
Tofacitinib May Inhibit Myofibroblast Differentiation from Rheumatoid-Fibroblast-like Synoviocytes Induced by TGF-β and IL-6

 ,
,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
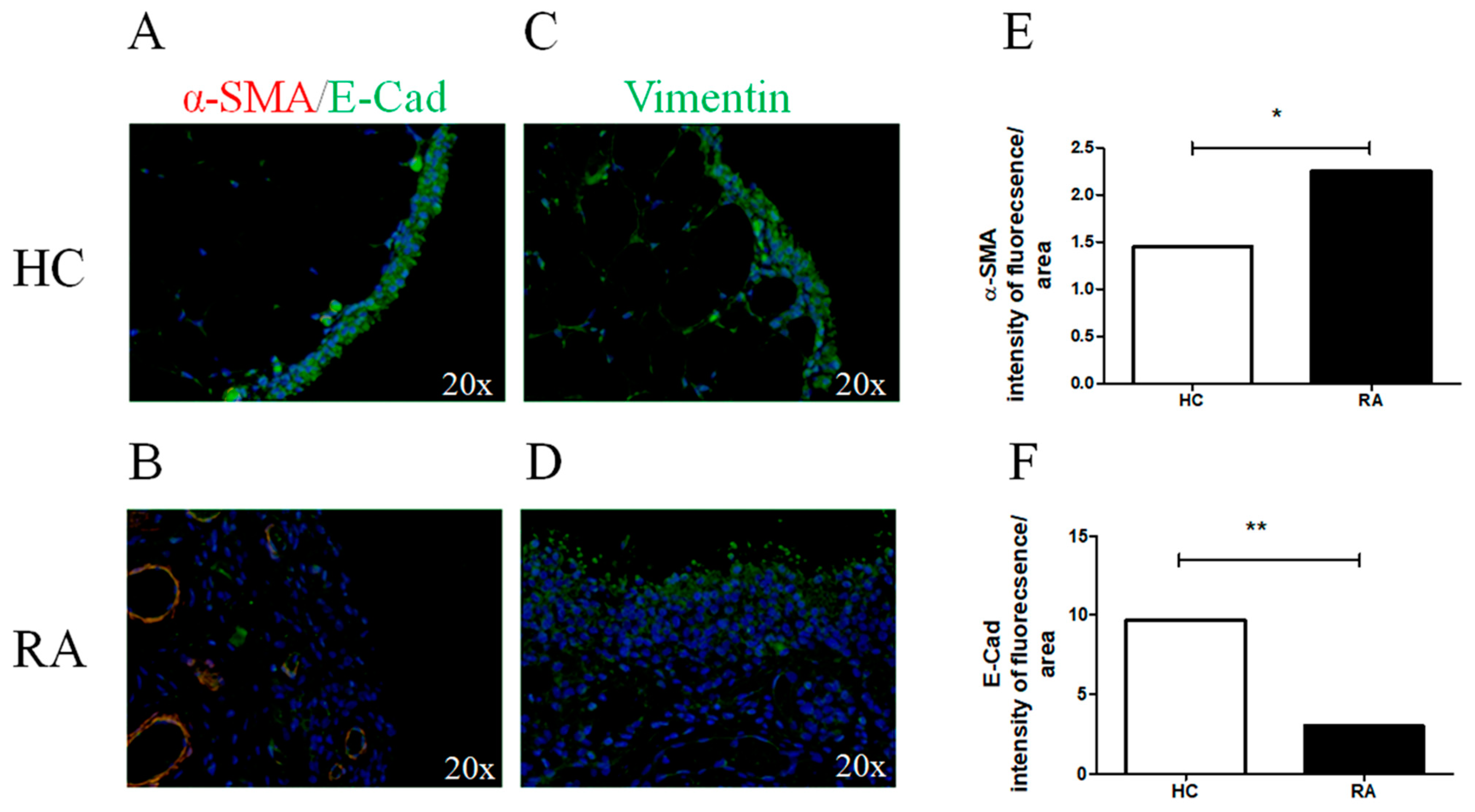
2.1. JAK-1, STAT-3, and Markers of Myofibroblast Differentiation in RA Synovial Tissues
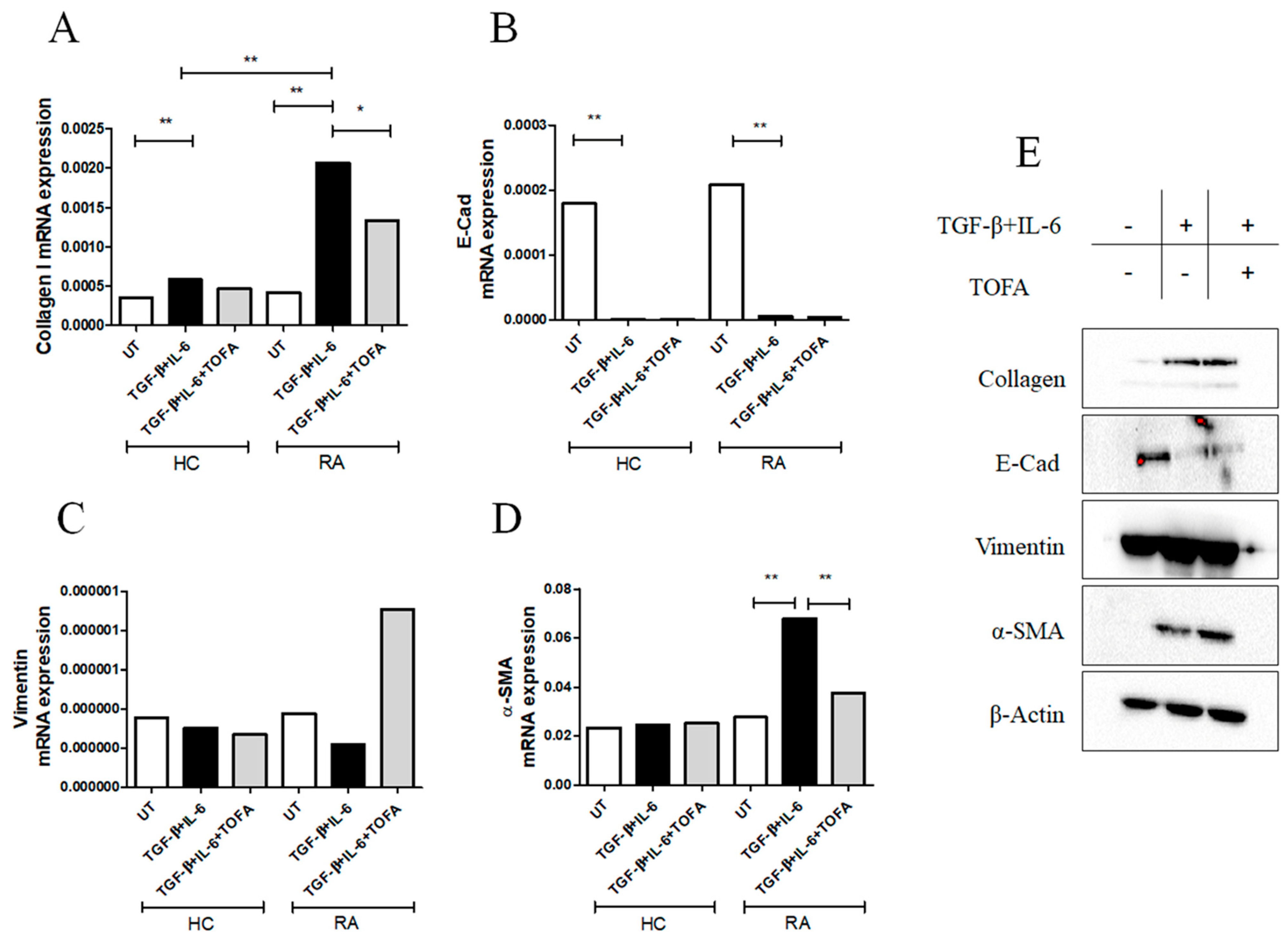
2.2. TGF-β and IL-6 Promoted Myofibroblast Differentiation in RA-FLSs
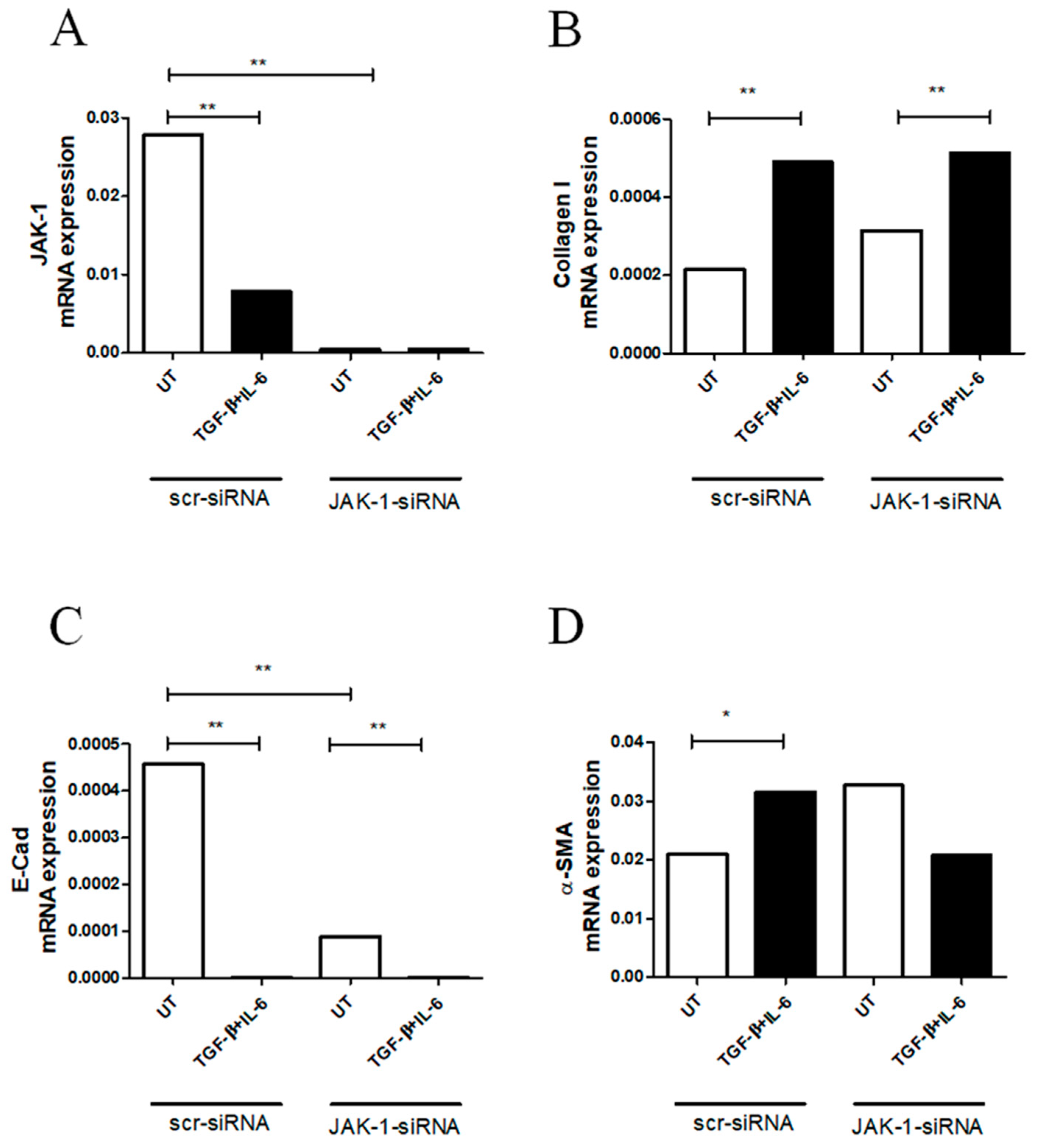
2.3. JAK-1 Gene Silencing Did Not Fully Affect Myofibroblast Differentiation in RA-FLSs
3. Discussion
4. Materials and Methods
4.1. Patients and Samples Collection
4.2. Reagents
4.3. Immunofluorescence
4.4. Immunohistochemistry
4.5. FLSs Isolation and Culture
4.6. FLSs stimulation with TGF-β + IL-6
4.7. FLSs Stimulation with Tofacitinib
4.8. qRT-PCR Analysisc
4.9. Western Blot
4.10. Chemoinvasion Assays
4.11. siRNA Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017, 389, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- Masoumi, M.; Bashiri, H.; Khorramdelazad, H.; Barzaman, K.; Hashemi, N.; Sereshki, H.A.; Sahebkar, A.; Karami, J. Destructive Roles of Fibroblast-like Synoviocytes in Chronic Inflammation and Joint Damage in Rheumatoid Arthritis. Inflammation 2021, 44, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, T.; Shikichi, M.; Kitamura, H.; Yanase, H.; Nozawa-Inoue, K. Morphology and functional roles of synoviocytes in the joint. Arch. Histol. Cytol. 2000, 63, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Steenvoorden, M.; Tolboom, T.C.; van der Pluijm, G.; Löwik, C.; Visser, C.P.; DeGroot, J.; Gittenberger-DeGroot, A.C.; DeRuiter, M.C.; Wisse, B.J.; Huizinga, T.W.; et al. Transition of healthy to diseased synovial tissue in rheumatoid arthritis is associated with gain of mesenchymal/fibrotic characteristics. Arthritis Res. Ther. 2006, 8, R165. [Google Scholar] [CrossRef] [Green Version]
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fi-brotic Disorders. Front. Cell Dev. Biol. 2020, 8, 607483. [Google Scholar] [CrossRef]
- Schuster, R.; Rockel, J.S.; Kapoor, M.; Hinz, B. The inflammatory speech of fibroblasts. Immunol. Rev. 2021, 302, 126–146. [Google Scholar] [CrossRef]
- Zhu, D.; Zhao, J.; Lou, A.; Huang, Q.; OuYang, Q.; Zhu, J.; Fan, M.; He, Y.; Ren, H.; Yang, M. Transforming growth factor β1 promotes fibroblast-like synoviocytes migration and invasion via TGF-β1/Smad signaling in rheumatoid arthritis. Mol. Cell Biochem. 2019, 459, 141–150. [Google Scholar] [CrossRef]
- Song, H.Y.; Kim, M.Y.; Kim, K.H.; Lee, I.H.; Shin, S.H.; Lee, J.S.; Kim, J.H. Synovial fluid of patients with rheumatoid arthritis induces alpha-smooth muscle actin in human adipose tissue-derived mesenchymal stem cells through a TGF-beta1-dependent mechanism. Exp. Mol. Med. 2010, 42, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.Y.; Zeng, Y.; Lei, Z.; Wang, L.; Yang, H.; Liu, Z.; Zhao, J.; Zhang, H.T. JAK/STAT3 signaling is required for TGF-β-induced epithelial-mesenchymal transition in lung cancer cells. Int. J. Oncol. 2014, 44, 1643–1651. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Liu, T.; Wu, J.C.; Luo, S.Z.; Chen, R.; Lu, L.G.; Xu, M.Y. STAT3 aggravates TGF-β1-induced hepatic epithelial-to-mesenchymal transition and migration. Biomed. Pharmacother. 2018, 98, 214–221. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, L.A.; Liongue, C.; Lewis, R.S.; Stephenson, S.E.; Ward, A.C. Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol. Immunol. 2007, 44, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Haan, C.; Kreis, S.; Margue, C.; Behrmann, I. Jaks and cytokine receptors—An intimate relationship. Biochem. Pharmacol. 2006, 72, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Laurence, A.; McInnes, I.B. Back to the future: Oral targeted therapy for RA and other autoimmune diseases. Nat. Rev. Rheumatol. 2013, 9, 173–182. [Google Scholar] [CrossRef]
- Shuai, K.; Liu, B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 2003, 3, 900–911. [Google Scholar] [CrossRef]
- Favoino, E.; Prete, M.; Catacchio, G.; Ruscitti, P.; Navarini, L.; Giacomelli, R.; Perosa, F. Working and safety profiles of JAK/STAT signaling inhibitors. Are these small molecules also smart? Autoimmun. Rev. 2021, 20, 102750. [Google Scholar] [CrossRef]
- Pohlers, D.; Beyer, A.; Koczan, D.; Wilhelm, T.; Thiesen, H.J.; Kinne, R.W. Constitutive upregulation of the transforming growth factor-beta pathway in rheumatoid arthritis synovial fibroblasts. Arthritis Res. Ther. 2007, 9, R59. [Google Scholar] [CrossRef] [Green Version]
- Di Benedetto, P.; Ruscitti, P.; Berardicurti, O.; Panzera, N.; Grazia, N.; Di Vito Nolfi, M.; Di Francesco, B.; Navarini, L.; Maurizi, A.; Rucci, N.; et al. Blocking Jak/STAT signalling using tofacitinib inhibits angiogenesis in ex-perimental arthritis. Arthritis Res. Ther. 2021, 23, 213. [Google Scholar] [CrossRef]
- Di Benedetto, P.; Liakouli, V.; Ruscitti, P.; Berardicurti, O.; Carubbi, F.; Panzera, N.; Di Bartolomeo, S.; Guggino, G.; Ciccia, F.; Triolo, G.; et al. Blocking CD248 molecules in perivascular stromal cells of patients with systemic sclerosis strongly inhibits their differentiation toward myofibroblasts and proliferation: A new potential target for antifibrotic therapy. Arthritis Res. Ther. 2018, 20, 223. [Google Scholar] [CrossRef] [Green Version]
- Sfikakis, P.P.; Vlachogiannis, N.I.; Christopoulos, P.F. Cadherin-11 as a therapeutic target in chronic, inflammatory rheumatic diseases. Clin. Immunol. 2017, 176, 107–113. [Google Scholar] [CrossRef]
- Cipriani, P.; Di Benedetto, P.; Ruscitti, P.; Liakouli, V.; Berardicurti, O.; Carubbi, F.; Ciccia, F.; Guggino, G.; Zazzeroni, F.; Alesse, E.; et al. Perivascular Cells in Diffuse Cutaneous Systemic Sclerosis Overexpress Activated ADAM12 and Are Involved in Myofibroblast Transdifferentiation and Development of Fibrosis. J. Rheumatol. 2016, 43, 1340–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cipriani, P.; Di Benedetto, P.; Ruscitti, P.; Capece, D.; Zazzeroni, F.; Liakouli, V.; Pantano, I.; Berardicurti, O.; Carubbi, F.; Pecetti, G.; et al. The Endothelial-mesenchymal Transition in Systemic Sclerosis Is Induced by Endothelin-1 and Transforming Growth Factor-β and May Be Blocked by Macitentan, a Dual Endothelin-1 Receptor Antagonist. J. Rheumatol. 2015, 42, 1808–1816. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Q.; Zhang, Y.; Liu, D.; Qian, Y.Y.; Zhang, H.; Guo, S.Y.; Sunagawa, M.; Hisamitsu, T.; Liu, Y.Q. PI3 kinase/Akt/HIF-1α pathway is associated with hypoxia-induced epithelial-mesenchymal transition in fibroblast-like synoviocytes of rheumatoid arthritis. Mol. Cell. Biochem. 2013, 372, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Zvaifler, N.J. Relevance of the stroma and epithelial-mesenchymal transition (EMT) for the rheumatic diseases. Arthritis Res. Ther. 2006, 8, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegertjes, R.; Van Caam, A.; Van Beuningen, H.; Koenders, M.; Van Lent, P.; Van Der Kraan, P.; van de Loo, F.; Davidson, E.B. TGF-β dampens IL-6 signaling in articular chondrocytes by decreasing IL-6 receptor expression. Osteoarthr. Cartil. 2019, 27, 1197–1207. [Google Scholar] [CrossRef]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Yu, Y.; Sun, C.; Liu, T.; Liang, T.; Zhan, L.; Lin, X.; Feng, X.H. STAT3 selectively interacts with Smad3 to antagonize TGF-β signalling. Oncogene 2016, 35, 4422. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, J.; Ruan, Y.; Sun, L.; Xu, C.; Jiang, H. Sialyltransferase ST3GAL1 promotes cell migration, invasion, and TGF-β1-induced EMT and confers paclitaxel resistance in ovarian cancer. Cell Death Dis. 2018, 9, 1102. [Google Scholar] [CrossRef] [Green Version]
- Boyle, D.L.; Soma, K.; Hodge, J.; Kavanaugh, A.; Mandel, D.; Mease, P.; Shurmur, R.; Singhal, A.K.; Wei, N.; Rosengren, S.; et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1311–1316. [Google Scholar] [CrossRef] [Green Version]
- Palmroth, M.; Kuuliala, K.; Peltomaa, R.; Virtanen, A.; Kuuliala, A.; Kurttila, A.; Kinnunen, A.; Leirisalo-Repo, M.; Silvennoinen, O.; Isomäki, P. Tofacitinib Suppresses Several JAK-STAT Pathways in Rheumatoid Arthritis In Vivo and Baseline Signaling Profile Associates with Treatment Response. Front. Immunol. 2021, 12, 738481. [Google Scholar] [CrossRef]
- Isailovic, N.; Ceribelli, A.; Cincinelli, G.; Vecellio, M.; Guidelli, G.; Caprioli, M.; Luciano, N.; Motta, F.; Selmi, C.; De Santis, M. Lymphocyte modulation by tofacitinib in patients with rheumatoid arthritis. Clin. Exp. Immunol. 2021, 205, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., III; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruscitti, P.; Liakouli, V.; Panzera, N.; Angelucci, A.; Berardicurti, O.; Di Nino, E.; Navarini, L.; Vomero, M.; Ursini, F.; Mauro, D.; et al. Tofacitinib May Inhibit Myofibroblast Differentiation from Rheumatoid-Fibroblast-like Synoviocytes Induced by TGF-β and IL-6. Pharmaceuticals 2022, 15, 622. https://doi.org/10.3390/ph15050622
Ruscitti P, Liakouli V, Panzera N, Angelucci A, Berardicurti O, Di Nino E, Navarini L, Vomero M, Ursini F, Mauro D, et al. Tofacitinib May Inhibit Myofibroblast Differentiation from Rheumatoid-Fibroblast-like Synoviocytes Induced by TGF-β and IL-6. Pharmaceuticals. 2022; 15(5):622. https://doi.org/10.3390/ph15050622
Chicago/Turabian StyleRuscitti, Piero, Vasiliki Liakouli, Noemi Panzera, Adriano Angelucci, Onorina Berardicurti, Elena Di Nino, Luca Navarini, Marta Vomero, Francesco Ursini, Daniele Mauro, and et al. 2022. "Tofacitinib May Inhibit Myofibroblast Differentiation from Rheumatoid-Fibroblast-like Synoviocytes Induced by TGF-β and IL-6" Pharmaceuticals 15, no. 5: 622. https://doi.org/10.3390/ph15050622
APA StyleRuscitti, P., Liakouli, V., Panzera, N., Angelucci, A., Berardicurti, O., Di Nino, E., Navarini, L., Vomero, M., Ursini, F., Mauro, D., Dolo, V., Ciccia, F., Giacomelli, R., & Cipriani, P. (2022). Tofacitinib May Inhibit Myofibroblast Differentiation from Rheumatoid-Fibroblast-like Synoviocytes Induced by TGF-β and IL-6. Pharmaceuticals, 15(5), 622. https://doi.org/10.3390/ph15050622