Google Scholar] [CrossRef]
- Sargin, B.; Choudhary, C.; Crosetto, N.; Schmidt, M.H.H.; Grundler, R.; Rensinghoff, M.; Thiessen, C.; Tickenbrock, L.; Schwäble, J.; Brandts, C.; et al. Flt3-Dependent transformation by inactivating c-Cbl mutations in AML. Blood 2007, 110, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Buchwald, M.; Pietschmann, K.; Müller, J.P.; Böhmer, F.D.; Heinzel, T.; Krämer, O. Ubiquitin conjugase UBCH8 targets active FMS-like tyrosine kinase 3 for proteasomal degradation. Leukemia 2010, 24, 1412–1421. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Grossman, S.R. Ubiquitin becomes ubiquitous in cancer: Emerging roles of ubiquitin ligases and deubiquitinases in tumorigenesis and as therapeutic targets. Cancer Biol. Ther. 2010, 10, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, E.L.; Schauer, N.; Yang, J.; Lamberto, I.; Doherty, L.; Bhatt, S.; Nonami, A.; Meng, C.; Letai, A.; Wright, R.; et al. Inhibition of USP10 induces degradation of oncogenic FLT3. Nat. Chem. Biol. 2017, 13, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Fang, Z.-X.; Wang, W.-W.; Zhang, Y.; Bu, Z.-L.; Liu, M.; Xiao, X.-H.; Zhang, Z.-L.; Zhang, X.-M.; Cao, Y.; et al. Wu-5, a novel USP10 inhibitor, enhances crenolanib-induced FLT3-ITD-positive AML cell death via inhibiting FLT3 and AMPK pathways. Acta Pharmacol. Sin. 2021, 42, 604–612. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| FLT3 Degrader | Mechanism | Reference |
|---|---|---|
| 17-AAG | Hsp90 inhibition | [18] |
| EGCG, EGC, ECG | Hsp90 inhibition | [19] |
| Bortezomib | Proteasome inhibition | [28] |
| Arsenic trioxide | FLT3-ITD ubiquitination | [29,30] |
| LAQ284 | HDAC inhibition | [33] |
| LBH589 | HDAC inhibition | [34] |
| Vandetanib | RET inhibition | [35] |
| Danusertib | RET inhibition | [35] |
| Wu-5 | USP10 inhibition | [45] |
| FLT3-PROTAC | PROTAC | [36,37,38] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, S.-Y. Small Molecule Induced FLT3 Degradation. Pharmaceuticals 2022, 15, 320. https://doi.org/10.3390/ph15030320
Han S-Y. Small Molecule Induced FLT3 Degradation. Pharmaceuticals. 2022; 15(3):320. https://doi.org/10.3390/ph15030320
Chicago/Turabian StyleHan, Sun-Young. 2022. "Small Molecule Induced FLT3 Degradation" Pharmaceuticals 15, no. 3: 320. https://doi.org/10.3390/ph15030320
APA StyleHan, S. -Y. (2022). Small Molecule Induced FLT3 Degradation. Pharmaceuticals, 15(3), 320. https://doi.org/10.3390/ph15030320