
Effect of Statins on Lung Cancer Molecular Pathways: A Possible Therapeutic Role

 , , ,
, , ,  ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Material and Methods
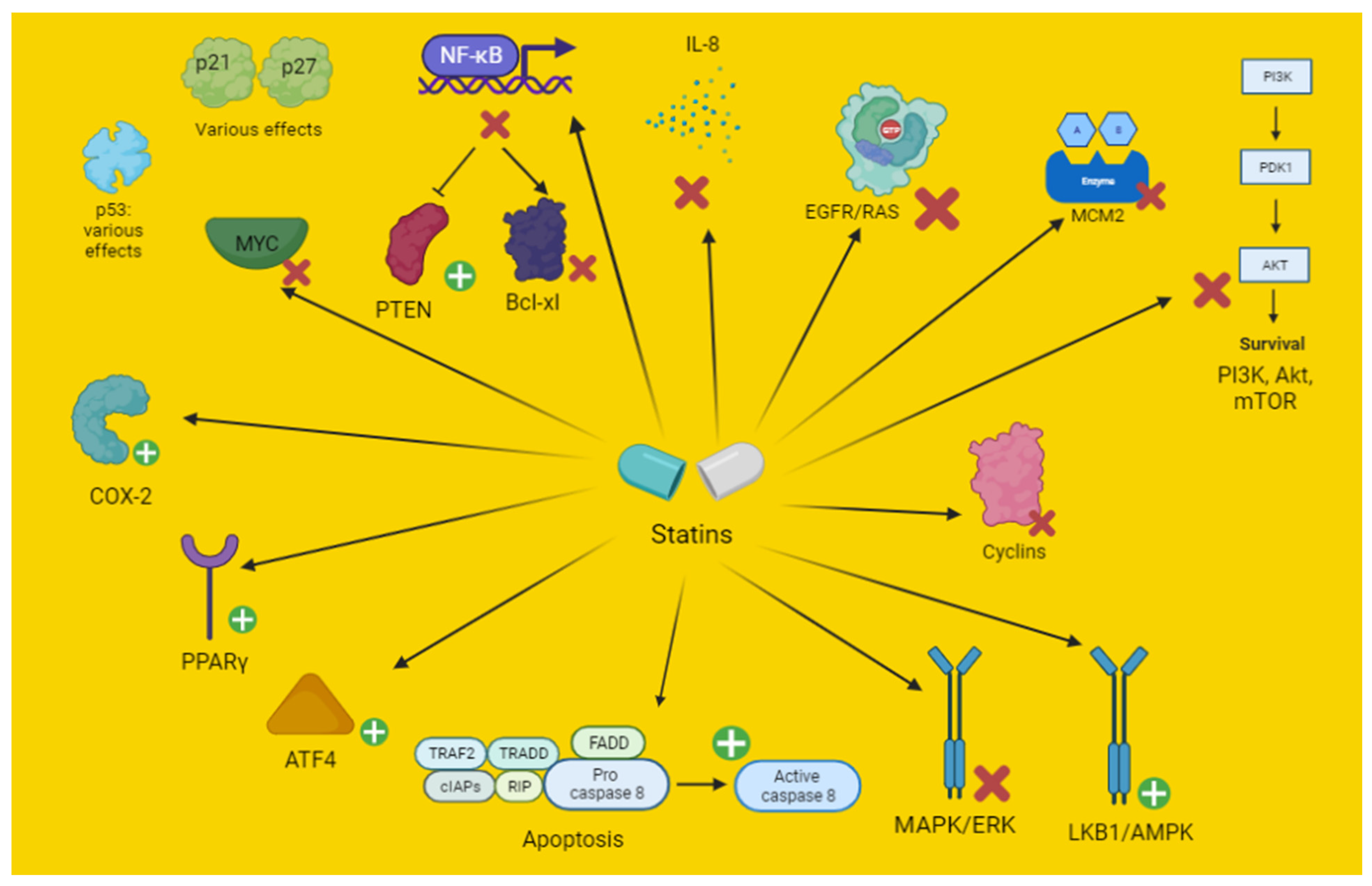
3. Effects of Statins on Lung Cancer Molecular Pathways
3.1. Antiproliferative and Pro-Apoptotic Effects
3.1.1. In Vitro and Preclinical Studies
3.1.2. Clinical Studies

{kind=link}
{kind=link}
{kind=link}
| Antiproliferative and Pro-Apoptotic Effects | ||
|---|---|---|
| In Vitro | ||
| Statin | Dosage | Mechanism and Effects |
| Fluvastatin | EGFR TKI-resistant NSCLC cell lines A549 and Calu6 (in combination with erlotinib). Minor effect in H1993 cells. 50–100 μM | Cells’ resistance related to K-RAS, EGFR or MET mutation. Inhibition of EGFR/K-RAS and then of Akt [30] |
| Lovastatin | 0–50 μM | A549 lung adenocarcinoma cells were treated with lovastatin alone or in combination with 0 to 8 Gy IR. Lovastatin reduced EGF-induced phosphorylation of EGFR and Akt, and IR-induced Akt phosphorylation. Furthermore, lovastatin enhanced AMPK expression, and reduced p53 and the cyclin-dependent kinase inhibitors p21cip1 and p27kip1 expression [32] |
| Study conducted on A549 and H358 lung carcinoma cells Effect on viability: 76.7 μM (A549) and 45.2 μM (H358). Apoptotic effect: 50 μM (A549) and 75 μM (H358). | Pro-apoptotic action in lung cancer cells upregulating COX-2 and PPAR-γ [40] | |
| Two NSCLC cell lines, A549 and GLC-82: various concentrations (0, 2.5, 5, 10 and 20 μmol/L) for 1 to 4 days, respectively | MCM 2 is targeted by lovastatin in NSCLC cells. This inhibition led to cellular cycle block, inhibiting cyclin D1, CDK4 and Rb, but increasing p21 and p53 expression [47]. | |
| Pitavastatin | EGFR TKI-resistant NSCLC cell lines A549 and Calu6 (in combination with erlotinib). Minor effect in H1993 cells 50–100 μM. Improved effect with erlotinib 5 μM, with a pitavastatin dosage of 1–50 μM (Calu6) and 1–10 μM (A549). | Cells’ resistance related to K-RAS, EGFR or MET mutation. Inhibition of EGFR/K-RAS and then of Akt, resulting in increased apoptosis [30] |
| Rosuvastatin | 1.25–30 μM | Reduction of RAS expression in an in vitro study on SCLC patients samples [21] |
| Simvastatin | 28 μM in Bm7 (R248W) p53 mutant cells (cytotoxicity). 7 μM (IC50) in Bm7 cells (cell growth inhibition). 10 or 50 μM increased apoptosis in a dose-dependent manner in Bm7 cells. 1 μM decreased lipid raft and cell motility. | Cytotoxic, apoptotic, effect in p53 mutated cells. Cell growth, motility and lipid rafts inhibition, alongside mutant p53 degradation [26] |
| 2.5–30 μM (30 μM best results) | Reduction of RAS expression in an in vitro study of SCLC patient samples [21] | |
| Human lung cancer cell line A549: 10 and 50 μM | Decreased Bcl-2, cyclin D1 and CDKs, Xiap, NF-kB; increased Bax, caspase-3, -8, and -9 mRNA [34]. | |
| H1975 NSCLC cells 2 μM for 48 h. | Expression of pro-apoptotic proteins was increased. Reduction of ERK 1/2 phosphorylation. Expression of BIM was blocked by gefitinib (1 μM), but significantly enhanced by simvastatin [48]. | |
| GLC-82 human lung adenocarcinoma cell line: 30 μM | Inhibition of stimulatory actions on ERK1/2 phosphorylation, NF-κB activation and IL-8 production. Simvastatin decreased H2O2-mediated induction of the cellular expression of MMP-2 and MMP-9, as well as of several components of the signaling complex activated by innate immune responses, including MyD88, TRAF2, TRAF6 and TRADD [19]. | |
| In vivo (animals) | ||
| Simvastatin | 5–10 mg/kg in Balb/C nude mice A459 cancer cells | Inhibition of MAPK/ERK pathway. Increase of p53 expression [29]. |
| In vivo (humans) | ||
| All statins | Dosage commonly used for hypercholesterolemia | Action on P53, improved prognosis in early stage patients (10,975 patients analyzed retrospectively) [26] |
| Atorvastatin | Atorvastatin (40–80 mg). Observational study performed in 253 patients with malignant pleural mesothelioma or advanced NSCLC treated with PD-1 inhibitors. | Better response and progression-free survival. These effects probably due to immune enhancement related to a prolonged retention of antigens on cell membrane and presentation increase [41]. |
| Rosuvastatin | Rosuvastatin (20–40 mg): high intensity. Observational study performed in 253 patients with malignant pleural mesothelioma or advanced NSCLC treated with PD-1 inhibitors. | Better response and progression-free survival. These effects probably due to immune enhancement related to prolonged retention of antigens on cell membrane and presentation increase [41]. |
| Antiproliferative and Pro-Apoptotic Effects | ||
|---|---|---|
| In Vitro | ||
| Statin | Dosage | Mechanism and Effects |
| Lovastatin | Mammary tumor cells: 0.25 μM | Combination low-dose statin and γ-tocotrienol induced cell cycle arrest at G1. Increased p27 and corresponding decrease in cyclin D1, CDK2, and hypophosphorylation of Rb protein [49] |
| HGT-1 gastric cancer cells: 12.5 μM alone or in combination with docetaxel. Lovastatin tested alone: 2.5, 5 μM for 48–72 h. | Apoptosis increase (better in combination). Increase of p21 and p27, with reduction of aurora kinases A and B, cyclins B1 and D1. Cleavage of procaspase-3, reduction of Mcl-1 protein, Poly-ADP-Ribose Polymerase and Bax. HGT-1 cell derivatives overexpressing the MDR-1 gene were more sensitive to lovastatin than docetaxel-sensitive cells [50]. | |
| 10–50 μM in HNSCC cells | Activation of integrated stress response through ATF4 stimulation [51] | |
| 1–25 μM SCC cells | Induction of LKB1 and AMPK activation [52] | |
| Mevastatin | Mammary tumor cells: 0.25 μM | Combination low-dose statin and γ-tocotrienol induced cell cycle arrest at G1. Increased p27 and corresponding decrease in cyclin D1, CDK2, and hypophosphorylation of Rb protein [49] |
| Pravastatin | Mammary tumor cells: 10 μM | Combination low-dose statin and γ-tocotrienol induced cell cycle arrest at G1. Increased p27 and corresponding decrease in cyclin D1, CDK2, and hypophosphorylation of Rb protein [49] |
| Simvastatin | Mammary tumor cells: 0.25 μM | Combination low-dose statin and γ-tocotrienol induced cell cycle arrest at G1. Increased p27 and corresponding decrease in cyclin D1, CDK2, and hypophosphorylation of Rb protein [49] |
| U251 and C6 glioma cell lines: 6 μM | AMPK, Raptor activation Downregulation of Akt, mTOR, p70 S6 kinase 1. These actions result in increased autophagy and cell survival reverted by autophagy inhibitors [53] | |
| In vivo (animals) | ||
| Atorvastatin | 100 mg/kg three times a week in transgenic mouse (HCC model) | The inhibition of HMG-CoA reductase suppresses MYC phosphorylation through Rac GTPase [54] |
| In vivo: 50 mg/kg/die for 21 days in BALB/c nude mice injected with HCC Huh7 cells. In vitro: CRC cells (HCT116), HCC cells (Huh7): 50 μmol/L atorvastatin for 2 and 5 days, respectively | Activation of AMPK, p21, promoting cell survival. A combination with an autophagy inhibitor may revert this effect [38]. | |
| Lovastatin | Daoy or D283 medulloblastoma cells: 10 and 40 μM Transfected mice: 1.0 mg/kg three times per week for 4 weeks | MYC inhibition through miR-33b increase [55] |
| Simvastatin | MDAMB-231 human breast cancer cell xenografts in mice: 5 mg/kg/die for 7 days | Inhibition of NFκB was associated with PTEN derepression and Bcl-xL reduction in breast cancer [35] |
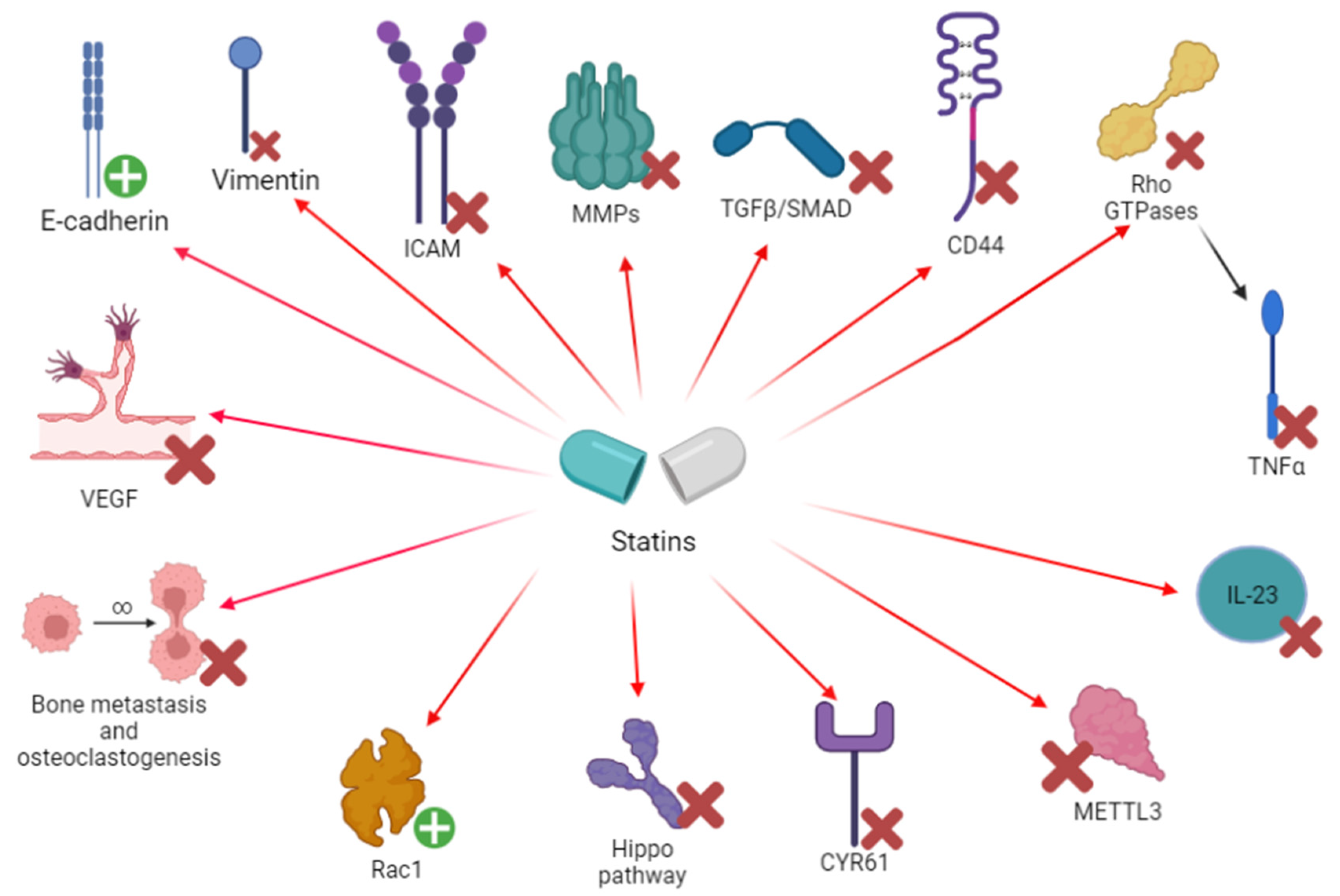
3.2. Chemotaxis, Invasion and Angiogenesis
3.2.1. Experimental Studies
3.2.2. Clinical Evidence
| In Vitro | ||
| Rosuvastatin | Lung cancer patient samples: 1.25–30 μM | Simvastatin reduced MMP2 and MMP9 (p < 0.01) expression in lung cancer tissue. Reduction of NFκB, both in normal and cancer tissue [21] |
| Simvastatin | 10–20 μg in A459 cells [56] 5–10 mg, analyzed in human samples (H1650 and H1975 cells) [58] | Inhibition of EMT, inhibiting TGF-β1 activity and SMAD pathway [56] 250 specimens from lung cancer patients, 51 of whom were treated with statins (mainly simvastatin), revealed an improved prognosis [58] |
| Lung cancer patient samples: 2.5–30 μM | Simvastatin reduced MMP2 and MMP9 (p < 0.01) expression in lung cancer tissue. Reduction of NFκB, both in normal and cancer tissue [21] | |
| A459 cells: 0, 5, 10 or 20 μM (the latter being the main dose) for 24 h. | METTL3 inhibition, thus reducing EZH2 activity in generating EMT in lung cancer cells. METTL3 may regulate the levels of EMT-associated genes, including JUNB [80]. | |
| Human lung cancer cell line A549: 10 and 50 μM | MMP-9 suppression [34] | |
| 5–10 mg/kg in Balb/C nude mice A459 cancer cells | Reduction of CD44, MMP-2 and MMP-9 [29]. Proliferation and bone metastasis inhibition | |
| In vivo (animals) | ||
| Atorvastatin | 10 mg/kg per day in mice | VEGF inhibition, through the blocking of ROS production, the suppression of Rac1/NADPH oxidase activity and the upregulation of glutathione peroxidase and catalase [81] |
| Endothelial cells: 0.005 to 0.01 μmol/L dose led to increased vascularization 0.05–1 μmol/L dose led to decreased vascularization Mice: In lung cancer murine models, inflammation-induced angiogenesis was enhanced with low-dose statin therapy (0.5 mg/kg/die), reduced with high dose (2.5 mg/kg/die) | Decreased VEGF at high dose [74] | |
| Cerivastatin | Endothelial cells: 0.005 to 0.01 μmol/L dose led to increased vascularization 0.05–1 μmol/L dose led to decreased vascularization Mice: In murine models, inflammation-induced angiogenesis was enhanced with low-dose statin therapy (0.5 mg/kg/die), reduced with high dose (2.5 mg/kg/die) | Decreased VEGF at high dose [74] |
| In vitro and in vivo murine models (1 mg/kg daily) | Cerivastatin was effective in inhibiting Hippo pathway, acting on YAP/TAZ and reducing expression of YAP-targeted oncogenes (EGFR, AXL, CYR61, and TGFbR2) [70] | |
| Fluvastatin | Mouse model: fluvastatin, 50 mg/kg | Fluvastatin may inhibit lung cancer bone metastasis [71]. Fluvastatin induced cell autophagy, preventing their spread (related to p53 increase) [72]. |
| Simvastatin | Inoculation of A459 cells in mice: 10 mg/kg simvastatin every day for 7 days | Reduction of CD44, MMP2 and MMP9 [29] |
| Chemotaxis and Invasion | ||
|---|---|---|
| In Vitro | ||
| Atorvastatin | In osteosarcoma cells: 10 μM | CYR61 silencing was an unfavorable setting for cancer proliferation, resulting in increased cell death [69] |
| Cerivastatin | 25 ng/mL in breast cancer MDA-MB-231 cells [60]. Aggressive breast cancer cell line, characterized by RAS and NFκB overactivation and RhoA high expression | Target: Rho GTPases (inhibition) Rho A inhibition may also result in NFκB and MMP downregulation |
| In osteosarcoma cells: dosage not available | CYR61 silencing was an unfavorable setting for cancer proliferation, resulting in increased cell death [69] | |
| Lovastatin | 0.1–1 μM in a study on colon carcinoma cells. Inhibitory concentration of lovastatin was in a physiologically relevant range (IC50 < 0.1 μM) | Lovastatin was effective in reducing E-selectin levels through Rho-mediated inhibition of TNFα [66] |
| Pravastatin | In osteosarcoma cells: dosage not available | CYR61 silencing was an unfavorable setting for cancer proliferation, resulting in increased cell death [69] |
| Simvastatin | HCT116 colorectal cancer cells: 10 μg | Paradoxical activation of RhoA, Cdc 42 and Rac1. Statins may also paradoxically activate Rho GTPases and their action is not easy to predict. Rho GDIα inhibition removed [61] |
| In seven human/murine osteosarcoma cells: dosage not available | CYR61 silencing was an unfavorable setting for cancer proliferation, resulting in increased cell death [69] | |
| In vivo (animals) | ||
| Simvastatin | In a mouse model of bone loss: 10 mg/kg Mouse derived macrophage-like cells were observed and mice bone mineral density | Simvastatin reduced osteoclastogenesis, a process enhanced by neoplastic cells, through RANK-L and IL-6 inhibition. Probable IRF4, NFκB and NFATc1 inhibition [73] |
| In vivo (humans) | ||
| All statins (mainly lipophilic statins) | Hypercholesterolemia dosage | Relevant reduction of VEGF in patients treated with statins. This effect was observed in patients treated with lipophilic statins, therapy duration ≥ 4 weeks, LDL-C reductions ≥ 50 mg/dL, and among people affected by a relevant comorbidity (in general population) [75] |
| Hypercholesterolemia dosage, various dosages | E-selectin and P-selectin reduction: statins (especially simvastatin) [77] | |
| Atorvastatin and Rosuvastatin | 184 aneurysmatic patients (two groups: statin and non-statin). Group III: non-aneurysmatic patients in statin treatment. ≤40 mg rosuvastatin, ≤80 mg atorvastatin | MMPs and NGAL reduction [39] |
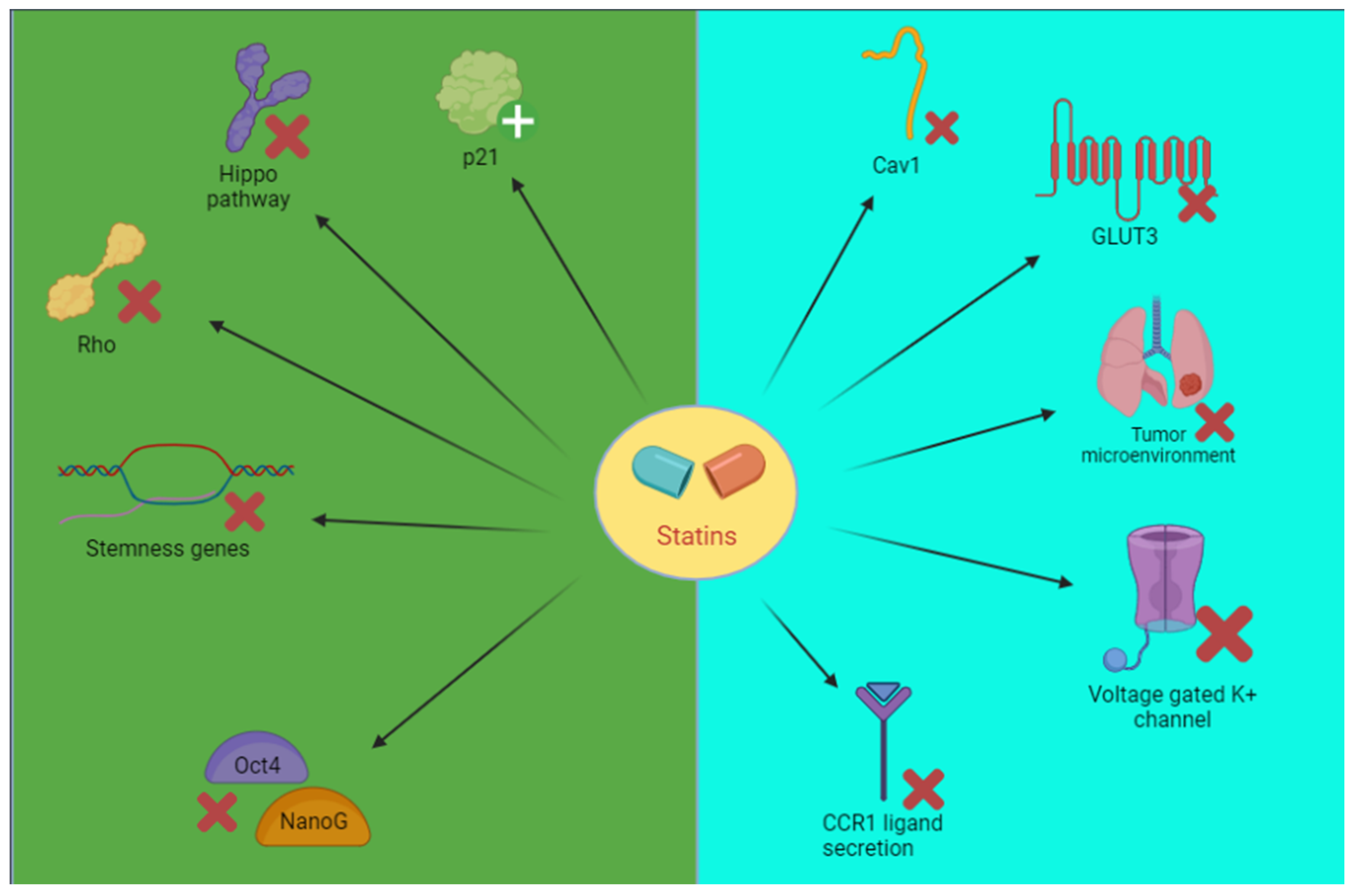
Cancer Stem Cells
Other Targets
| Cancer Stem Cells | ||
|---|---|---|
| In vitro | ||
| Cerivastatin | 1 μM in H1299 NSCLC cells | YAP/TAZ inhibition, MCM7 inhibition, p21 restoration [82] |
| Other targets | ||
| In vitro | ||
| Simvastatin | 5 or 10 μM in human SCC cells. Action on microenvironment | Simvastatin inhibits the MSCs–PCCs crosstalk. Pleiotropic effects on cell metabolism, suppression of IL-6 and CCL2 production by MSCs and CCL3 secretion by PCCs [85] |
| In vivo (animals) | ||
| Atorvastatin | In vitro/in vivo study: Mice: 30 mg/kg Cells (TKI-sensitive (PC-9 and HCC827) and resistant (PC-9GR, H1975, and H1703)): 0.2, 1 or 5 μM | Atorvastatin (in combination with tyrosine kinase inhibitors, TKI) showed a specific in vitro/in vivo action against TKI resistant NSCLC cells. Inhibition of Cav1 and GLUT 3 [83] |
| Lung adenocarcinoma mice: 10 mg/kg/day | Inhibition of pro-tumorigenic macrophages in the tumor microenvironment. Inhibition of Rac-mediated CCR1 ligand secretion [86] | |
| Cancer Stem Cells | ||
|---|---|---|
| In Vitro | ||
| Lovastatin | Studied in hESC (HES3), karyotypically abnormal hESC (BG0IV), embryonal carcinoma (NTERA-2), ovarian (TOV-112D) and colorectal cancer (HT-29) cells 1−20 μmol/L (significant at 20 μmol/L) | Ineffective in hESC. However, BG01V, NTERA-2, TOV-112D and HT-29 were inhibited (apoptosis in karyotypically abnormal cancer cells, suppression of stemness-genes on chromosome 12 and 17) [87] |
| Abnormal hESCs (BG01V) and breast adenocarcinoma cells (MCF-7) | Downregulation of Oct4 and NanoG, stemness gene reduction [88] | |
| Mevastatin | Studied in hESC (HES3), karyotypically abnormal hESC (BG0IV), embryonal carcinoma (NTERA-2), ovarian (TOV-112D) and colorectal cancer (HT-29) cells 1−20 μmol/L (significant at 10 μmol/L) | Inhibition of cell proliferation [87] |
| Abnormal hESCs (BG01V) and breast adenocarcinoma cells (MCF-7) | Downregulation of Oct4 and NanoG, stemness gene reduction [88] | |
| Simvastatin | Mouse embryonic stem cells (J1, D3, and RW.4): 10 μM | RhoA and YAP/TAZ inhibition. Oct4 and NanoG downregulation [88,89] |
| Abnormal hESCs (BG01V) and breast adenocarcinoma cells (MCF-7):5, 10, 20 mmol/L | Downregulation of Oct4 and NanoG, stemness genes reduction [88] | |
| Studied in hESC (HES3), karyotypically abnormal hESC (BG0IV), embryonal carcinoma (NTERA-2), ovarian (TOV-112D) and colorectal cancer (HT-29) cells 1−20 μmol/L (significant at 20 μmol/L) | Simvastatin was the most potent inhibitor. Ineffective in hESC. However, BG01V, NTERA-2, TOV-112D and HT-29 were inhibited (apoptosis in karyotypically abnormal cancer cells, suppression of stemness-genes on chromosome 12 and 17) [87] | |
| Other targets | ||
| In vitro | ||
| Mevastatin, pravastatin and simvastatin | Neoplastic T cells Jurkat: 1.5 μM to 50 μM 30 μM (mevastatin and simvastatin), 50 μM (pravastatin) being the main dosages | Target: voltage gated potassium channel of the Kv1.3 type [84] Simvastatin was the more potent drug, pravastatin the less. Inhibitory effect was partially irreversible with simvastatin and completely reversible with pravastatin and mevastatin |
4. Statins’ Effectiveness in Lung Cancer
5. Gender Influence on Statins’ Effect in Lung Cancer
6. Discussion and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABL | abelson tyrosine-protein kinase |
| ACLY | ATP citrate lyase |
| ACSL | Acyl-CoA synthetase long chain family member |
| ADP | adenosine diphosphate |
| Akt | protein kinase B |
| ALK | anaplastic lymphoma kinase |
| AMPK | 5′ adenosine monophosphate-activated protein kinase |
| ApoB | apolipoprotein B |
| ARIC | atherosclerosis risk in communities |
| ATF | activation of transcription factor |
| AXL | anexelekto |
| BA | bioavailability |
| BAX | bcl-2-like protein 4 |
| BCL | B-cell lymphoma (apoptosis gene) |
| Bcl-XL | B-cell lymphoma–extra large |
| BMI | body mass index |
| Cav | caveolin |
| CCI | Charlson comorbidity index |
| CCL | CC chemokine ligands |
| CCR | chemokine receptor type |
| CD | cluster of differentiation |
| CDC | cell division control |
| CDK | cyclin-dependent kinase |
| COPD | chronic obstructive pulmonary disease |
| COX | cyclooxygenase |
| CVD | cardiovascular disease |
| CYP450 | cytochrome 450 |
| CYR61 | cysteine-rich angiogenic inducer 61 |
| DNA | deoxyribonucleic acid |
| EGFR | epithelial growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| ERK | extracellular signal-regulated kinases |
| EZH | enhancer of zeste homolog |
| FADD | Fas-associated protein with death domain |
| FAS | fatty acid synthase |
| FPPS | farnesyl pyrophosphate synthase |
| FN | fibronectin |
| FOXP3 | forkhead box P3 |
| FTI | farnesyl transferase inhibitors |
| G1 | Gap1 |
| GDI | GDP-dissociation inhibitor |
| GGTI | geranylgeranyl transferase inhibitors |
| GLUT | glucose transporters |
| GTP | guanosine triphosphate |
| Gy | gray |
| H2O2 | hydrogen peroxide |
| HDAC | histone deacetylase |
| hESC | human embryonic stem cells |
| HDL | high-density lipoprotein |
| HMG-CoA | hydroxy-3-methylglutaryl-coenzyme A |
| HNSCC | head and neck squamous cell carcinomas |
| HPV | human papillomavirus |
| HRT | hormone replacement therapy |
| hs-CRP | high sensitivity C-reactive protein |
| IAP | inhibitor of apoptosis |
| ICAM | intercellular adhesion molecule |
| ICB | immune checkpoint blockade |
| IC50 | half maximal inhibitory concentration |
| IDOs | Indoleamine 2,3-dioxygenases |
| IL | interleukin |
| IR | ionizing radiation |
| IRF | interferon regulatory factor |
| KRAS | kirsten rat sarcoma virus |
| LDL | low density lipoprotein |
| LKB1 | liver kinase B1 |
| MAPK | mitogen-activated protein kinase |
| Mcl | myeloid leukemia cell differentiation protein |
| MCM | minichromosome maintenance |
| MDR | multidrug resistance |
| MEK | mitogen-activated protein kinase kinase |
| mESC | mouse embryonic stem cells |
| MET | mesenchymal epithelial transition tyrosine kinase |
| METTL3 | methyltransferase 3 |
| miRNA | micro-RNA |
| MMP | metalloproteinase |
| mRNA | messenger ribonucleic acid |
| MSC | mesenchymal stem cells |
| mTOR | mechanistic target of rapamycin |
| MyD88 | myeloid differentiation primary response 88 |
| MYC | myelocytomatosis (similar oncogene) |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NBP | nitrogen containing bisphosphonates |
| NFκB | nuclear factor kappa B |
| NGAL | neutrophil gelatinase-associated lipocalin |
| NNAL | nicotine-derived nitrosamine alcohol |
| NNK | nicotine-derives nitrosamine ketone |
| NOS | nitric oxide synthase |
| NSCLC | non-small cell lung cancer |
| OATP | organic anion transporting polypeptide |
| Oct4 | octamer-binding transcription factor 4 |
| PCOS | polycystic ovary syndrome |
| PDGFR | platelet-derived growth factor receptors |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | Programmed Death-Ligand 1 |
| PFS | progression free survival |
| PI3K | phosphoinositide 3-kinases |
| PPAR-γ | peroxisome proliferator-activated receptors-gamma |
| PCCs | primary cancer cells |
| PTEN | phosphatase and tensin homolog |
| PTI | prenyltransferase inhibitors |
| p21 | cyclin-dependent kinase inhibitor 1 |
| p27 | cyclin-dependent kinase inhibitor 1 B |
| Rac | ras-related C3 botulinum toxin substrate |
| RANKL | receptor activator of nuclear factor kappa-B ligand |
| RB | retinoblastoma protein |
| RCT | randomized controlled trial |
| RECK | reversion-inducing cysteine-rich protein with kazal motif |
| Rho | ras homologous protein |
| RIP | receptor interacting protein |
| ROS | receptor tyrosine kinase |
| RR | response rate |
| RT-PCR | real-time polymerase chain reaction |
| SCC | squamous cell lung carcinoma |
| SCLC | small cell lung cancer |
| SLCO | solute carrier organic anion transporter family member |
| SMA | smooth muscle actin |
| SMAD | small mother against decapentaplegic |
| SmPC | summary of product characteristics |
| SOX | SRY-related HMG-box genes |
| SREBP | sterol regulatory element-binding proteins |
| STAT | signal transducer and activator of transcription |
| TAZ | transcriptional coactivator with pdz-binding motif |
| TGF | transforming growth factor |
| TKI | tyrosine kinase inhibitor |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| TRADD | tumor necrosis factor receptor type 1-associated death domain protein |
| TRAF | TNF-receptor associated factors |
| t1/2 | half-life |
| UGT | uridine diphosphate-glucuronosyltransferase |
| Vi | vimentin |
| XIAP | X-linked inhibitor of apoptosis protein |
| YAP | yes-associated protein |
| VEGF | vascular endothelial growth factor |
References
- Zalcman, G.; Bergot, E.; Lechapt, E. Update on nonsmall cell lung cancer. Eur. Respir. Rev. 2010, 19, 173–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatehi Hassanabad, A.; McBride, S.A. Statins as Potential Therapeutics for Lung Cancer: Molecular Mechanisms and Clinical Outcomes. Am. J. Clin. Oncol. Cancer Clin. Trials 2019, 42, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol. Biomarkers Prev. 2019, 28, 1563–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Ireland, A.S.; Micinski, A.M.; Kastner, D.W.; Guo, B.; Wait, S.J.; Spainhower, K.B.; Conley, C.C.; Chen, O.S.; Guthrie, M.R.; Qiao, Y.; et al. MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer Cell 2021, 38, 60–78. [Google Scholar] [CrossRef]
- Cristea, S.; Coles, G.L.; Hornburg, D.; Gershkovitz, M.; Arand, J.; Cao, S.; Sen, T.; Williamson, S.C.; Kim, J.W.; Drainas, A.P.; et al. The MEK5-ERK5 kinase axis controls lipid metabolism in small-cell lung cancer. Cancer Res. 2020, 80, 1293–1303. [Google Scholar] [CrossRef] [Green Version]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef]
- Marino, F.Z.; Bianco, R.; Accardo, M.; Ronchi, A.; Cozzolino, I.; Morgillo, F.; Rossi, G.; Franco, R. Molecular heterogeneity in lung cancer: From mechanisms of origin to clinical implications. Int. J. Med. Sci. 2019, 16, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Merino Salvador, M.; Gómez de Cedrón, M.; Merino Rubio, J.; Falagán Martínez, S.; Sánchez Martínez, R.; Casado, E.; Ramírez de Molina, A.; Sereno, M. Lipid metabolism and lung cancer. Crit. Rev. Oncol. Hematol. 2017, 112, 31–40. [Google Scholar] [CrossRef]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef]
- Kucharska-Newton, A.M.; Rosamond, W.D.; Schroeder, J.C.; McNeill, A.M.; Coresh, J.; Folsom, A.R. HDL-cholesterol and the incidence of lung cancer in the Atherosclerosis Risk in Communities (ARIC) study. Lung Cancer 2008, 61, 292–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Hu, J.W.; He, X.R.; Jin, W.L.; He, X.Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; La Vecchia, C.; de Groh, M.; Negri, E.; Morrison, H.; Mery, L.; Paulse, B.; Dewar, R.; Dryer, D.; Kreiger, N.; et al. Dietary cholesterol intake and cancer. Ann. Oncol. 2012, 23, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Grabarek, B.O.; Boroń, D.; Morawiec, E.; Michalski, P.; Palazzo-Michalska, V.; Pach, Ł.; Dziuk, B.; Świder, M.; Zmarzły, N. Crosstalk between statins and cancer prevention and therapy: An update. Pharmaceuticals 2021, 14, 1220. [Google Scholar] [CrossRef]
- Climent, E.; Benaiges, D.; Pedro-Botet, J. Hydrophilic or Lipophilic Statins? Front. Cardiovasc. Med. 2021, 8, 687585. [Google Scholar] [CrossRef]
- Schachter, M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: An update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic Effects of Statins on the Cardiovascular System. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Dong, J.; Yu, Z. Pleiotropic use of statins as non-lipid-lowering drugs. Int. J. Biol. Sci. 2020, 16, 2704–2711. [Google Scholar] [CrossRef]
- Gallelli, L.; Falcone, D.; Scaramuzzino, M.; Pelaia, G.; D’Agostino, B.; Mesuraca, M.; Terracciano, R.; Spaziano, G.; Maselli, R.; Navarra, M.; et al. Effects of simvastatin on cell viability and proinflammatory pathways in lung adenocarcinoma cells exposed to hydrogen peroxide. BMC Pharmacol. Toxicol. 2014, 15, 67. [Google Scholar] [CrossRef] [Green Version]
- Pelaia, G.; Gallelli, L.; Renda, T.; Fratto, D.; Falcone, D.; Caraglia, M.; Busceti, M.T.; Terracciano, R.; Vatrella, A.; Maselli, R.; et al. Effects of statins and farnesyl transferase inhibitors on ERK phosphorylation, apoptosis and cell viability in non-small lung cancer cells. Cell Prolif. 2012, 45, 557–565. [Google Scholar] [CrossRef]
- Falcone, D.; Gallelli, L.; Di Virgilio, A.; Tucci, L.; Scaramuzzino, M.; Terracciano, R.; Pelaia, G.; Savino, R. Effects of simvastatin and rosuvastatin on RAS protein, Matrix metalloproteinases and NF-κB in lung cancer and in normal pulmonary tissues. Cell Prolif. 2013, 46, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Palleria, C.; Di Paolo, A.; Giofrè, C.; Caglioti, C.; Leuzzi, G.; Siniscalchi, A.; De Sarro, G.; Gallelli, L. Pharmacokinetic drug-drug interaction and their implication in clinical management. J. Res. Med. Sci. 2013, 18, 601–610. [Google Scholar] [PubMed]
- Staltari, O.; Leporini, C.; Caroleo, B.; Russo, E.; Siniscalchi, A.; De Sarro, G.; Gallelli, L. Drug-drug interactions: Antiretroviral drugs and recreational drugs. Recent Pat. CNS Drug Discov. 2014, 9, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Di Mizio, G.; Marcianò, G.; Palleria, C.; Muraca, L.; Rania, V.; Roberti, R.; Spaziano, G.; Piscopo, A.; Ciconte, V.; Di Nunno, N.; et al. Drug–Drug Interactions in Vestibular Diseases, Clinical Problems, and Medico-Legal Implications. Int. J. Environ. Res. Public Health 2021, 18, 12936. [Google Scholar] [CrossRef]
- Iannelli, F.; Lombardi, R.; Milone, M.R.; Pucci, B.; De Rienzo, S.; Budillon, A.; Bruzzese, F. Targeting Mevalonate Pathway in Cancer Treatment: Repurposing of Statins. Recent Pat. Anticancer Drug Discov. 2018, 13, 184–200. [Google Scholar] [CrossRef]
- Chou, C.W.; Lin, C.H.; Hsiao, T.H.; Lo, C.C.; Hsieh, C.Y.; Huang, C.C.; Sher, Y.P. Therapeutic effects of statins against lung adenocarcinoma via p53 mutant-mediated apoptosis. Sci. Rep. 2019, 9, 20403. [Google Scholar] [CrossRef] [Green Version]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrell, F.K.; Kerr, E.M.; Gao, M.; Thorpe, H.; Doherty, G.J.; Cridge, J.; Shorthouse, D.; Speed, A.; Samarajiwa, S.; Hall, B.A.; et al. Lung tumors with distinct p53 mutations respond similarly to p53 targeted therapy but exhibit genotype-specific statin sensitivity. Genes Dev. 2017, 31, 1339–1353. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wang, Z.; Li, Y.; Li, W.; Chen, Y. Simvastatin prevents proliferation and bone metastases of lung adenocarcinoma in vitro and in vivo. Neoplasma 2013, 60, 607–616. [Google Scholar] [CrossRef]
- Otahal, A.; Aydemir, D.; Tomasich, E.; Minichsdorfer, C. Delineation of cell death mechanisms induced by synergistic effects of statins and erlotinib in non-small cell lung cancer cell (NSCLC) lines. Sci. Rep. 2020, 10, 959. [Google Scholar] [CrossRef]
- Podmirseg, S.R.; Vosper, J.; Hengst, L. p27 Kip1–p(RhoB)lematic in lung cancer. J. Pathol. 2019, 248, 3–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanli, T.; Liu, C.; Rashid, A.; Hopmans, S.N.; Tsiani, E.; Schultz, C.; Farrell, T.; Singh, G.; Wright, J.; Tsakiridis, T. Lovastatin sensitizes lung cancer cells to ionizing radiation: Modulation of molecular pathways of radioresistance and tumor suppression. J. Thorac. Oncol. 2011, 6, 439–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Liu, N.; Ma, M.; Huang, H.; Handley, M.; Bai, X.; Shan, F. Methionine enkephalin (MENK) suppresses lung cancer by regulating the Bcl-2/Bax/caspase-3 signaling pathway and enhancing natural killer cell-driven tumor immunity. Int. Immunopharmacol. 2021, 98, 107837. [Google Scholar] [CrossRef]
- Yu, X.; Pan, Y.; Ma, H.; Li, W. Simvastatin inhibits proliferation and induces apoptosis in human lung cancer cells. Oncol. Res. 2013, 20, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Choudhury, N.; Mandal, C.C.; Ghosh-Choudhury, N.; Ghosh-Choudhury, G. Simvastatin induces derepression of PTEN expression via NFkappaB to inhibit breast cancer cell growth. Cell. Signal. 2010, 22, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichner, L.J.; Brun, S.N.; Herzig, S.; Young, N.P.; Curtis, S.D.; Shackelford, D.B.; Shokhirev, M.N.; Leblanc, M.; Vera, L.I.; Hutchins, A.; et al. Genetic Analysis Reveals AMPK Is Required to Support Tumor Growth in Murine Kras-Dependent Lung Cancer Models. Cell Metab. 2019, 29, 285–302. [Google Scholar] [CrossRef] [Green Version]
- Henin, N.; Vincent, M.-F.; Gruber, H.E.; Van Den Berghe, G. Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP-activated protein kinase. FASEB J. 1995, 9, 541–546. [Google Scholar] [CrossRef]
- Ming-Yang, P.; Liu, Y.-L.; Lin, Y.-C.; Shun, C.-T.; Wu, M.-S.; Chen, C.-C. Inhibition of autophagy enhances anticancer effects of atorvastatin in digestive malignancies. Cancer Res. 2010, 70, 7699–7709. [Google Scholar] [CrossRef] [Green Version]
- Cione, E.; Piegari, E.; Gallelli, G.; Caroleo, M.C.; Lamirata, E.; Curcio, F.; Colosimo, F.; Cannataro, R.; Ielapi, N.; Colosimo, M.; et al. Expression of MMP-2, MMP-9, and NGAL in tissue and serum of patients with vascular aneurysms and their modulation by statin treatment: A pilot study. Biomolecules 2020, 10, 359. [Google Scholar] [CrossRef] [Green Version]
- Walther, U.; Emmrich, K.; Ramer, R.; Mittag, N.; Hinz, B. Lovastatin lactone elicits human lung cancer cell apoptosis via a COX-2/PPARγ-dependent pathway. Oncotarget 2016, 7, 10345–10362. [Google Scholar] [CrossRef] [Green Version]
- Cantini, L.; Pecci, F.; Hurkmans, D.P.; Belderbos, R.A.; Lanese, A.; Copparoni, C.; Aerts, S.; Cornelissen, R.; Dumoulin, D.W.; Fiordoliva, I.; et al. High-intensity statins are associated with improved clinical activity of PD-1 inhibitors in malignant pleural mesothelioma and advanced non-small cell lung cancer patients. Eur. J. Cancer 2021, 144, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Tanaka, F.; Takata, T.; Yanagihara, K.; Otake, Y.; Hanaoka, N.; Miyahara, R.; Nakagawa, T.; Kawano, Y.; Ishikawa, S.; et al. Clinical significance of p21 expression in non-small-cell lung cancer. J. Clin. Oncol. 2002, 20, 3865–3871. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.Y.; Kang, H.K.; Choi, J.E.; Jang, J.S.; Kim, E.J.; Cha, S.I.; Lee, W.K.; Kam, S.; Kim, C.H.; Han, S.B.; et al. Comprehensive assessment of P21 polymorphisms and lung cancer risk. J. Hum. Genet. 2008, 53, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, D.; Lan, L.; Huang, K.; Chen, L.; Xu, C.; Wang, R.; Shi, Y.; Wu, X.; Wang, L.; Liu, Y.; et al. Association of p53/p21 expression and cigarette smoking with tumor progression and poor prognosis in non-small cell lung cancer patients. Oncol. Rep. 2014, 32, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Yin, H.T.; Yin, X.L.; Wang, J.; Zhang, D.P. High p27 expression is associated with a better prognosis in East Asian non-small cell lung cancer patients. Clin. Chim. Acta 2011, 412, 2228–2231. [Google Scholar] [CrossRef]
- Yang, L.; Yang, J. Expression and clinical significance of microRNA-21, PTEN and p27 in cancer tissues of patients with non-small cell lung cancer. Oncol. Lett. 2020, 20, 49. [Google Scholar] [CrossRef]
- Zhang, X.; Teng, Y.; Yang, F.; Wang, M.; Hong, X.; Ye, L.G.; Gao, Y.N.; Chen, G.Y. MCM2 is a therapeutic target of lovastatin in human non-small cell lung carcinomas. Oncol. Rep. 2015, 33, 2599–2605. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Y.; Kim, I.K.; Lee, H.I.; Mo, J.Y.; Yeo, C.D.; Kang, H.H.; Moon, H.S.; Lee, S.H. The apoptotic effect of simvastatin via the upregulation of BIM in nonsmall cell lung cancer cells. Exp. Lung Res. 2016, 42, 14–23. [Google Scholar] [CrossRef]
- Wali, V.B.; Bachawal, S.V.; Sylvester, P.W. Combined treatment of γ-tocotrienol with statins induce mammary tumor cell cycle arrest in G1. Exp. Biol. Med. 2009, 234, 639–650. [Google Scholar] [CrossRef]
- Follet, J.; Corcos, L.; Baffet, G.; Ezan, F.; Morel, F.; Simon, B.; Le Jossic-Corcos, C. The association of statins and taxanes: An efficient combination trigger of cancer cell apoptosis. Br. J. Cancer 2012, 106, 685–692. [Google Scholar] [CrossRef] [Green Version]
- Niknejad, N.; Morley, M.; Dimitroulakos, J. Activation of the integrated stress response regulates lovastatin-induced apoptosis. J. Biol. Chem. 2007, 282, 29748–29756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Niknejad, N.; Gorn-Hondermann, I.; Dayekh, K.; Dimitroulakos, J. Lovastatin Induces Multiple Stress Pathways Including LKB1/AMPK Activation That Regulate Its Cytotoxic Effects in Squamous Cell Carcinoma Cells. PLoS ONE 2012, 7, e46055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misirkic, M.; Janjetovic, K.; Vucicevic, L.; Tovilovic, G.; Ristic, B.; Vilimanovich, U.; Harhaji-Trajkovic, L.; Sumarac-Dumanovic, M.; Micic, D.; Bumbasirevic, V.; et al. Inhibition of AMPK-dependent autophagy enhances in vitro antiglioma effect of simvastatin. Pharmacol. Res. 2012, 65, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Fan-Minogue, H.; Bellovin, D.I.; Yevtodiyenko, A.; Yang, Q.; Gambhir, S.S.; Felsher, D.W. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res 2011, 71, 2286–2297. [Google Scholar] [CrossRef] [Green Version]
- Takwi, A.A.L.; Li, Y.; Becker Buscaglia, L.E.; Zhang, J.; Choudhury, S.; Park, A.K.; Liu, M.; Young, K.H.; Park, W.Y.; Martin, R.C.G.; et al. A statin-regulated microRNA represses human c-Myc expression and function. EMBO Mol. Med. 2012, 4, 896–909. [Google Scholar] [CrossRef]
- Yang, T.; Chen, M.; Sun, T. Simvastatin attenuates TGF-β1-induced epithelial-mesenchymal transition in human alveolar epithelial cells. Cell. Physiol. Biochem. 2013, 31, 863–874. [Google Scholar] [CrossRef]
- Xu, X.; Cao, L.; Zhang, Y.; Lian, H.; Sun, Z.; Cui, Y. MicroRNA-1246 inhibits cell invasion and epithelial mesenchymal transition process by targeting CXCR4 in lung cancer cells. Cancer Biomark. 2018, 21, 251–260. [Google Scholar] [CrossRef]
- Nishikawa, S.; Menju, T.; Takahashi, K.; Miyata, R.; Chen-Yoshikawa, T.F.; Sonobe, M.; Yoshizawa, A.; Sabe, H.; Sato, T.; Date, H. Statins may have double-edged effects in patients with lung adenocarcinoma after lung resection. Cancer Manag. Res. 2019, 11, 3419–3432. [Google Scholar] [CrossRef] [Green Version]
- McCormack, J.; Welsh, N.J.; Braga, V.M.M. Cycling around cell-cell adhesion with rho GTPase regulators. J. Cell Sci. 2013, 126, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Denoyelle, C.; Vasse, M.; Körner, M.; Mishal, Z.; Ganné, F.; Vannier, J.P.; Soria, J.; Soria, C. Cerivastatin, an inhibitor of HMG-CoA reductase, inhibits the signaling pathways involved in the invasiveness and metastatic properties of highly invasive breast cancer cell lines: An in vitro study. Carcinogenesis 2001, 22, 1139–1148. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Casey, P.J.; Kumar, A.P.; Pervaiz, S. Deciphering the signaling networks underlying simvastatin-induced apoptosis in human cancer cells: Evidence for non-canonical activation of RhoA and. Cell Death Dis. 2013, 4, e568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayachandran, G.; Sazaki, J.I.; Nishizaki, M.; Xu, K.; Girard, L.; Minna, J.D.; Roth, J.A.; Ji, L. Fragile histidine triad-mediated tumor suppression of lung cancer by targeting multiple components of the Ras/Rho GTPase molecular switch. Cancer Res. 2007, 67, 10379–10388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooke, M.; Baker, M.J.; Kazanietz, M.G.; Casado-Medrano, V. PKCε regulates Rho GTPases and actin cytoskeleton reorganization in non-small cell lung cancer cells. Small GTPases 2021, 12, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Mao, X.; Yin, J.; Li, X.; Chen, J.; Zhu, T.; Li, Q.; Zhou, H.; Liu, Z. Emerging roles of RAC1 in treating lung cancer patients. Clin. Genet. 2017, 91, 520–528. [Google Scholar] [CrossRef]
- Zeng, R.J.; Xie, W.J.; Zheng, C.W.; Chen, W.X.; Wang, S.M.; Li, Z.; Cheng, C.B.; Zou, H.Y.; Xu, L.Y.; Li, E.M. Role of Rho guanine nucleotide exchange factors in non-small cell lung cancer. Bioengineered 2021, 12, 11169–11187. [Google Scholar] [CrossRef]
- Nübel, T.; Dippold, W.; Kleinert, H.; Kaina, B.; Fritz, G. Lovastatin inhibits Rho-regulated expression of E-selectin by TNFalpha and attenuates tumor cell adhesion. FASEB J. 2004, 18, 140–142. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; O’Kelly, J.; Xie, D.; Mori, A.; Lemp, N.; McKenna, R.; Miller, C.W.; Koeffler, H.P. Cyr61 suppresses the growth of non-small-cell lung cancer cells via the β-catenin-c-myc-p53 pathway. Oncogene 2004, 23, 4847–4855. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.P.; Li, W.J.; Wang, Y.; Zhao, S.; Li, D.Y.; Feng, L.Y.; Shi, X.L.; Koeffler, H.P.; Tong, X.J.; Xie, D. Expression of Cyr61, CTGF, and WISP-1 Correlates with Clinical Features of Lung Cancer. PLoS ONE 2007, 2, e534. [Google Scholar] [CrossRef]
- Fromigue, O.; Hamidouche, Z.; Vaudin, P.; Lecanda, F.; Patino, A.; Barbry, P.; Mari, B.; Marie, P.J. CYR61 downregulation reduces osteosarcoma cell invasion, migration, and metastasis. J. Bone Miner. Res. 2011, 26, 1533–1542. [Google Scholar] [CrossRef]
- Yun, M.R.; Choi, H.M.; Lee, Y.W.; Joo, H.S.; Park, C.W.; Choi, J.W.; Kim, D.H.; Kang, H.N.; Pyo, K.; Shin, E.J.; et al. Targeting YAP to overcome acquired resistance to ALK inhibitors in ALK -rearranged lung cancer. EMBO Mol. Med. 2019, 11, e10581. [Google Scholar] [CrossRef]
- Sarkar, D. Statins as Inhibitors of Lung Cancer Bone Metastasis. EBioMedicine 2017, 19, 6–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Su, Z.; DeWitt, J.P.; Xie, L.; Chen, Y.; Li, X.; Han, L.; Li, D.; Xia, J.; Zhang, Y.; et al. Fluvastatin Prevents Lung Adenocarcinoma Bone Metastasis by Triggering Autophagy. EBioMedicine 2017, 19, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, Y.; Haneji, T. Stimulation of osteoclast formation by RANKL requires interferon regulatory factor-4 and is inhibited by simvastatin in a mouse model of bone loss. PLoS ONE 2013, 8, e72033. [Google Scholar] [CrossRef] [PubMed]
- Weis, M.; Heeschen, C.; Glassford, A.J.; Cooke, J.P. Statins have biphasic effects on angiogenesis. Circulation 2002, 105, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Sahebkar, A.; Ponziani, M.C.; Goitre, I.; Bo, S. Does statin therapy reduce plasma VEGF levels in humans? A systematic review and meta-analysis of randomized controlled trials. Metabolism 2015, 64, 1466–1476. [Google Scholar] [CrossRef]
- Kiziltunc Ozmen, H.; Simsek, M. Serum IL-23, E-selectin and sICAM levels in non-small cell lung cancer patients before and after radiotherapy. J. Int. Med. Res. 2020, 48, 300060520923493. [Google Scholar] [CrossRef]
- Zinellu, A.; Mangoni, A.A. Systematic Review and Meta-Analysis of the Effect of Statins on Circulating E-Selectin, L-Selectin, and P-Selectin. Biomedicines 2021, 9, 1707. [Google Scholar] [CrossRef]
- Mori, A.; Desmond, J.C.; Komatsu, N.; O’Kelly, J.; Miller, C.W.; Legaspi, R.; Marchevsky, A.M.; McKenna, R.J.; Koeffler, H.P. CYR61: A new measure of lung cancer outcome. Cancer Invest. 2007, 25, 738–741. [Google Scholar] [CrossRef]
- Ačkar, L.; Casjens, S.; Andreas, A.; Raiko, I.; Brüning, T.; Geffken, M.; Peine, S.; Kollmeier, J.; Johnen, G.; Bartkowiak, K.; et al. Blood-based detection of lung cancer using cysteine-rich angiogenic inducer 61 (CYR61) as a circulating protein biomarker: A pilot study. Mol. Oncol. 2021, 15, 2877–2890. [Google Scholar] [CrossRef]
- Chen, W.W.; Qi, J.W.; Hang, Y.; Wu, J.X.; Zhou, X.X.; Chen, J.Z.; Wang, J.; Wang, H.H. Simvastatin is beneficial to lung cancer progression by inducing METTL3-induced m6A modification on EZH2 mRNA. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4263–4270. [Google Scholar] [CrossRef]
- Chen, J.; Liu, B.; Yuan, J.; Yang, J.; Zhang, J.; An, Y.; Tie, L.; Pan, Y.; Li, X. Atorvastatin reduces vascular endothelial growth factor (VEGF) expression in human non-small cell lung carcinomas (NSCLCs) via inhibition of reactive oxygen species (ROS) production. Mol. Oncol. 2012, 6, 62–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Sardo, F.; Forcato, M.; Sacconi, A.; Capaci, V.; Zanconato, F.; di Agostino, S.; Del Sal, G.; Pandolfi, P.P.; Strano, S.; Bicciato, S.; et al. MCM7 and its hosted miR-25, 93 and 106b cluster elicit YAP/TAZ oncogenic activity in lung cancer. Carcinogenesis 2017, 38, 64–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.; Levantini, E.; Fhu, C.W.; Teo, J.T.; Clohessy, J.G.; Goggi, J.L.; Wu, C.S.; Chen, L.; Chin, T.M.; Tenen, D.G. CAV1-GLUT3 signaling is important for cellular energy and can be targeted by Atorvastatin in Non-Small Cell Lung Cancer. Theranostics 2019, 9, 6157–6174. [Google Scholar] [CrossRef]
- Teisseyre, A.; Uryga, A.; Michalak, K. Statins as inhibitors of voltage-gated potassium channels Kv1.3 in cancer cells. J. Mol. Struct. 2021, 1230, 129905. [Google Scholar] [CrossRef] [PubMed]
- Galland, S.; Martin, P.; Fregni, G.; Letovanec, I.; Stamenkovic, I. Attenuation of the pro-inflammatory signature of lung cancer-derived mesenchymal stromal cells by statins. Cancer Lett. 2020, 484, 50–64. [Google Scholar] [CrossRef]
- Kamata, T.; Al Dujaily, E.; Alhamad, S.; So, T.Y.; Margaritaki, O.; Giblett, S.; Pringle, J.H.; Le Quesne, J.; Pritchard, C. Statins mediate anti- and pro-tumourigenic functions by remodelling the tumour microenvironment. Dis. Model. Mech. 2022, 15, dmm049148. [Google Scholar] [CrossRef]
- Gauthaman, K.; Richards, M.; Wong, J.; Bongso, A. Comparative evaluation of the effects of statins on human stem and cancer cells in vitro. Reprod. Biomed. Online 2007, 15, 566–581. [Google Scholar] [CrossRef]
- Gauthaman, K.; Manasi, N.; Bongso, A. Statins inhibit the growth of variant human embryonic stem cells and cancer cells in vitro but not normal human embryonic stem cells. Br. J. Pharmacol. 2009, 157, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Cho, Y.S.; Han, Y. Simvastatin Suppresses Self-Renewal of Mouse Embryonic Stem Cells by Inhibiting RhoA Geranylgeranylation. Stem Cells 2007, 25, 1654–1663. [Google Scholar] [CrossRef]
- Cardwell, C.R.; Menamin, M.; Hughes, C.M.; Murray, L.J. Statin Use and Survival from Lung Cancer: A Population-Based Cohort Study. Cancer Epidemiol. Biomark. Prev. 2015, 24, 833–841. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.J.; Ezer, N.; Sigel, K.; Mhango, G.; Wisnivesky, J.P. The effect of statins on survival in patients with stage IV lung cancer. Lung Cancer 2016, 99, 137–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin Use and Reduced Cancer-Related Mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, V.; Bejjanki, H.R.; Caldito, G.; Owens, M.W. Statins reduce the risk of lung cancer in humans: A large case-control study of US Veterans. Chest 2007, 131, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Jeong, S.M.; Shin, D.W.; Cho, M.; Cho, J.H.; Kim, J. The Associations of Aspirin, Statins, and Metformin With Lung Cancer Risk and Related Mortality: A Time-Dependent Analysis of Population-Based Nationally Representative Data. J. Thorac. Oncol. 2021, 16, 76–88. [Google Scholar] [CrossRef]
- Nguyen, P.A.; Chang, C.C.; Galvin, C.J.; Wang, Y.C.; An, S.Y.; Huang, C.W.; Wang, Y.H.; Hsu, M.H.; Li, Y.C.; Yang, H.C. Statins use and its impact in EGFR-TKIs resistance to prolong the survival of lung cancer patients: A Cancer registry cohort study in Taiwan. Cancer Sci. 2020, 111, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Yang, T.Y.; Hsu, Y.P.; Hao, W.R.; Kao, P.F.; Sung, L.C.; Chen, C.C.; Wu, S.Y. Statins dose-dependently exert a chemopreventive effect against lung cancer in COPD patients: A population-based cohort study. Oncotarget 2016, 7, 59618–59629. [Google Scholar] [CrossRef] [Green Version]
- Leigh, D.; Eken, J.; Beal, J.R.; Ganti, A.K.; Sahmoun, A.E. Statins use and risk for brain metastasis from lung cancer. Cancer Invest. 2011, 29, 68–72. [Google Scholar] [CrossRef]
- Fiala, O.; Pesek, M.; Finek, J.; Minarik, M.; Benesova, L.; Bortlicek, Z.; Topolcan, O. Statins augment efficacy of EGFR-TKIs in patients with advanced-stage non-small cell lung cancer harbouring KRAS mutation. Tumor Biol. 2015, 36, 5801–5805. [Google Scholar] [CrossRef]
- Seckl, M.J.; Ottensmeier, C.H.; Cullen, M.; Schmid, P.; Ngai, Y.; Muthukumar, D.; Thompson, J.; Harden, S.; Middleton, G.; Fife, K.M.; et al. Multicenter, phase III, randomized, double-blind, placebo-controlled trial of pravastatin added to first-line standard chemotherapy in small-cell lung cancer (LUNGSTAR). J. Clin. Oncol. 2017, 35, 1506–1514. [Google Scholar] [CrossRef]
- Han, J.Y.; Lee, S.H.; Yoo, N.J.; Lee, S.H.; Moon, Y.J.; Yun, T.; Kim, H.T.; Lee, J.S. A randomized phase II study of gefitinib plus simvastatin versus gefitinib alone in previously treated patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2011, 17, 1553–1560. [Google Scholar] [CrossRef] [Green Version]
- Han, J.Y.; Lim, K.Y.; Yu, S.Y.; Yun, T.; Kim, H.T.; Lee, J.S. A phase 2 study of irinotecan, cisplatin, and simvastatin for untreated extensive-disease small cell lung cancer. Cancer 2011, 117, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, K.H.; Lee, G.K.; Lee, S.H.; Lim, K.Y.; Joo, J.; Go, Y.J.; Lee, J.S.; Han, J.Y. Randomized phase II study of afatinib plus simvastatin versus afatinib alone in previously treated patients with advanced nonadenocarcinomatous non-small cell lung cancer. Cancer Res. Treat. 2017, 49, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Zhang, S.; Yi, L.; Chen, S. Can statins reduce risk of lung cancer, especially among elderly people? A meta-analysis. Chin. J. Cancer Res. 2013, 25, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.K.; Hu, Z.G.; Tian, Y.F.; Zeng, F.J. Statin use and prognosis of lung cancer: A systematic review and meta-analysis of observational studies and randomized controlled trials. Drug Des. Dev. Ther. 2019, 13, 405–422. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, C.; Tao, H.; Cheng, Y.; Han, L.; Li, X.; Hu, Y. Statin Use and Risk of Lung Cancer: A Meta-Analysis of Observational Studies and Randomized Controlled Trials. PLoS ONE 2013, 8, e77950. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Song, X.; Zhang, G.; Peng, A.; Li, X.; Li, M.; Liu, Y.; Wang, C. Statins and the Risk of Lung Cancer: A Meta-Analysis. PLoS ONE 2013, 8, e57349. [Google Scholar] [CrossRef]
- Mederos, N.; Friedlaender, A.; Peters, S.; Addeo, A. Gender-specific aspects of epidemiology, molecular genetics and outcome: Lung cancer. ESMO Open 2020, 5, e000796. [Google Scholar] [CrossRef]
- Stapelfeld, C.; Dammann, C.; Maser, E. Sex-specificity in lung cancer risk. Int. J. Cancer 2020, 146, 2376–2382. [Google Scholar] [CrossRef]
- Stabile, L.P.; Lyker, J.S.; Gubish, C.T.; Zhang, W.; Grandis, J.R.; Siegfried, J.M. Combined Targeting of the Estrogen Receptor and the Epidermal Growth Factor Receptor in Non–Small Cell Lung Cancer Shows Enhanced Antiproliferative Effects. Cancer Res. 2005, 65, 1459–1471. [Google Scholar] [CrossRef] [Green Version]
- Hammoud, Z.; Tan, B.; Badve, S.; Bigsby, R.M. Estrogen promotes tumor progression in a genetically defined mouse model of lung adenocarcinoma. Endocr. Relat. Cancer 2008, 15, 475–483. [Google Scholar] [CrossRef]
- Vavalà, T.; Catino, A.; Pizzutilo, P.; Longo, V.; Galetta, D. Gender differences and immunotherapy outcome in advanced lung cancer. Int. J. Mol. Sci. 2021, 22, 11942. [Google Scholar] [CrossRef] [PubMed]
- Araujo, J.M.; Prado, A.; Cardenas, N.K.; Zaharia, M.; Dyer, R.; Doimi, F.; Bravo, L.; Pinillos, L.; Morante, Z.; Aguilar, A.; et al. Repeated observation of immune gene sets enrichment in women with non-small cell lung cancer. Oncotarget 2016, 7, 20282–20292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Jing, Y.; Li, L.; Mills, G.B.; Diao, L.; Liu, H.; Han, L. Sex-associated molecular differences for cancer immunotherapy. Nat. Commun. 2020, 11, 1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, S.; Bayer, A.J.; O’Mahony, M.S. Age and gender bias in statin trials. QJM-Mon. J. Assoc. Physicians 2001, 94, 127–132. [Google Scholar] [CrossRef]
- Dale, K.M.; Coleman, C.I.; Shah, S.A.; Patel, A.A.; Kluger, J.; White, C.M. Impact of gender on statin efficacy. Curr. Med. Res. Opin. 2007, 23, 565–574. [Google Scholar] [CrossRef]
- Abramson, B.L.; Benlian, P.; Hanson, M.E.; Lin, J.; Shah, A.; Tershakovec, A.M. Response by sex to statin plus ezetimibe or statin monotherapy: A pooled analysis of 22,231 hyperlipidemic patients. Lipids Health Dis. 2011, 10, 146. [Google Scholar] [CrossRef] [Green Version]
- Cangemi, R.; Romiti, G.F.; Campolongo, G.; Ruscio, E.; Sciomer, S.; Gianfrilli, D.; Raparelli, V. Gender related differences in treatment and response to statins in primary and secondary cardiovascular prevention: The never-ending debate. Pharmacol. Res. 2017, 117, 148–155. [Google Scholar] [CrossRef]
- Zucker, I.; Prendergast, B.J. Sex differences in pharmacokinetics predict adverse drug reactions in women. Biol. Sex Differ. 2020, 11, 32. [Google Scholar] [CrossRef]
- Faubion, S.S.; Kapoor, E.; Moyer, A.M.; Hodis, H.N.; Miller, V.M. Statin therapy: Does sex matter? Menopause 2019, 26, 1425–1435. [Google Scholar] [CrossRef]
- Luttman, J.H.; Hoj, J.P.; Lin, K.H.; Lin, J.; Gu, J.J.; Rouse, C.; Nichols, A.G.; MacIver, N.J.; Wood, K.C.; Pendergast, A.M. ABL allosteric inhibitors synergize with statins to enhance apoptosis of metastatic lung cancer cells. Cell Rep. 2021, 37, 109880. [Google Scholar] [CrossRef]
- Hwang, K.E.; Park, C.; Kwon, S.J.; Kim, Y.S.; Park, D.S.; Lee, M.K.; Kim, B.R.; Park, S.H.; Yoon, K.H.; Jeong, E.T.; et al. Synergistic induction of apoptosis by sulindac and simvastatin in A549 human lung cancer cells via reactive oxygen species-dependent mitochondrial dysfunction. Int. J. Oncol. 2013, 43, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Qi, H.; Zhang, H.; Ding, L.; Huang, Q.; Zhao, D.; Wu, B.J.; Li, X. Targeting SREBP-2-Regulated Mevalonate Metabolism for Cancer Therapy. Front. Oncol. 2020, 10, 1510. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.H.; Loo, C.Y.; Ghadiri, M.; Leong, C.R.; Young, P.M.; Traini, D. The potential to treat lung cancer via inhalation of repurposed drugs. Adv. Drug Deliv. Rev. 2018, 133, 107–130. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liu, L.; Fu, Y.; Gao, J.; He, Y.; Wu, Y.; Lian, X. Dietary cholesterol intake and risk of lung cancer: A meta-analysis. Nutrients 2018, 10, 185. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.-J.; Lee, M.; Oh, Y.; Fang, X.-Q.; Lee, S.; Lim, C.-H.; Park, J.; Lim, J.-H. Statins Decrease Programmed Death-Ligand 1 (PD-L1) by inhibiting AKT and β-Catenin signaling. Cells 2021, 10, 2488. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Leonardi, G.C.; Puccetti, P.; Fallarino, F.; Bianconi, V.; Sahebkar, A.; Baglivo, S.; Chiari, R.; Pirro, M. Targeting indoleamine-2,3-dioxygenase in cancer: Scientific rationale and clinical evidence. Pharmacol. Ther. 2019, 196, 105–116. [Google Scholar] [CrossRef]
- Fernández, L.P.; Merino, M.; Colmenarejo, G.; Moreno-Rubio, J.; Sánchez-Martínez, R.; Quijada-Freire, A.; de Cedrón, M.G.; Reglero, G.; Casado, E.; Sereno, M.; et al. Metabolic enzyme ACSL3 is a prognostic biomarker and correlates with anticancer effectiveness of statins in non- small cell lung cancer. Mol. Oncol. 2020, 14, 3135–3152. [Google Scholar] [CrossRef]
- Vishwanathan, K.; Cantarini, M.; So, K.; Masson, E.; Fetterolf, J.; Ramalingam, S.S.; Harvey, R.D. Impact of Disease and Treatment Response in Drug–Drug Interaction Studies: Osimertinib and Simvastatin in Advanced Non-Small Cell Lung Cancer. Clin. Transl. Sci. 2020, 13, 41–46. [Google Scholar] [CrossRef]
- Tomaszewski, M.; Stȩpień, K.M.; Tomaszewska, J.; Czuczwar, S.J. Statin-induced myopathies. Pharmacol. Rep. 2011, 63, 859–866. [Google Scholar] [CrossRef]
- Chindapasirt, J. Sarcopenia in cancer patients. Asian Pac. J. Cancer Prev. 2015, 16, 8075–8077. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.R.; Dunne, R.F.; Giri, S.; Shachar, S.S.; Caan, B.J. Sarcopenia in the Older Adult With Cancer. J. Clin. Oncol. 2021, 39, 2068–2078. [Google Scholar] [CrossRef] [PubMed]



| BA [16] | t1/2 | Hydrophile/Lipophile | Metabolism | Protein Binding | |
|---|---|---|---|---|---|
| Simvastatin | <5% | 2 h | Lipophile | Hepatic (first-pass, CYP3A4) | >95% |
| Pravastatin | 17–18% | 1.8 h | Hydrophile | Hepatic (first-pass) | 50% |
| Atorvastatin | 12% | 14 h | Lipophile | Hepatic (CYP3A4) | ≥98% |
| Lovastatin | ≤5% | 3 h | Lipophile | Hepatic (first-pass, CYP3A4) | ≥95% |
| Fluvastatin | 24% | 1.2 h | Lipophile | Hepatic (CYP2C9) | ≥98% |
| Rosuvastatin | 20% | 19 h | Hydrophile/lipophile | Scarcely metabolized (10% by liver: CYP2C9, with minor involvement of 2C19, 3A4 e 2D6 isoforms | ~90% |
| Pitavastatin | 51–80% | 5.7–8.9 h | Lipophile | Mainly unmodified Liver has a minor role (UGT1A3 and 2B7; CYP2C8/9) | >99% |
| Dosage | Elimination | Dose Adjustment Kidney Impairment | Dose Adjustment Hepatic Disease | |
|---|---|---|---|---|
| Simvastatin | 5–80 mg | 13% urine 60% feces | eGFR < 30 mL/min: dosage superior to 10 mg/die should be evaluated carefully | Contraindicated in active hepatic disease or persistent transaminases increase |
| Pravastatin | 10–40 mg | 20% urine 70% feces | Moderate–severe kidney impairment: 10 mg starting dose, with follow-up | Contraindicated in active hepatic disease or persistent transaminases increase (3 times superior to upper limit) In some cases, 10 mg dosage is a possible option, with follow-up |
| Atorvastatin | 10–80 mg | 95% bile/feces <5% urine | No dose adjustment needed | Contraindicated in active hepatic disease or persistent transaminases increase (3 times superior to upper limit) Use carefully in other patients with hepatic impairment |
| Lovastatin | 20–40 mg | 83% feces 10% urine | Severe kidney impairment (eGFR ≤ 30 mL/min): doses superior to 20 mg must be evaluated carefully | Contraindicated in active hepatic disease, transaminases increase, cholestasis |
| Fluvastatin | 20–80 mg | Major quote excreted in feces ≤6% urine | Mild–severe kidney impairment: no expected pharmacokinetics variation. However, carefully administer doses > 40 mg/die, in case of severe kidney impairment | Contraindicated in active hepatic disease, transaminases increase |
| Rosuvastatin | 5–40 mg | 90% feces (unmodified) 10% urine | Moderate kidney impairment (eGFR < 60 mL/min): starting dose 5 mg 40 mg dose is contraindicated Severe kidney impairment: do not administrate | Contraindicated in active hepatic disease, transaminases increase Child–Pugh 8–9: consider evaluation of kidney function Child–Pugh > 9: no data available |
| Pitavastatin | 1–4 mg | 95% feces (unmodified, enterohepatic circulation) 5% urine | No dose adjustment in patients with mild kidney impairment. However, caution is needed 4 mg dose is contraindicated in patients with severe kidney impairment | Contraindicated in active hepatic disease or persistent transaminases increase (3 times superior to upper limit) Child Pugh A and B: max 2 mg Child Pugh C: contraindicated |
| In Vivo (Humans) | ||
|---|---|---|
| Retrospective Studies | ||
| Statin | Dosage and Patients | Effectiveness |
| All statins | Dosage commonly used for hypercholesterolemia | Improved prognosis in early-stage patients (10,975 patients analyzed retrospectively) [26] |
| NSCLC stage IV patients: hypercholesterolemia dosage | A cohort of 5118 patients was examined and the statin group had a better survival rate [91] | |
| 3638 lung cancer patients (after diagnosis) 11,051 lung cancer patients (before diagnosis) Hypercholesterolemia dosage | Prognosis improvement in patients consuming statins before (better outcome) and after diagnosis. Lipophilic statins and patients with at least 12 prescriptions had better results [90] | |
| 295,925 patients with 13 different cancer types, 18,721 used statins regularly before the cancer diagnosis: hypercholesterolemia dosage | Reduced cancer mortality [92] | |
| 7280 patients receiving statins and affected by lung cancer in a larger court. Hypercholesterolemia dosage | Statin use > 6 months was associated with a risk reduction of lung cancer of 55% [93] | |
| 5990 lung cancer patients in a larger cohort. Hypercholesterolemia dosage | Statins showed mortality reduction, especially in combination with metformin and aspirin [94] | |
| 41 lung cancer patients (statin group) compared to 792 non-statin group. All patients treated with EGFR-TKIs. Hypercholesterolemia dosage | Better mortality in statin group, especially in tumors < 3 cm and with a CCI score < 3 [95] | |
| 43,802 COPD patients: 10,086 used statins, whereas 33,716 did not Hypercholesterolemia dosage | Liu et al. retrospectively found that the risk of COPD evolution in lung cancer was reduced by statins [96] | |
| Atorvastatin | Atorvastatin (40–80 mg) Observational study performed in 253 patients with malignant pleural mesothelioma or advanced NSCLC treated with PD-1 inhibitors | Better response and progression-free survival. These effects probably due to immune enhancement related to a prolonged retention of antigens on cell membrane and presentation increase [41] |
| 252 patients NSCLC, with 73 statin users (46 atorvastatin): hypercholesterolemia dosage | Evaluation of brain metastasis risk in lung cancer patients treated with statins: no significant results [97] | |
| Rosuvastatin | Rosuvastatin (20–40 mg): high intensity | Better response and progression-free survival. These effects probably due to immune enhancement related to a prolonged retention of antigens on cell membrane and presentation increase [41] |
| Simvastatin | 250 adenocarcinoma tissues (51 statin users) 5–10 mg Low-dose rosuvastatin, pitavastatin, fluvastatin and pravastatin were used, but with minor effects compared to simvastatin | Reduction of EMT, improved sensibility to EGFR-TKI and improved prognosis in adenocarcinoma patients holding p53 mutation. However, a worse outcome was described in wild-type p53 population. Survival of statin users was generally better [58] |
| 252 NSCLC patients, with 73 statin users (18 atorvastatin): hypercholesterolemia dosage | Evaluation of brain metastasis risk in lung cancer patients treated with statins: no significant results [97] | |
| Clinical trials | ||
| Atorvastatin | 67 patients with advanced NSCLC (holding KRAS mutation): 20 mg | Better outcomes in those treated with EGFR-TKI plus simvastatin/atorvastatin than EGFR-TKI alone [98] |
| Pravastatin | The multicenter and randomized phase III trial LUNGSTAR: 846 SCLC patients: 40 mg | The first group received pravastatin plus chemotherapy (etoposide + cisplatin/carboplatin) vs. chemotherapy alone. No significant improvement in outcomes were reported [99] |
| Simvastatin | Phase II trial in 106 NSCLC patients: 40 mg. | No superiority of gefitinib + simvastatin compared to gefitinib alone. However, the combination therapy resulted in better RR and progression-free survival PFS in patients with EGFR adenocarcinomas (wild-type) [100] |
| Phase 2 trial in 61 SCLC patients: 40 mg | Irinotecan + cisplatin + simvastatin showed no significant results [101] Ever-smokers showed a better overall survival compared to never smokers | |
| 68 patients with non-adenocarcinomatous NSCLC (phase II): 40 mg | No significant results with simvastatin plus afatinib [102] | |
| 67 patients with advanced NSCLC (holding KRAS mutation): 20 mg | Better outcomes in those treated with EGFR-TKI plus simvastatin/atorvastatin than EGFR-TKI alone [98] | |
| Metanalysis | ||
| All statins | A total of 23 studies were selected, including 15 observational studies and 8 RCTs. Various dosages, mainly hypercholesterolemia dosage | No protective effect of statins on lung cancer risk [103] |
| Seventeen studies involving 98,445 patients. Various dosages, mainly hypercholesterolemia dosage | Decreased mortality in cohort studies, but not in clinical trials or case-control studies. Enhanced effect of EGFR-TKI [104] | |
| Twenty studies examined. Various dosages, mainly hypercholesterolemia dosage | No correlation between statin use and lung cancer risk [105] | |
| Nineteen studies involving 38,013 lung cancer patients. Various dosages, mainly hypercholesterolemia dosage | No correlation between statin use and lung cancer risk [106] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcianò, G.; Palleria, C.; Casarella, A.; Rania, V.; Basile, E.; Catarisano, L.; Vocca, C.; Bianco, L.; Pelaia, C.; Cione, E.; et al. Effect of Statins on Lung Cancer Molecular Pathways: A Possible Therapeutic Role. Pharmaceuticals 2022, 15, 589. https://doi.org/10.3390/ph15050589
Marcianò G, Palleria C, Casarella A, Rania V, Basile E, Catarisano L, Vocca C, Bianco L, Pelaia C, Cione E, et al. Effect of Statins on Lung Cancer Molecular Pathways: A Possible Therapeutic Role. Pharmaceuticals. 2022; 15(5):589. https://doi.org/10.3390/ph15050589
Chicago/Turabian StyleMarcianò, Gianmarco, Caterina Palleria, Alessandro Casarella, Vincenzo Rania, Emanuele Basile, Luca Catarisano, Cristina Vocca, Luigi Bianco, Corrado Pelaia, Erika Cione, and et al. 2022. "Effect of Statins on Lung Cancer Molecular Pathways: A Possible Therapeutic Role" Pharmaceuticals 15, no. 5: 589. https://doi.org/10.3390/ph15050589
APA StyleMarcianò, G., Palleria, C., Casarella, A., Rania, V., Basile, E., Catarisano, L., Vocca, C., Bianco, L., Pelaia, C., Cione, E., D’Agostino, B., Citraro, R., De Sarro, G., & Gallelli, L. (2022). Effect of Statins on Lung Cancer Molecular Pathways: A Possible Therapeutic Role. Pharmaceuticals, 15(5), 589. https://doi.org/10.3390/ph15050589