
Design and Synthesis of Novel Bis-Imidazolyl Phenyl Butadiyne Derivatives as HCV NS5A Inhibitors

 , ,
, ,  , ,
, ,  and
and
Abstract
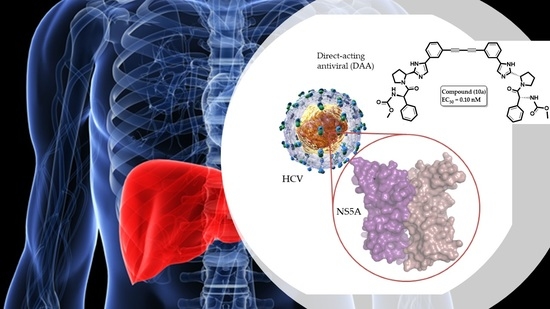
:
1. Introduction
2. Results and Discussion
2.1. Design Concept
2.2. Synthesis
2.3. Biological Activity
2.3.1. Structure–Activity Relationship
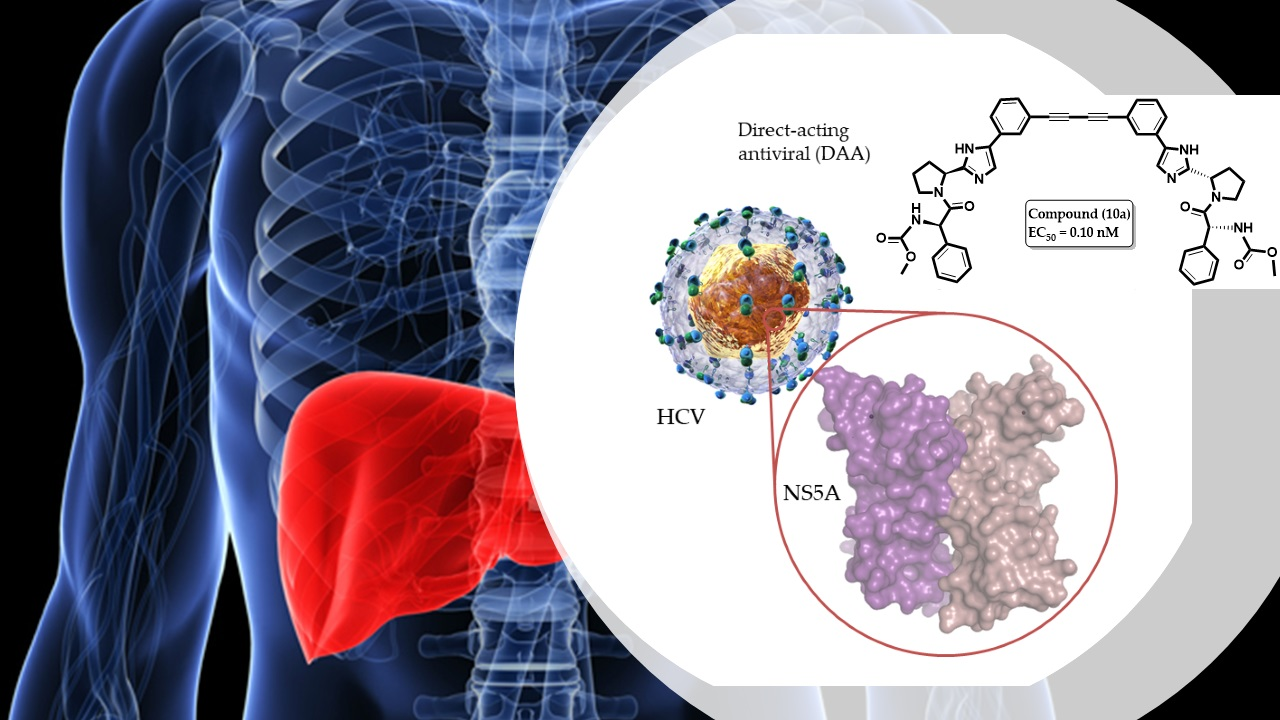
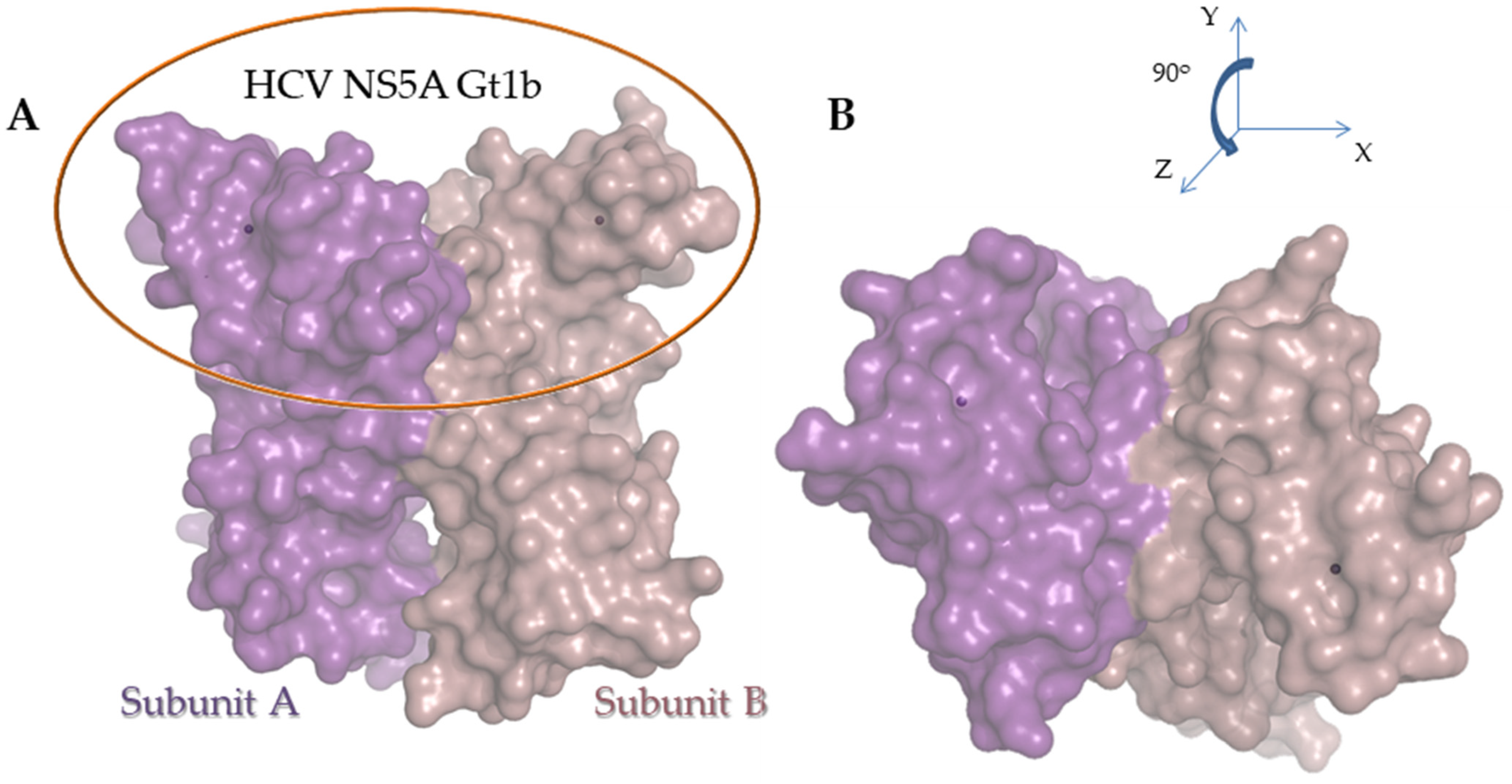
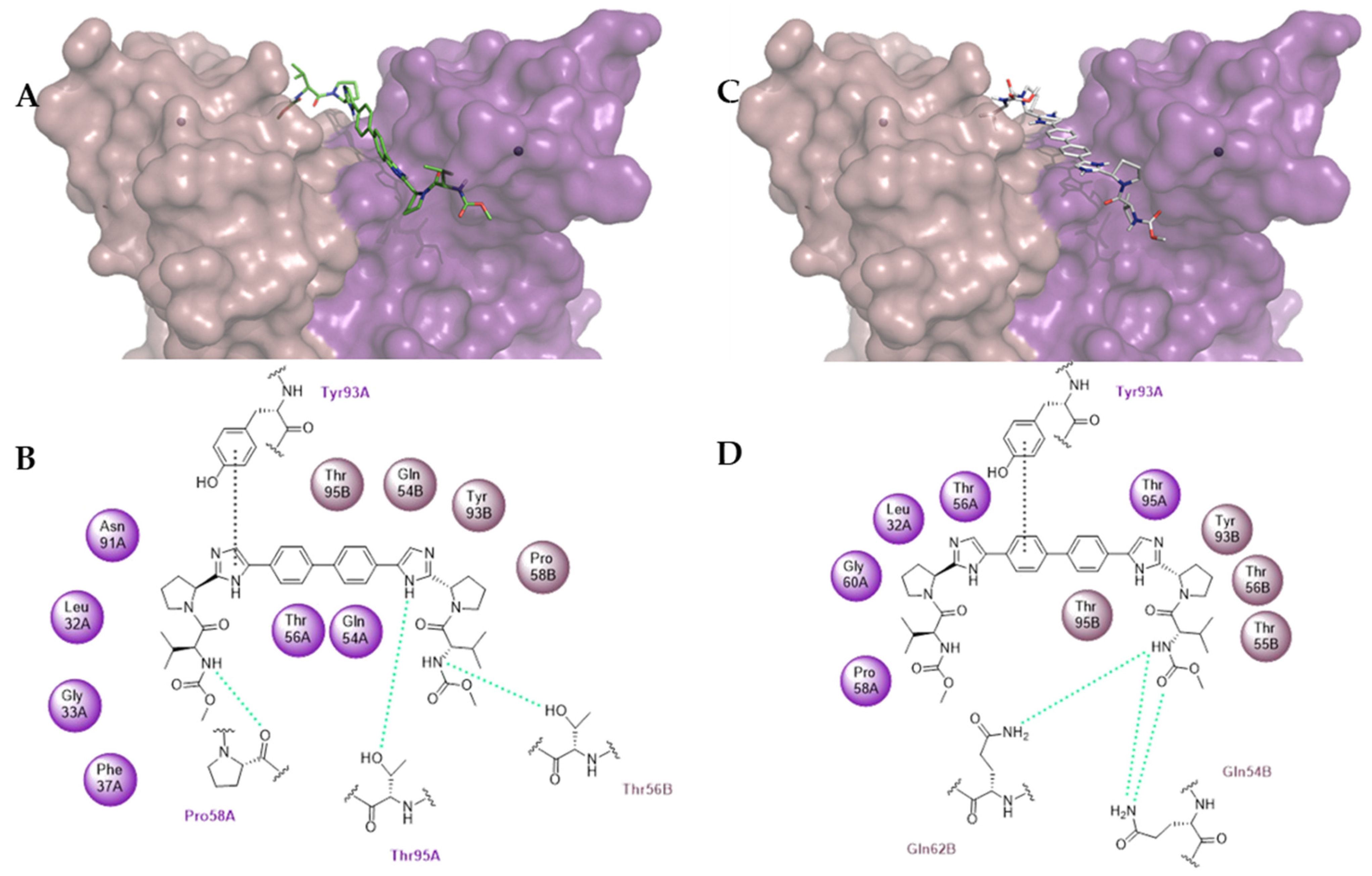
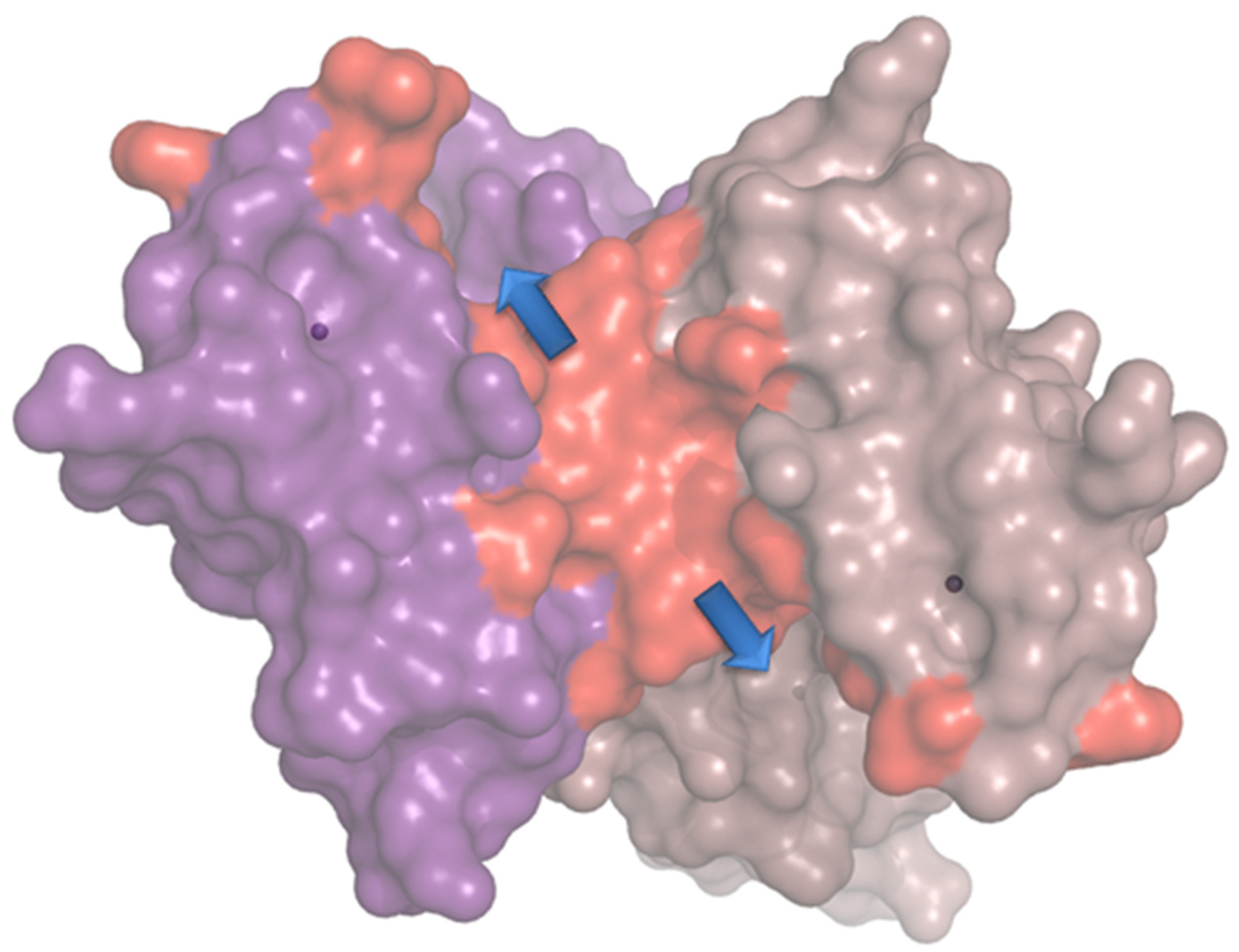
2.3.2. Molecular Modeling
2.3.3. Pharmacochemical Evaluation and Drug-Likeness
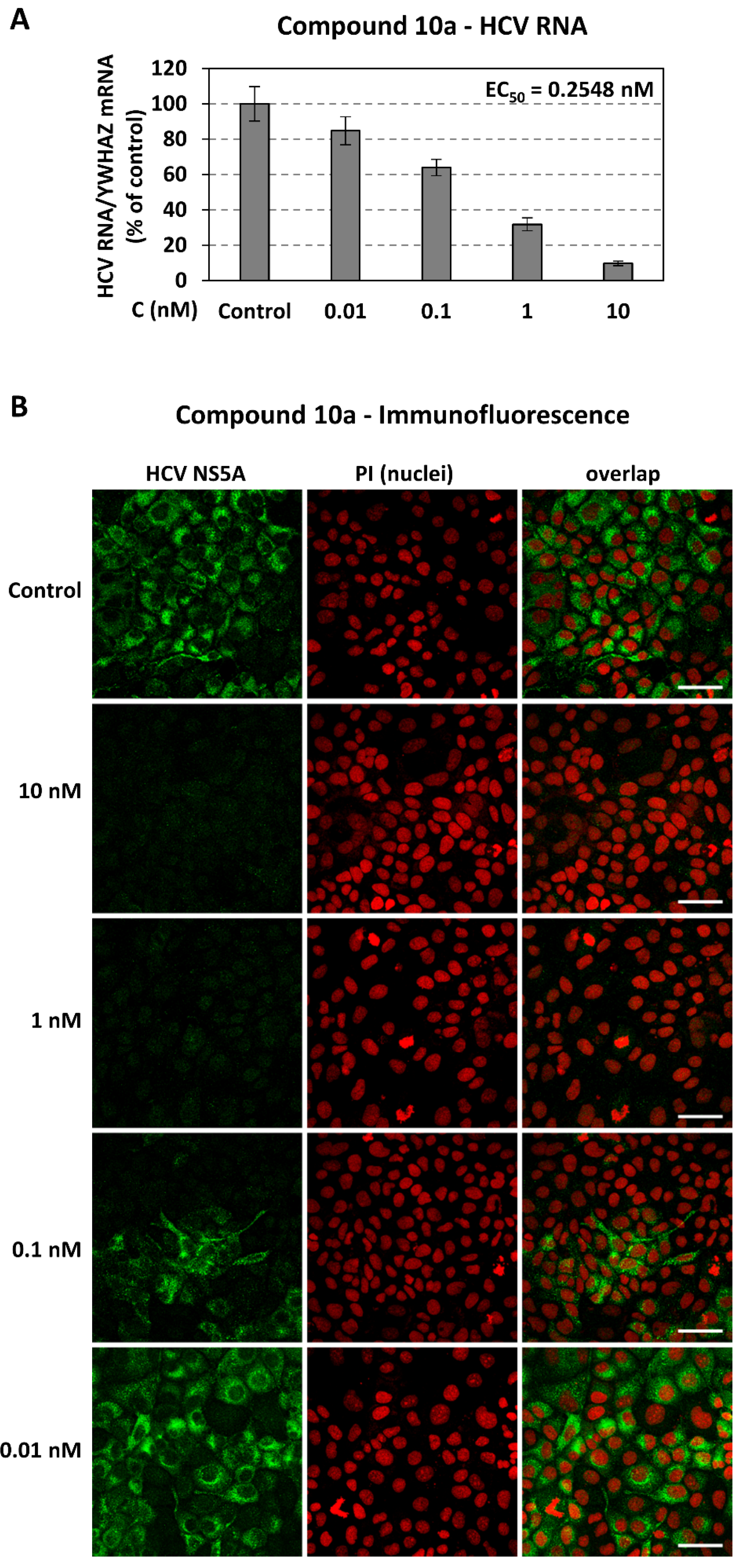
2.3.4. Activity of Compound 10a in Other HCV Genotypes
2.3.5. Validation of Compound Activity with Additional Assays
2.3.6. Evaluation of Metabolic Stability
3. Materials and Methods
3.1. Molecular Modeling
3.1.1. Ligand and Protein Preparation
3.1.2. Molecular Docking Replication Experiments
3.2. Pharmacochemical Profiling
3.3. Chemistry
3.3.1. General Synthetic Methods and Experimental Details for All Compounds
General Procedure for Carbamate Synthesis
General Procedure for Alpha Carbon Bromination
General Procedure for Pyrrolidine Dicarboxylate Formation. Boc-L-proline (4.6 mmol, 1.5 g) was added to Compound A1 or A2 in Acetonitrile (15 mL)
General Procedure for the Formation of the Imidazole Ring
General Procedure for Boc Deprotection
General Procedure for Amide Coupling, Compounds 1–20
Procedures of Sonogashira Reaction Followed by Desilylation and Dimerization, Compounds (1a–10a) and Compounds (1b–10b)
3.4. Biological Assays
3.4.1. Cell Lines and Plasmids
3.4.2. In Vitro Transcription
3.4.3. Transfection with In Vitro Transcribed RNA
3.4.4. Anti-HCV Assay
3.4.5. Luciferase and Bradford Assays
3.4.6. Cytotoxicity Assay
3.4.7. Indirect Immunofluorescence
3.4.8. Total RNA Extraction and Quantification of Viral Replicons
3.4.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kazmierski, W.M.; Baskaran, S.; Walker, J.T.; Miriyala, N.; Meesala, R.; Beesu, M.; Adjabeng, G.; Grimes, R.M.; Hamatake, R.; Leivers, M.R.; et al. GSK2818713, a Novel Biphenylene Scaffold-Based Hepatitis C NS5A Replication Complex Inhibitor with Broad Genotype Coverage. J. Med. Chem. 2020, 63, 4155–4170. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Tan, J.; Wang, Y.; Chen, J.; Li, J.; Jiang, Z.; Quan, Y.; Jin, J.; Li, Y.; Cen, S.; et al. 2-((4-Arylpiperazin-1-yl)methyl)benzonitrile Derivatives as Orally Available Inhibitors of Hepatitis C Virus with a Novel Mechanism of Action. J. Med. Chem. 2020, 63, 5972–5989. [Google Scholar] [CrossRef] [PubMed]
- The Lancet Gastroenterology Hepatology. The hunt for a vaccine for hepatitis C virus continues. Lancet Gastroenterol. Hepatol. 2021, 6, 253. [Google Scholar] [CrossRef]
- Spearman, C.W.; Dusheiko, G.M.; Hellard, M.; Sonderup, M. Hepatitis C. Lancet 2019, 394, 1451–1466. [Google Scholar] [CrossRef]
- Lohmann, V.; Bartenschlager, R. On the history of hepatitis C virus cell culture systems. J. Med. Chem. 2014, 57, 1627–1642. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Lohmann, V.; Penin, F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 2013, 11, 482–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef]
- Fernandez-Caso, B.; Fernandez-Caballero, J.A.; Chueca, N.; Rojo, E.; de Salazar, A.; Garcia Buey, L.; Cardenoso, L.; Garcia, F. Infection with multiple hepatitis C virus genotypes detected using commercial tests should be confirmed using next generation sequencing. Sci. Rep. 2019, 9, 9264. [Google Scholar] [CrossRef]
- Borgia, S.M.; Hedskog, C.; Parhy, B.; Hyland, R.H.; Stamm, L.M.; Brainard, D.M.; Subramanian, M.G.; McHutchison, J.G.; Mo, H.; Svarovskaia, E.; et al. Identification of a Novel Hepatitis C Virus Genotype from Punjab, India: Expanding Classification of Hepatitis C Virus into 8 Genotypes. J. Infect. Dis. 2018, 218, 1722–1729. [Google Scholar] [CrossRef] [Green Version]
- Tsukiyama-Kohara, K.; Kohara, M. Hepatitis C Virus: Viral Quasispecies and Genotypes. Int. J. Mol. Sci. 2018, 19, 23. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Seeff, L.B. Natural history of chronic hepatitis C. Hepatology 2002, 36, S35–S46. [Google Scholar] [CrossRef] [PubMed]
- Jazwinski, A.B.; Muir, A.J. Direct-acting antiviral medications for chronic hepatitis C virus infection. Gastroenterol. Hepatol. 2011, 7, 154–162. [Google Scholar]
- Li, G.; De Clercq, E. Current therapy for chronic hepatitis C: The role of direct-acting antivirals. Antivir. Res. 2017, 142, 83–122. [Google Scholar] [CrossRef]
- Wilby, K.J.; Partovi, N.; Ford, J.A.; Greanya, E.; Yoshida, E.M. Review of boceprevir and telaprevir for the treatment of chronic hepatitis C. Can. J. Gastroenterol. 2012, 26, 205–210. [Google Scholar] [CrossRef]
- European Association for Study of the Liver. EASL Recommendations on Treatment of Hepatitis C 2015. J. Hepatol. 2015, 63, 199–236. [Google Scholar] [CrossRef] [Green Version]
- Maasoumy, B.; Port, K.; Markova, A.A.; Serrano, B.C.; Rogalska-Taranta, M.; Sollik, L.; Mix, C.; Kirschner, J.; Manns, M.P.; Wedemeyer, H.; et al. Eligibility and safety of triple therapy for hepatitis C: Lessons learned from the first experience in a real world setting. PLoS ONE 2013, 8, e55285. [Google Scholar] [CrossRef] [Green Version]
- Pearlman, B.L. Protease inhibitors for the treatment of chronic hepatitis C genotype-1 infection: The new standard of care. Lancet Infect. Dis. 2012, 12, 717–728. [Google Scholar] [CrossRef]
- Asselah, T.; Marcellin, P.; Schinazi, R.F. Treatment of hepatitis C virus infection with direct-acting antiviral agents: 100% cure? Liver Int. 2018, 38, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.S., Jr.; Buti, M.; Rodrigues, L.; Chulanov, V.; Chuang, W.L.; Aguilar, H.; Horvath, G.; Zuckerman, E.; Carrion, B.R.; Rodriguez-Perez, F.; et al. Glecaprevir/pibrentasvir for 8weeks in treatment-naive patients with chronic HCV genotypes 1–6 and compensated cirrhosis: The EXPEDITION-8 trial. J. Hepatol. 2020, 72, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Ramdas, V.; Talwar, R.; Banerjee, M.; Joshi, A.A.; Das, A.K.; Walke, D.S.; Borhade, P.; Dhayagude, U.; Loriya, R.; Gote, G.; et al. Discovery and Characterization of Potent Pan-Genotypic HCV NS5A Inhibitors Containing Novel Tricyclic Central Core Leading to Clinical Candidate. J. Med. Chem. 2019, 62, 10563–10582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elazar, M.; Glenn, J.S. HCV NS5A inhibitors: The devil is in the details. Gastroenterology 2014, 147, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Ross-Thriepland, D.; Harris, M. Hepatitis C virus NS5A: Enigmatic but still promiscuous 10 years on! J. Gen. Virol. 2015, 96, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Link, J.O.; Taylor, J.G.; Xu, L.; Mitchell, M.; Guo, H.; Liu, H.; Kato, D.; Kirschberg, T.; Sun, J.; Squires, N.; et al. Discovery of ledipasvir (GS-5885): A potent, once-daily oral NS5A inhibitor for the treatment of hepatitis C virus infection. J. Med. Chem. 2014, 57, 2033–2046. [Google Scholar] [CrossRef]
- Belema, M.; Lopez, O.D.; Bender, J.A.; Romine, J.L.; St. Laurent, D.R.; Langley, D.R.; Lemm, J.A.; O’Boyle, D.R., 2nd; Sun, J.H.; Wang, C.; et al. Discovery and development of hepatitis C virus NS5A replication complex inhibitors. J. Med. Chem. 2014, 57, 1643–1672. [Google Scholar] [CrossRef]
- Yu, W.; Tong, L.; Selyutin, O.; Chen, L.; Hu, B.; Zhong, B.; Hao, J.; Ji, T.; Zan, S.; Yin, J.; et al. Discovery of MK-6169, a Potent Pan-Genotype Hepatitis C Virus NS5A Inhibitor with Optimized Activity against Common Resistance-Associated Substitutions. J. Med. Chem. 2018, 61, 3984–4003. [Google Scholar] [CrossRef]
- Abdallah, M.; Hamed, M.M.; Frakolaki, E.; Katsamakas, S.; Vassilaki, N.; Bartenschlager, R.; Zoidis, G.; Hirsch, A.K.H.; Abdel-Halim, M.; Abadi, A.H. Redesigning of the cap conformation and symmetry of the diphenylethyne core to yield highly potent pan-genotypic NS5A inhibitors with high potency and high resistance barrier. Eur. J. Med. Chem. 2022, 229, 114034. [Google Scholar] [CrossRef]
- Leila, A.R.S.; Mousa, M.H.A.; Frakolaki, E.; Vassilaki, N.; Bartenschlager, R.; Zoidis, G.; Abdel-Halim, M.; Abadi, A.H. Symmetric Anti-HCV Agents: Synthesis, Antiviral Properties, and Conformational Aspects of Core Scaffolds. ACS Omega 2019, 4, 11440–11454. [Google Scholar] [CrossRef] [Green Version]
- Kumari, S.; Carmona, A.V.; Tiwari, A.K.; Trippier, P.C. Amide Bond Bioisosteres: Strategies, Synthesis, and Successes. J. Med. Chem. 2020, 63, 12290–12358. [Google Scholar] [CrossRef]
- Kumar, D.; Mariappan, G.; Husain, A.; Monga, J.; Kumar, S. Design, synthesis and cytotoxic evaluation of novel imidazolone fused quinazolinone derivatives. Arab. J. Chem. 2017, 10, 344–350. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Ma, Z.; Zhang, D. Synthesis of imidazole-based medicinal molecules utilizing the van leusen imidazole synthesis. Pharmaceuticals 2020, 13, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishnan, A.K.; Angamaly, S.A.; Velayudhan, M.P. An Insight into the Biological Properties of Imidazole-Based Schiff Bases: A Review. ChemistrySelect 2021, 6, 10918–10947. [Google Scholar] [CrossRef]
- Zhang, L.; Peng, X.M.; Damu, G.L.; Geng, R.X.; Zhou, C.H. Comprehensive review in current developments of imidazole-based medicinal chemistry. Med. Res. Rev. 2014, 34, 340–437. [Google Scholar] [CrossRef] [PubMed]
- Ivanenkov, Y.A.; Aladinskiy, V.A.; Bushkov, N.A.; Ayginin, A.A.; Majouga, A.G.; Ivachtchenko, A.V. Small-molecule inhibitors of hepatitis C virus (HCV) non-structural protein 5A (NS5A): A patent review (2010–2015). Expert Opin. Ther. Pat. 2017, 27, 401–414. [Google Scholar] [CrossRef]
- Bae, I.H.; Kim, H.S.; You, Y.; Chough, C.; Choe, W.; Seon, M.K.; Lee, S.G.; Keum, G.; Jang, S.K.; Moon Kim, B. Novel benzidine and diaminofluorene prolinamide derivatives as potent hepatitis C virus NS5A inhibitors. Eur. J. Med. Chem. 2015, 101, 163–178. [Google Scholar] [CrossRef]
- Bae, I.H.; Choi, J.K.; Chough, C.; Keum, S.J.; Kim, H.; Jang, S.K.; Kim, B.M. Potent Hepatitis C Virus NS5A Inhibitors Containing a Benzidine Core. ACS Med. Chem. Lett. 2014, 5, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Love, R.A.; Brodsky, O.; Hickey, M.J.; Wells, P.A.; Cronin, C.N. Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. J. Virol. 2009, 83, 4395–4403. [Google Scholar] [CrossRef] [Green Version]
- Kazmierski, W.M.; Miriyala, N.; Johnson, D.K.; Baskaran, S. The Discovery of Conformationally Constrained Bicyclic Peptidomimetics as Potent Hepatitis C NS5A Inhibitors. ACS Med. Chem. Lett. 2021, 12, 1649–1655. [Google Scholar] [CrossRef]
- Berger, C.; Romero-Brey, I.; Radujkovic, D.; Terreux, R.; Zayas, M.; Paul, D.; Harak, C.; Hoppe, S.; Gao, M.; Penin, F.; et al. Daclatasvir-like inhibitors of NS5A block early biogenesis of hepatitis C virus-induced membranous replication factories, independent of RNA replication. Gastroenterology 2014, 147, 1094–1105.e25. [Google Scholar] [CrossRef]
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 2017, 33, 3658–3660. [Google Scholar] [CrossRef] [Green Version]
- Darwish, S.S.; Abdel-Halim, M.; Salah, M.; Abadi, A.H.; Becker, W.; Engel, M. Development of novel 2, 4-bispyridyl thiophene–based compounds as highly potent and selective Dyrk1A inhibitors. Part I: Benzamide and benzylamide derivatives. Eur. J. Med. Chem. 2018, 157, 1031–1050. [Google Scholar] [CrossRef] [PubMed]
- Darwish, S.S.; Abdel-Halim, M.; ElHady, A.K.; Salah, M.; Abadi, A.H.; Becker, W.; Engel, M. Development of novel amide–derivatized 2, 4-bispyridyl thiophenes as highly potent and selective Dyrk1A inhibitors. Part II: Identification of the cyclopropylamide moiety as a key modification. Eur. J. Med. Chem. 2018, 158, 270–285. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, P.C.; Nicholls, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef]
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef]
- McGaughey, G.B.; Sheridan, R.P.; Bayly, C.I.; Culberson, J.C.; Kreatsoulas, C.; Lindsley, S.; Maiorov, V.; Truchon, J.F.; Cornell, W.D. Comparison of topological, shape, and docking methods in virtual screening. J. Chem. Inf. Model. 2007, 47, 1504–1519. [Google Scholar] [CrossRef]
- McGann, M.R.; Almond, H.R.; Nicholls, A.; Grant, J.A.; Brown, F.K. Gaussian docking functions. Biopolymers 2003, 68, 76–90. [Google Scholar] [CrossRef] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. In Chemical Biology; Springer: Cham, Switzerland, 2015; pp. 243–250. [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.4.1. Available online: https://pymol-molecular-graphics-system.soft112.com/ (accessed on 1 March 2021).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Kelley, B.P.; Brown, S.P.; Warren, G.L.; Muchmore, S.W. POSIT: Flexible Shape-Guided Docking For Pose Prediction. J. Chem. Inf. Model. 2015, 55, 1771–1780. [Google Scholar] [CrossRef]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Allinger, N.L. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 2002, 99, 8127–8134. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannhold, R.; Poda, G.I.; Ostermann, C.; Tetko, I.V. Calculation of molecular lipophilicity: State-of-the-art and comparison of log P methods on more than 96,000 compounds. J. Pharm. Sci. 2009, 98, 861–893. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Oprea, T.I.; Davis, A.M.; Teague, S.J.; Leeson, P.D. Is there a difference between leads and drugs? A historical perspective. J. Chem. Inf. Comput. Sci. 2001, 41, 1308–1315. [Google Scholar] [CrossRef]
- Oprea, T.I. Property distribution of drug-related chemical databases. J. Comput. Aided Mol. Des. 2000, 14, 251–264. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Park, B.K.; Boobis, A.; Clarke, S.; Goldring, C.E.; Jones, D.; Kenna, J.G.; Lambert, C.; Laverty, H.G.; Naisbitt, D.J.; Nelson, S.; et al. Managing the challenge of chemically reactive metabolites in drug development. Nat. Rev. Drug Discov. 2011, 10, 292–306. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ElHady, A.K.; Shih, S.-P.; Chen, Y.-C.; Liu, Y.-C.; Ahmed, N.S.; Keeton, A.B.; Piazza, G.A.; Engel, M.; Abadi, A.H.; Abdel-Halim, M. Extending the use of tadalafil scaffold: Development of novel selective phosphodiesterase 5 inhibitors and histone deacetylase inhibitors. Bioorganic Chem. 2020, 98, 103742. [Google Scholar] [CrossRef] [PubMed]
- Vrolijk, J.M.; Kaul, A.; Hansen, B.E.; Lohmann, V.; Haagmans, B.L.; Schalm, S.W.; Bartenschlager, R. A replicon-based bioassay for the measurement of interferons in patients with chronic hepatitis C. J. Virol. Methods 2003, 110, 201–209. [Google Scholar] [CrossRef]
- Jo, J.; Aichele, U.; Kersting, N.; Klein, R.; Aichele, P.; Bisse, E.; Sewell, A.K.; Blum, H.E.; Bartenschlager, R.; Lohmann, V.; et al. Analysis of CD8+ T-cell-mediated inhibition of hepatitis C virus replication using a novel immunological model. Gastroenterology 2009, 136, 1391–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lougiakis, N.; Frakolaki, E.; Karmou, P.; Pouli, N.; Marakos, P.; Madan, V.; Bartenschlager, R.; Vassilaki, N. Novel nucleoside analogues targeting HCV replication through an NS5A-dependent inhibition mechanism. Chem. Biol. Drug Des. 2017, 90, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.; Scheel, T.K.; Gottwein, J.M.; Marukian, S.; Dustin, L.B.; Bukh, J.; Rice, C.M. Efficient replication of genotype 3a and 4a hepatitis C virus replicons in human hepatoma cells. Antimicrob. Agents Chemother. 2012, 56, 5365–5373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimakami, T.; Welsch, C.; Yamane, D.; McGivern, D.R.; Yi, M.; Zeuzem, S.; Lemon, S.M. Protease inhibitor-resistant hepatitis C virus mutants with reduced fitness from impaired production of infectious virus. Gastroenterology 2011, 140, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Kaul, A.; Woerz, I.; Meuleman, P.; Leroux-Roels, G.; Bartenschlager, R. Cell culture adaptation of hepatitis C virus and in vivo viability of an adapted variant. J. Virol. 2007, 81, 13168–13179. [Google Scholar] [CrossRef] [Green Version]
- Vassilaki, N.; Friebe, P.; Meuleman, P.; Kallis, S.; Kaul, A.; Paranhos-Baccala, G.; Leroux-Roels, G.; Mavromara, P.; Bartenschlager, R. Role of the hepatitis C virus core+1 open reading frame and core cis-acting RNA elements in viral RNA translation and replication. J. Virol. 2008, 82, 11503–11515. [Google Scholar] [CrossRef] [Green Version]
- Van den Hoff, M.J.B.; Christoffels, V.M.; Labruyère, W.T.; Moorman, A.F.M.; Lamers, W.H. Electrotransfection with “Intracellular” Buffer. In Animal Cell Electroporation and Electrofusion Protocols; Nickoloff, J.A., Ed.; Humana Press: Totowa, NJ, USA, 1995; pp. 185–197. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
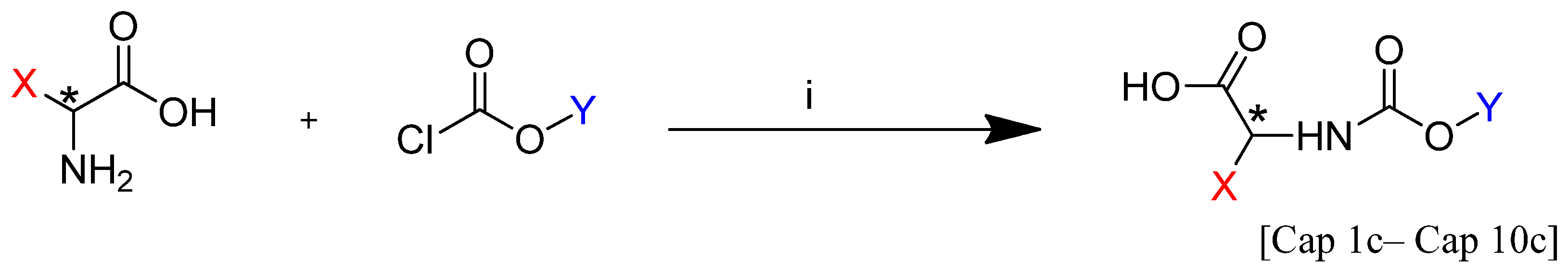{kind=link}
| Compound | Attachment to Core | Terminal Cap Stereochemistry | X | Y |
|---|---|---|---|---|
| 1a | meta | S | –CH(CH3)2 | –CH3 |
| 2a | meta | R | –CH(CH3)2 | –CH3 |
| 3a | meta | S | –CH(CH3)2 | –C2H5 |
| 4a | meta | S | –CH(CH3)2 | –C4H9 |
| 5a | meta | S | –CH2CH(CH3)2 | –CH3 |
| 6a | meta | R | –CH2CH(CH3)2 | –CH3 |
| 7a | meta | S | –CH2CH(CH3)2 | –C2H5 |
| 8a | meta | R | –CH2CH(CH3)2 | –C2H5 |
| 9a | meta | S | –C6H5 | –CH3 |
| 10a | meta | R | –C6H5 | –CH3 |
| 1b | para | S | –CH(CH3)2 | –CH3 |
| 2b | para | R | –CH(CH3)2 | –CH3 |
| 3b | para | S | –CH(CH3)2 | –C2H5 |
| 4b | para | S | –CH(CH3)2 | –C4H9 |
| 5b | para | S | –CH2CH(CH3)2 | –CH3 |
| 6b | para | R | –CH2CH(CH3)2 | –CH3 |
| 7b | para | S | –CH2CH(CH3)2 | –C2H5 |
| 8b | para | R | –CH2CH(CH3)2 | –C2H5 |
| 9b | para | S | –C6H5 | –CH3 |
| 10b | para | R | –C6H5 | –CH3 |
| Cap | * | X | Y |
|---|---|---|---|
| 1c | S | -CH(CH3)2 | Methyl |
| 2c | R | -CH(CH3)2 | Methyl |
| 3c | S | -CH(CH3)2 | Ethyl |
| 4c | S | -CH(CH3)2 | Butyl |
| 5c | S | -CH2CH(CH3)2 | Methyl |
| 6c | R | -CH2CH(CH3)2 | Methyl |
| 7c | S | -CH2CH(CH3)2 | Ethyl |
| 8c | R | -CH2CH(CH3)2 | Ethyl |
| 9c | S | -C6H5 | Methyl |
| 10c | R | -C6H5 | Ethyl |
 | ||||||
|---|---|---|---|---|---|---|
| Compound | Cap * | X | Y | HCV Replicon 1b | ||
| EC50 (nM) b | CC50 (nM) b | SI50 | ||||
| 1a | S | –CH(CH3)2 | –CH3 | 166.40 | >100,000 | >601.96 |
| 2a | R | –CH(CH3)2 | –CH3 | 399.90 | >100,000 | >250.06 |
| 3a | S | –CH(CH3)2 | –C2H5 | 158.80 | >100,000 | >629.72 |
| 4a | S | –CH(CH3)2 | –C4H9 | 901.80 | >100,000 | >110.89 |
| 5a | S | –CH2CH(CH3)2 | –CH3 | 113.70 | >100,000 | >879.51 |
| 6a | R | –CH2CH(CH3)2 | –CH3 | 96.69 | >100,000 | >1034.23 |
| 7a | S | –CH2CH(CH3)2 | –C2H5 | 97.89 | >100,000 | >1021.55 |
| 8a | R | –CH2CH(CH3)2 | –C2H5 | 85.41 | >100,000 | >1170.82 |
| 9a | S | –C6H6 | –CH3 | 62.04 | >100,000 | >1611.86 |
| 10a | R | –C6H6 | –CH3 | 0.1001 | >100,000 | >999,001.00 |
| 1b | S | –CH(CH3)2 | –CH3 | 89.80 | >100,000 | >1113.58 |
| 2b | R | –CH(CH3)2 | –CH3 | 402.90 | >100,000 | >248.20 |
| 3b | S | –CH(CH3)2 | –C2H5 | 336.20 | >100,000 | >297.44 |
| 4b | S | –CH(CH3)2 | –C4H9 | >1000 | >100,000 | ND |
| 5b | S | –CH2CH(CH3)2 | –CH3 | 16.12 | >100,000 | >6203.47 |
| 6b | R | –CH2CH(CH3)2 | –CH3 | 42.11 | >100,000 | >1512.63 |
| 7b | S | –CH2CH(CH3)2 | –C2H5 | 58.85 | >100,000 | >1699.23 |
| 8b | R | –CH2CH(CH3)2 | –C2H5 | 77.52 | >100,000 | >1289.98 |
| 9b | S | –C6H6 | –CH3 | 184.30 | >100,000 | >542.59 |
| 10b | R | –C6H6 | –CH3 | 84.69 | >100,000 | >1180.77 |
| Daclatasvir | 0.027 | 17,700 | 655,556 | |||
| Gt1a | Gt2a | Gt3a | Gt4a | ||||||
|---|---|---|---|---|---|---|---|---|---|
| CC50 (nM) | EC50 (nM) | SI | EC50 (nM) | SI | EC50 (nM) | SI | EC50 (nM) | SI | |
| Comp. 10a | >100,000 | 35.81 | >2792.52 | 28.03 | >3567.61 | 128.40 | >777.00 | 65.04 | >1537.52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamdy, J.; Emadeldin, N.; Hamed, M.M.; Frakolaki, E.; Katsamakas, S.; Vassilaki, N.; Zoidis, G.; Hirsch, A.K.H.; Abdel-Halim, M.; Abadi, A.H. Design and Synthesis of Novel Bis-Imidazolyl Phenyl Butadiyne Derivatives as HCV NS5A Inhibitors. Pharmaceuticals 2022, 15, 632. https://doi.org/10.3390/ph15050632
Hamdy J, Emadeldin N, Hamed MM, Frakolaki E, Katsamakas S, Vassilaki N, Zoidis G, Hirsch AKH, Abdel-Halim M, Abadi AH. Design and Synthesis of Novel Bis-Imidazolyl Phenyl Butadiyne Derivatives as HCV NS5A Inhibitors. Pharmaceuticals. 2022; 15(5):632. https://doi.org/10.3390/ph15050632
Chicago/Turabian StyleHamdy, Jehad, Nouran Emadeldin, Mostafa M. Hamed, Efseveia Frakolaki, Sotirios Katsamakas, Niki Vassilaki, Grigoris Zoidis, Anna K. H. Hirsch, Mohammad Abdel-Halim, and Ashraf H. Abadi. 2022. "Design and Synthesis of Novel Bis-Imidazolyl Phenyl Butadiyne Derivatives as HCV NS5A Inhibitors" Pharmaceuticals 15, no. 5: 632. https://doi.org/10.3390/ph15050632
APA StyleHamdy, J., Emadeldin, N., Hamed, M. M., Frakolaki, E., Katsamakas, S., Vassilaki, N., Zoidis, G., Hirsch, A. K. H., Abdel-Halim, M., & Abadi, A. H. (2022). Design and Synthesis of Novel Bis-Imidazolyl Phenyl Butadiyne Derivatives as HCV NS5A Inhibitors. Pharmaceuticals, 15(5), 632. https://doi.org/10.3390/ph15050632