1. Introduction
Biomedical imaging, one of the pillars of modern cancer management, is tightly integrated into clinical decision making. Imaging is involved in all stages of the diagnostic process including screening, staging, therapy planning and management. Early diagnosis is known to be a major factor in the reduction of mortality, treatment costs and hospital stays. Since cancer is a multi-factorial disease multiple imaging approaches need to be combined to correspond to various mechanisms and phases of development. With the use of highly specific probes, it is now possible to visualize cancer development at a very early stage [
1,
2].
In the last two decades, monoclonal antibodies (mAbs) have become an increasingly successful treatment modality in a wide range of diseases due to their high specificity and affinity. The same properties of mAbs are also explored in molecular imaging to target antigens of interest in vivo [
3]. Imaging probes are the key elements of molecular imaging and have to offer high sensitivity, low background noise, low toxicity and relative stability [
4]. A number of studies indicate that, in certain approaches, aptamers are superior to mAbs offering reduced immunogenicity, structural stability and smaller size (~ 3 nm compared to 10–15 nm for antibodies) [
5]. However, to date the use of aptamers in imaging has not been sufficiently explored.
Aptamer-based probes were shown to provide superior images as compared to mAbs in stimulated emission depletion microscopy (STED) [
6] and were able to recognize more epitopes than antibodies; thus, providing a denser labeling of the structures of interest. In addition, a study comparing the utility of
111In-labeled aptamers and antibodies targeting EGFR in μSPECT/CT imaging demonstrated better tumor-specific uptake of aptamers in nude mice bearing the highly malignant human OSC-19 tumors [
7]. These two studies clearly demonstrated the superior quality of aptamer-based probes over mAbs in terms of obtaining accurate images for cell biology and in vivo. Consequently, aptamers are evaluated as diagnostic tools in a number of clinical trials [
8]. Nonetheless, the diversity of available diagnostic imaging techniques employed in tumor detection, characterization and evaluation of therapeutic interventions (optical imaging, fluorescence and bioluminescence, magnetic resonance imaging (MRI), positron-emission tomography (PET), single-photon emission computed tomography (SPECT), computed tomography (CT), and ultrasound (US) techniques and vast landscape of possible markers opens a significantly broader and yet unexplored perspective of the utility of aptamer-based probes [
8].
Here, we describe the development of a single-stranded aptamer-based probe specific for human Programmed death-ligand 1 (PD-L1). SELEX methodology allowed to identify functional oligonucleotide sequence, which was then examined for specificity and ability to selectively label tumor cells in several in vitro and in vivo models. Most interestingly, our probe demonstrated the potential of in vivo tumor imaging in two unrelated mouse models providing a starting point for the development of the noninvasive diagnostic method of tumor classification according to the PD-L1 status.
3. Discussion
The spectacular success of PD-L1 targeting monoclonal antibodies in cancer therapy has kindled significant interest in investigating the possible utility of PD-L1 as a diagnostic marker for imaging, a selection marker for antibody therapy [
15] and for monitoring of treatment. Antibodies provide suitable tools in some imaging approaches, but aptamer-based detection methods could provide superior in vivo properties and significantly lower production costs.
Aptamer probes are synthetic oligonucleotides that can be easily modified and chemically coupled to reporters suited for various imaging techniques. Positron emission tomography (PET), computed tomography (CT), magnetic resonance imaging (MRI) and fluorescence imaging all require specific probes. Near-infrared imaging (NIR) is especially attractive in this context as it does not require expensive instrumentation and does not expose the patients and the personnel to harmful radiation. Aptamer-based NIR probes could selectively highlight lesions to facilitate diagnosis or surgical resection [
16]. Targeting probes might also serve to direct delivery of bioactive agents (photosensitizers, drugs).
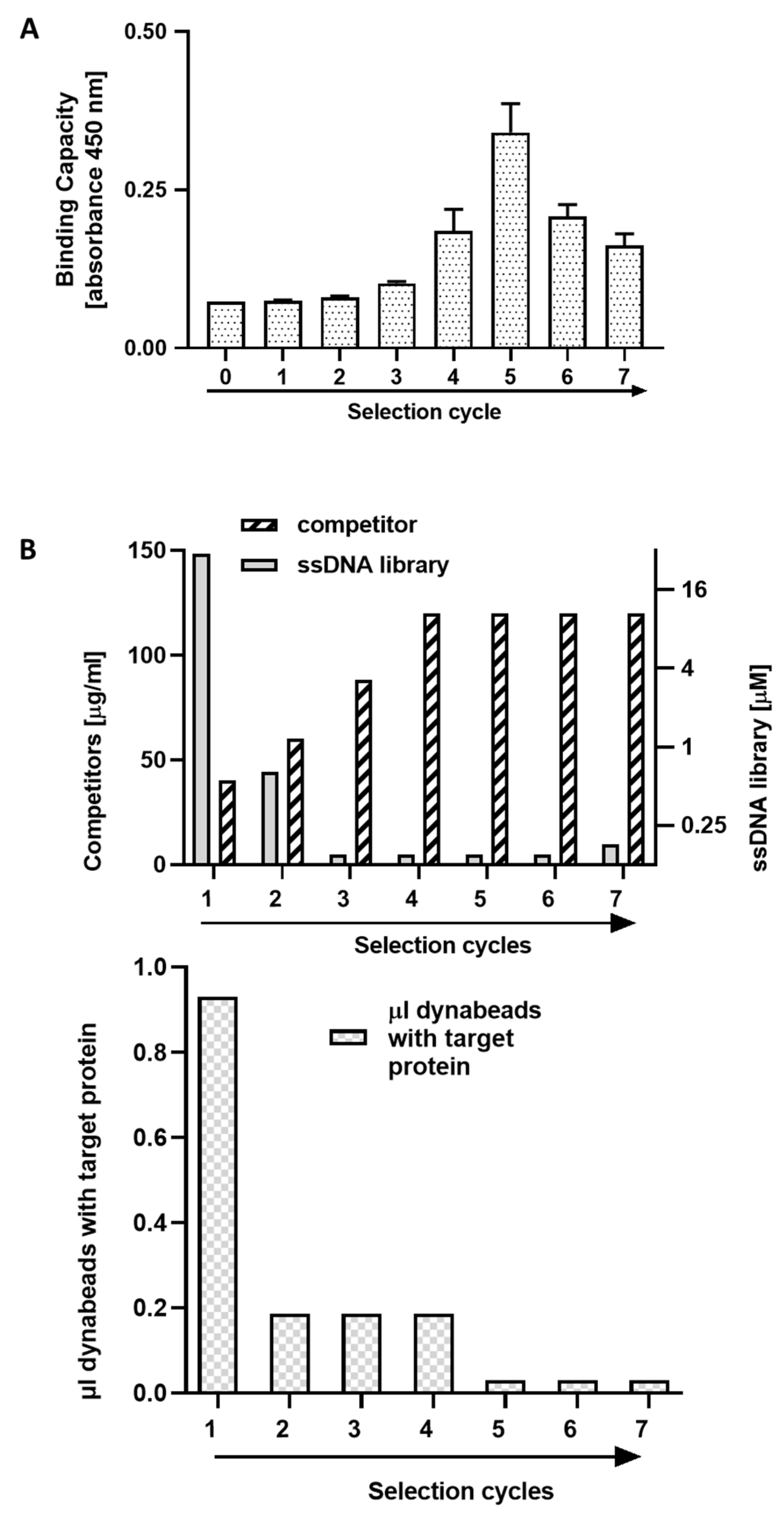
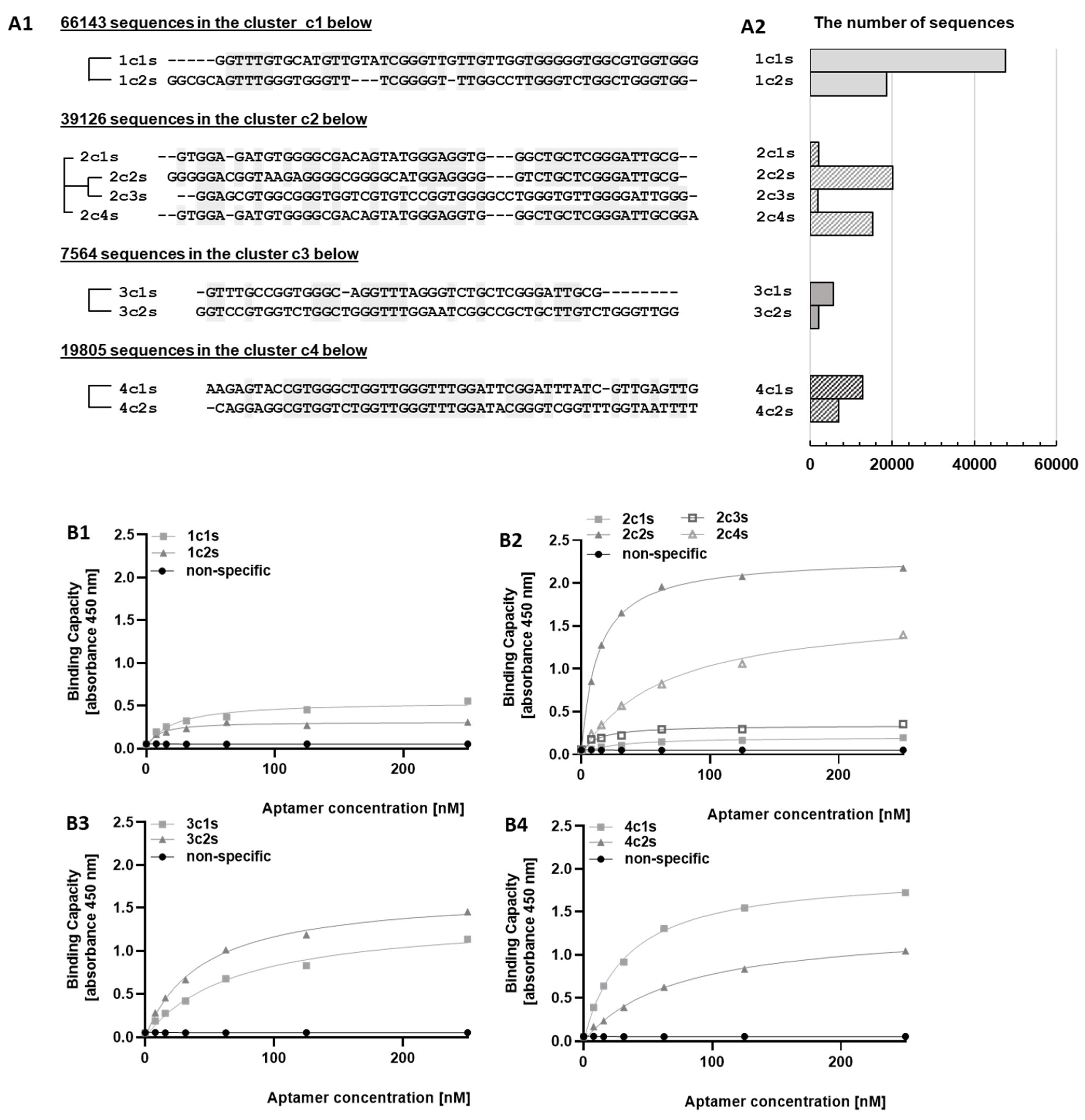
In this work, using the SELEX procedure we identified DNA aptamers specifically recognizing the human extracellular domain of PD-L1. During selection, we observed an unexpected profile of affinity maturation. The first five selection steps gradually increased the affinity of the DNA pool, but further steps resulted in decreased affinity. Deep sequencing of aptamer pools at consecutive selection rounds allowed to rationalize the observation. Starting from cycle 6, sequences characterized by weak binding to the target protein prevail. We believe that preferential amplification of aptamers characterized by less stable secondary structures may provide a likely explanation; however, this was not tested systematically. Regardless of the mechanistic explanation, the results allow us to conclude that, paradoxically, limiting the number of selection cycles may be beneficial in selecting high affinity binders. Therefore, fast convergence of selection was sought by manipulating the selection pressure to avoid progressing into cycles favoring highly amplifiable low-affinity binders. The selection of binders from early selection cycles was further facilitated through deep sequencing and sequence cluster analysis.
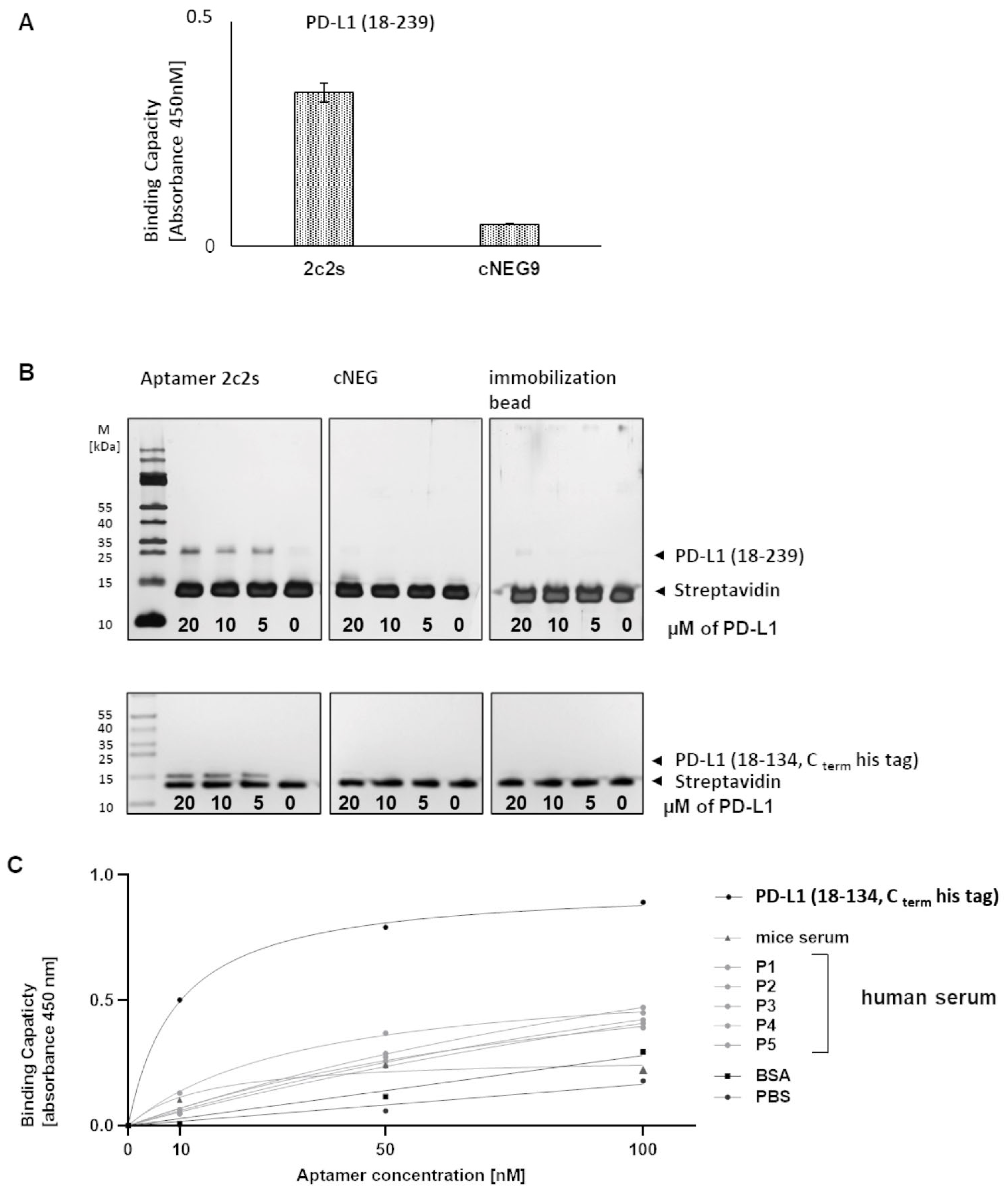
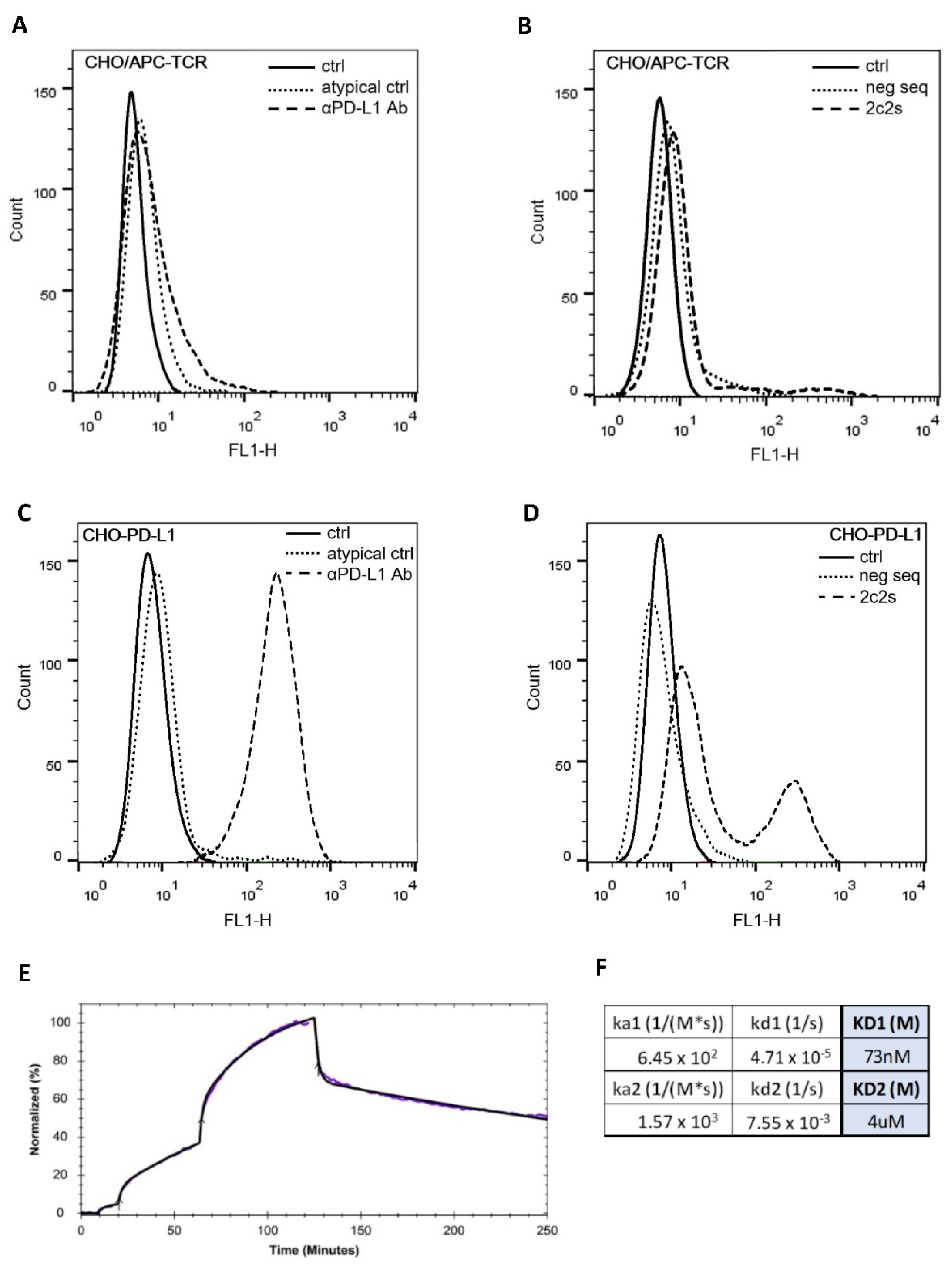
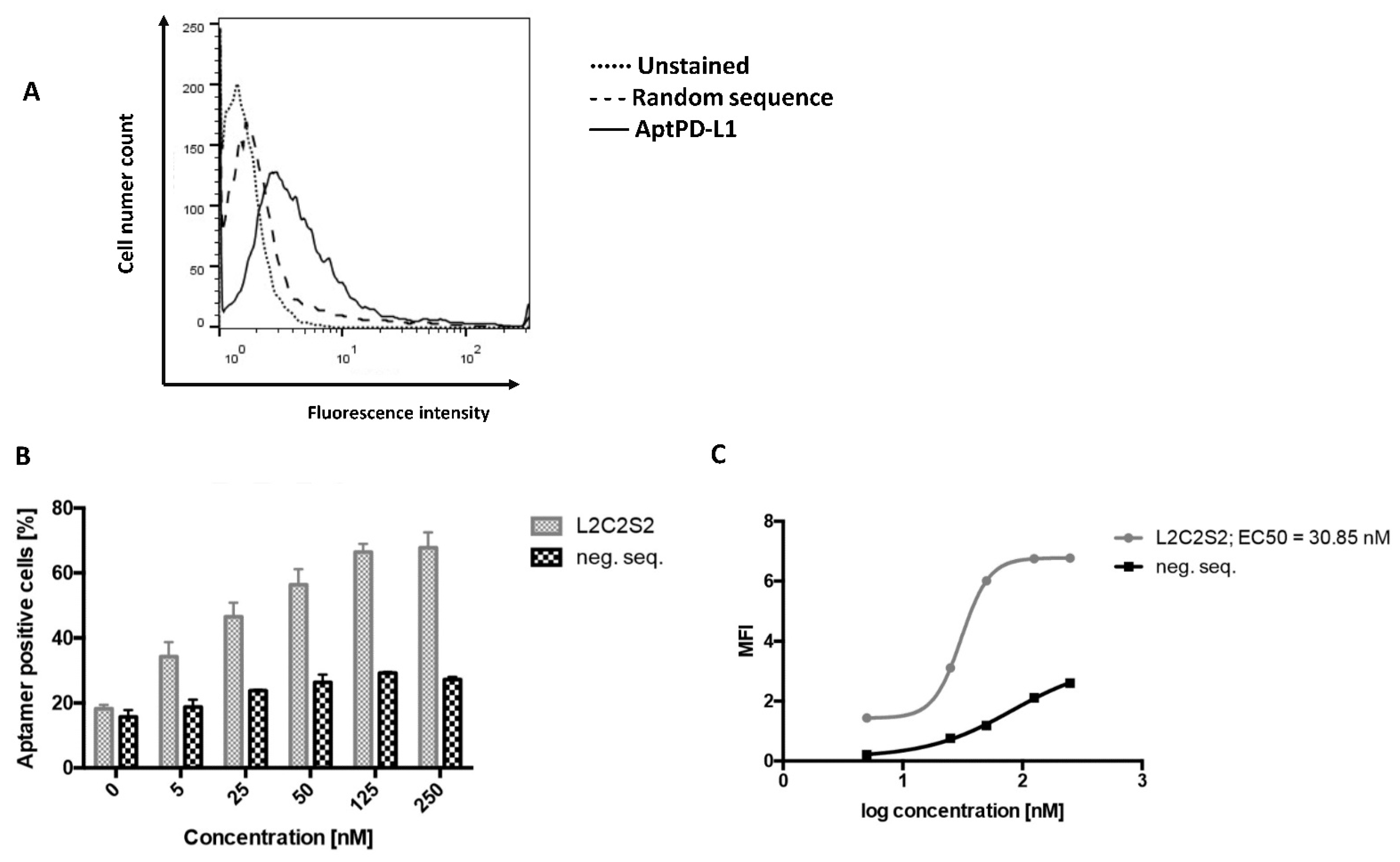
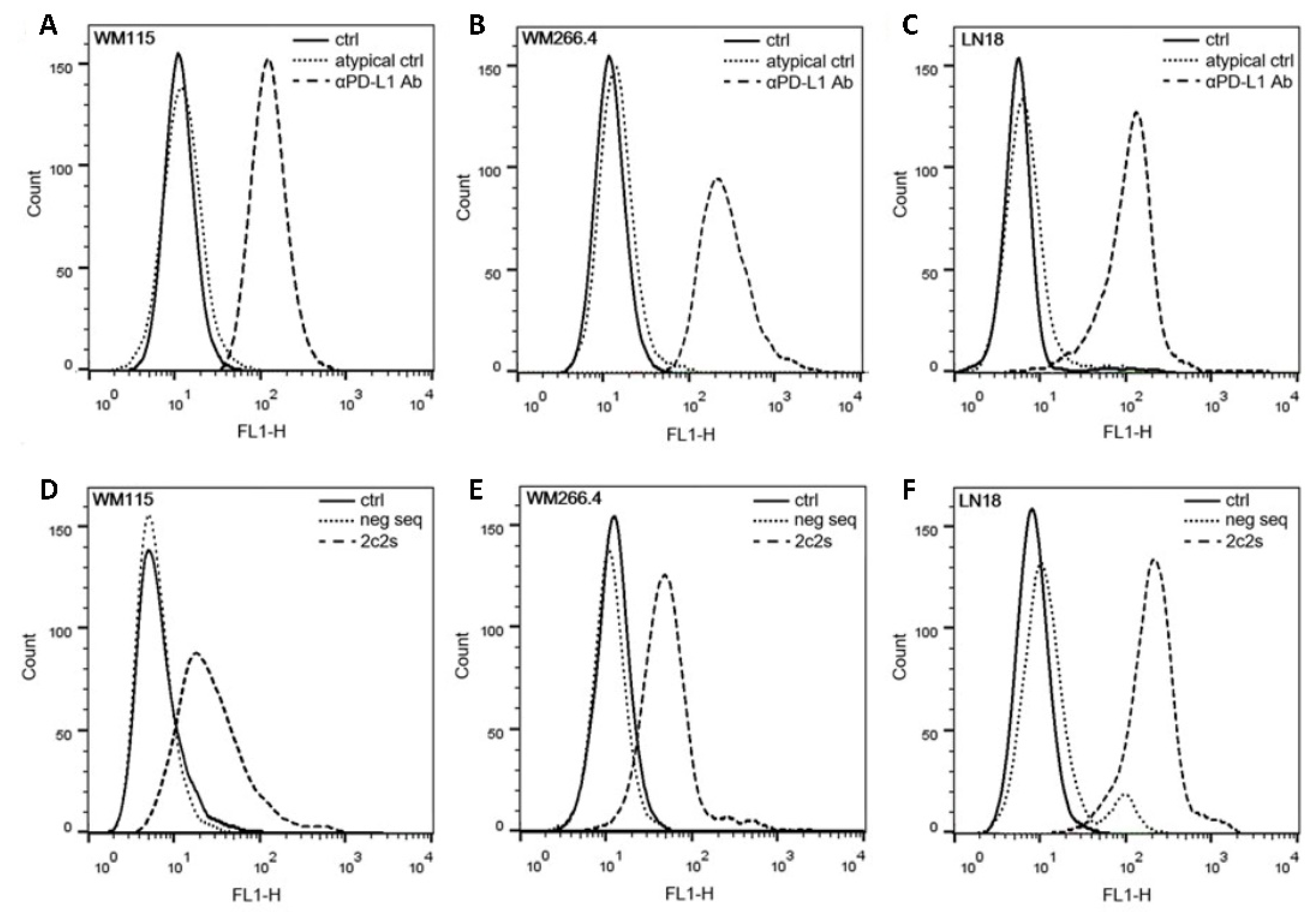
The selected aptamer, 2c2s, demonstrated an affinity for both the short (single domain) construct used in selection, but also for a two-domain construct of PD-L1 which it did not encounter during selection signifying that we selected a universal PD-L1 detecting binder, rather than an assay-specific molecule. Switching to the cellular environment, the simple system where hPD-L1 is expressed at the surface of CHO cells demonstrated partial masking of the antigen recognized by 2c2s. This could significantly limit the utility of the 2c2s molecule in tumor labeling, was it not for the fact that masking was only characteristic for that particular cell line and possibly related to its hamster origin. All human cancer cell lines tested were characterized by uniform labeling and demonstrated the correlation between α-PD-L1 mAb and 2c2s labeling, indicating that 2c2s is suitable for detection of PD-L1 at the surface of cancer cells.
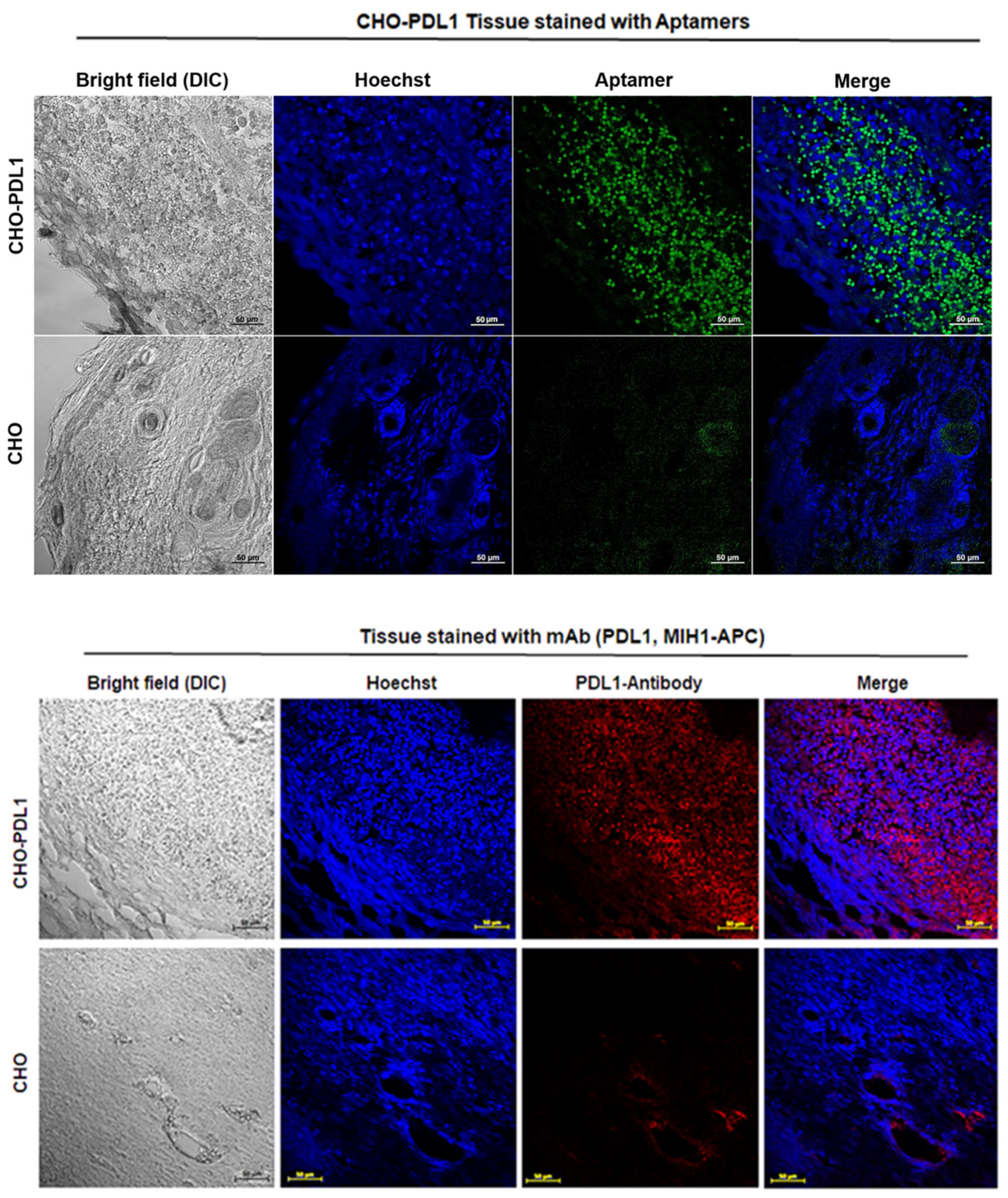
Given the limited accessibility of the 2c2s epitope on PD-L1 aAPC/CHO-K1 cells, the aptamer was surprisingly efficient in staining the PD-L1 aAPC/CHO-K1 tumor areas in biopsy samples. One could easily distinguish the tumor from the surrounding tissue demonstrating the utility of 2c2s in ex vivo sample analysis.
Earlier reports have already provided aptamers targeting the PD-1/PD-L1 immune checkpoint. Tian Gao et al. [
17] obtained an aptamer binding to the PD-1 and antagonizing its interaction with PD-L1. Wei-Yun Lai et al. [
18] developed anti-PD-L1 molecules antagonizing the interaction between human PD-L1 and PD-1. In contrast to the prior developments, aptamers delivered in this study do not antagonize the PD-1/PD-L1 interaction. In vitro, anti-PD-L1 aptamers were earlier shown to effectively recognize PD-L1 at the surface of the MDA-MB-231 cell line [
19]. Moreover, Jiyuan Li et al. [
20] reported an anti-PD-L1 aptamer able to detect the target protein ex vivo in normal human tonsils and non-small cell lung cancer tissue. However, to our knowledge, no earlier study reported in vivo effects.
The success of our ex vivo imaging prompted us to evaluate 2c2s in vivo. PD-L1 overexpression has been documented in both primary [
21,
22] and metastatic tumors [
22,
23,
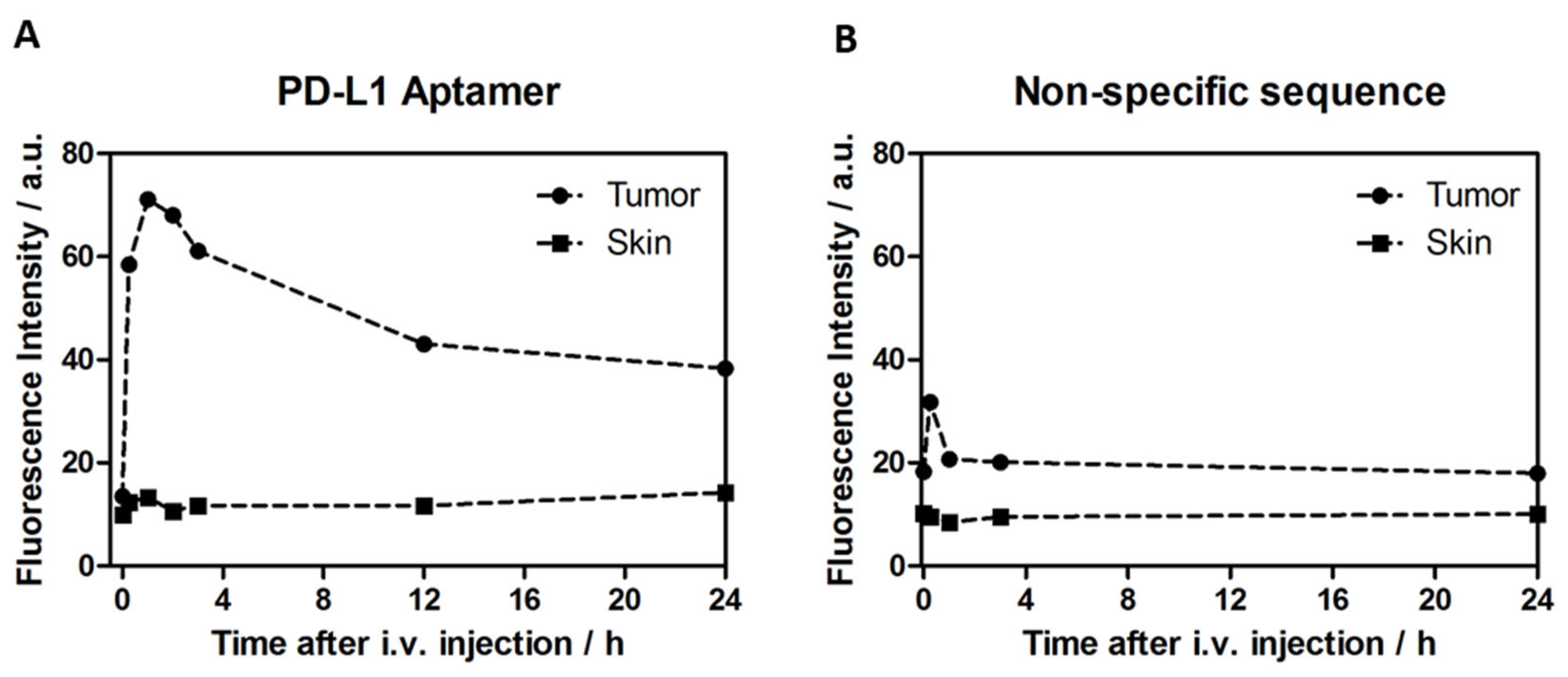
24]. In vitro detection is well performed with antibodies, and aptamers provide little advantage. In vivo, however, the small size of aptamers compared to antibodies and versatile labeling possibilities promise wider opportunity for tailoring the response. Upon systemic application, 2c2s aptamer labeled with visible light sensitive fluorescent probe allowed for selective detection of the subcutaneous tumor with very simple instrumentation (optic fiber spectrophotometer). The specificity window of 3-times the signal from tumor adjacent skin, obtained without significant optimization of the probe label or dose, demonstrates the significant potential of our approach in diagnostic imaging.
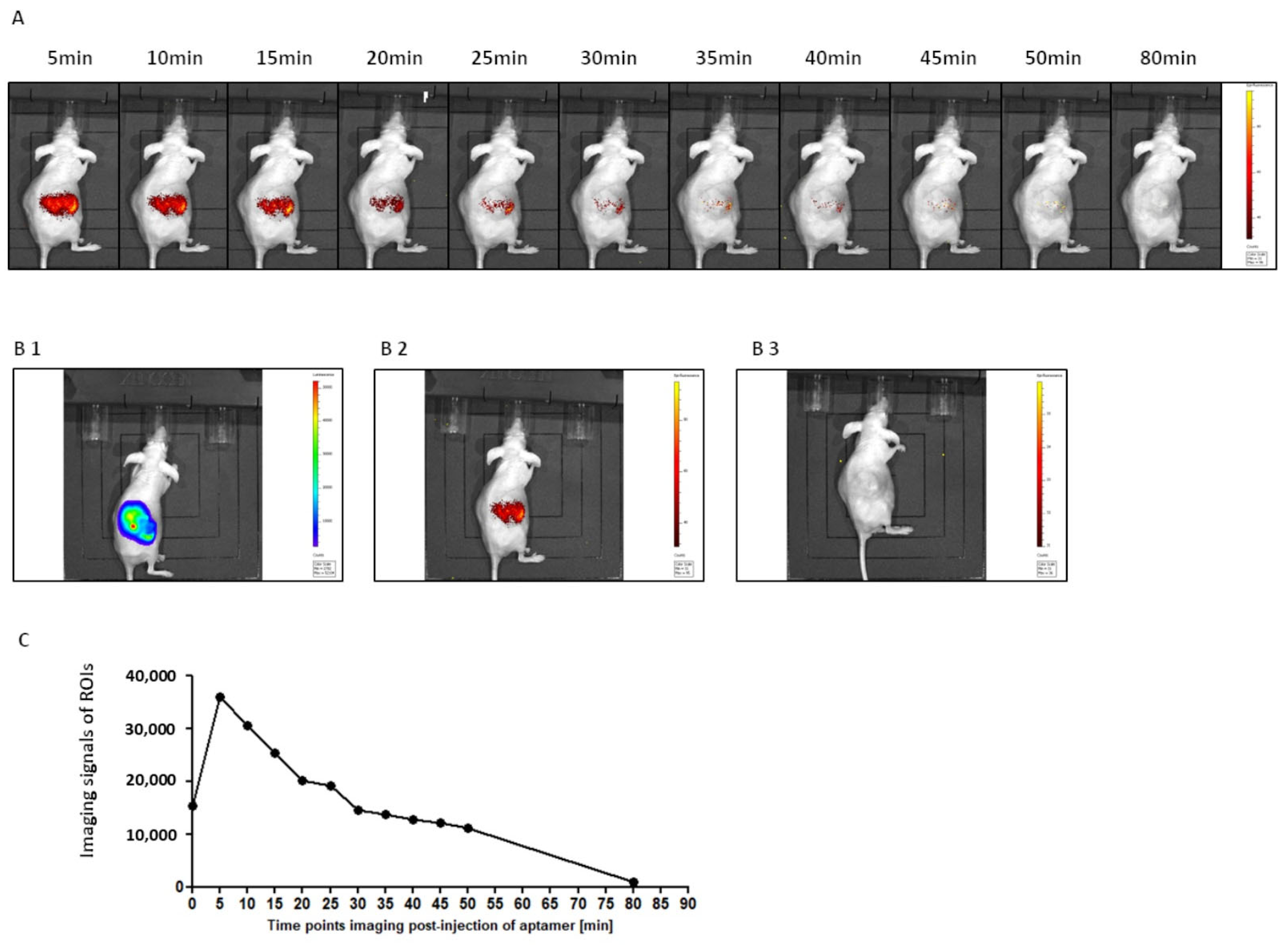
Detection of malignancy located in an internal organ required adjustment of the detection probe for a red-shifted fluorescence. The label adjustment procedure was successful within a week, demonstrating the flexibility of aptamer design for tailoring the labeling applications. Upon systemic application, a tumor-restricted imaging signal was obtained already at a few minutes after administration and lasted up to 20 min, providing enough time for accurate imaging. The result demonstrated the potential of the approach even in the deeper buried tissue.
Neither intravenous nor intraperitoneal probe application resulted in detectable acute toxicity. Aptamers are generally regarded as safe given their chemical nature identical to the body physiological components. Multiple metabolically neutral labels are available, and it is expected that safe aptamer/label combinations may be achieved via optimization. Further testing is necessary to select the best labeling techniques, assess the long-term effects, evaluate metabolism and excretion, and test the utility in clinical settings, but our study has paved this exciting road by providing a universal PD-L1 probe characterized by low molecular weight compared to previously available antibody probes.
4. Materials and Methods
4.1. Protein Purification
The IgV like domain of human PD-L1 (residues 18–134, C-terminal His-tag) and full extracellular domain of human PD-L1 (residues 18–239) were expressed and purified using previously described protocols [
25]. In short,
E.coli BL21 (DE3) were cultured at 37 °C in LB medium until OD
600 1.0, induced with 1 mM IPTG, and further incubated at 37 °C. Bacteria were collected by centrifugation, resuspended in PBS and sonicated. Inclusion bodies were collected by centrifugation, washed twice with 50 mM Tris-HCl pH 8.0, 200 mM NaCl, 0.5% Triton X-100, 10 mM EDTA, 10 mM 2-mercaptoethanol, and once with the same buffer without Triton X-100. Purified inclusion bodies were then solubilized in 50 mM Tris pH 8.0, 6M GuHCl, 200 mM NaCl and 10 mM 2-mercaptoethanol and refolded at 4 °C by dilution into 0.1 M Tris pH 8.0, 1 M L-Arg hydrochloride, 2 mM EDTA, 0.25 mM oxidized glutathione and 0.25 mM reduced glutathione. After refolding, the protein was dialyzed 3 times against 10 mM Tris pH 8.0, 20 mM NaCl, concentrated, and purified by gel filtration on Superdex 75 in 25 mM Na
2HPO
4, 25 mM NaH
2PO
4 pH 6.4, 100 mM NaCl, 5 mM MgCl
2 and 10 mM KCl or alternatively in 10 mM Tris pH 8.0 containing 20 mM NaCl. Purified protein was flash frozen in liquid nitrogen and stored for further experiments as 0.2 mg/mL stocks at −80 °C.
4.2. In Vitro Selection of Aptamers
Selection of aptamers was performed using a single-stranded DNA library (5’-CATGCTTCCCCAGGGAGATG-N50-GAGGAACATGCGTCGCAAAC-3′; 50-nucleotide random sequence), synthesized at 0.2 μM scale and purified by HPLC (IBA, Göttingen, Germany). Aptamers were selected for their specific binding to PD-L1 extracellular domain (residues 18–134, C-terminal His-tag). PD-L1 was immobilized at Dynabeads ™ (Thermo Fisher Scientific, Waltham, MA, USA) in a binding buffer (100 mM Sodium Phosphate, pH 8.0, 600 mM NaCl, 0.02% Tween™-20) and washed with the selection buffer (PBS containing 5 mM MgCl2, 10 mM KCl and 0.01% Tween 20, pH 7.4). The ssDNA library or ssDNA pool before each subsequent selection cycle were renatured (5 min at 92 °C, 10 min 4 °C, 15 min RT) and suspended in binding buffer (selection buffer supplemented with 40–120 µg/mL yeast tRNA (Invitrogen, Waltham, MA, USA) and 125 μg/ml BSA (BioShop Canada Inc., Burlington, VT, Canada)). Beads with immobilized target protein were added and incubated for 20 min with shaking at 24 °C. To increase the selection pressure during the selection process, the ssDNA concentration (30 to 0.18 µM) and the immobilized protein bead amount (3 to 0.3 ul) were gradually reduced while increasing the competitor concentration (40 to 120 μg/mL yeast tRNA). After incubation, unbound aptamers were removed by washing with a selection buffer using a magnetic concentrator (Invitrogen, Waltham, MA, USA). Subsequently, the beads with the immobilized protein and associated DNA were suspended in 400 μL of PCR mix containing: 1 μM primers (unmodified ss50_For: 5’-CATGCTTCCCCAGGGAGATG-3′ and 5’-phosphorylated ss50_Rev: 5’-GTTTGCGACGCATGTTCCTC-3’), 5 mM dNTP, 2.5 mM MgCl2 and 1.25 U/100 uL polymerase Taq (Thermo Fisher Scientific, Waltham, MA, USA). PCR was carried out for 35 cycles consisting of the following steps: 30 s in 95 °C, 30 s in 53 °C and 30 s in 72 °C. The final elongation was carried out at 72 °C for 5 min. The PCR products were then extracted with a phenol-chloroform-isoamyl alcohol (Sigma-Aldrich, St. Louis, MO, USA) and precipitated overnight with ethanol at −20 °C. The DNA pellet was washed twice with 70% ethanol, dried and dissolved in dH2O. The obtained dsDNA was digested with 100U of λ exonuclease (Thermo Fisher Scientific, Waltham, MA, USA) to recover the relevant single strand. Digestion was carried out for 1 h at 37 °C with gentle shaking. Digested products (ssDNA) were again extracted with a phenol-chloroform-isoamyl alcohol mixture, precipitated, and dissolved in 100 μL of dH2O.
To eliminate the nonspecific binding, the negative selection was performed by incubating ssDNA pool/library with free Dynabeads prior the aptamer selection cycles. During the selection, an enrichment of the aptamer pool with PD-L1 binding sequences was monitored by ELISA (see below). The aptamer pools after the 5th, 6th and 7th selection cycle were analyzed by Next-Generation Sequencing (NGS; Genomed S. A., Warszawa, Poland).
4.3. Sequence Analysis
FASTQ files containing demultiplexed sequences after barcode trimming were further processed. Adapters (flanking sequences) were excluded at both ends and, if needed, sequences were reversed. The obtained aptamer sequences were clustered in groups of identical sequences while only considering sequences at least 10 nucleotides long. The groups were compressed to common sequences (later referred to as compressed_seq) and identified by their cardinality. Compressed_seqs were represented as their nucleotide sequences and 10 most frequent compressed_seqs were clustered with the use of k-means algorithm. The optimal number of clusters was determined employing the silhouette method, and silhouette score was computed for 2 ≤ k ≤ 4. Compressed_seqs in each of the obtained clusters were aligned with ClustalW. The alignments were subsequently processed with dnadist and neighbour programs from PHYLIP package to generate (pseudo-) phylogenetic trees representing the relationships between compressed_seqs (process illustrated in
Supplementary Figure S1).
4.4. ELISA
Affinity and binding specificity to PD-L1 was assessed by ELISA. A 96-well microtiter plate (Nunc, Rochester, NY, USA) was coated with 100 μL of human PD-L1, BSA, or mouse or human serum and incubated overnight, at 4 °C. Unbound protein was removed by washing with selection buffer. Biotinylated aptamers and nonspecific sequences (in the concentration range of 0–2.5 µM) were added to the wells and incubated for 30 min. Unbound aptamers were removed by extensive washing with selection buffer. Then, 100 μL HRP conjugated streptavidin was added to the wells in 1:200 dilution (R&D Systems, Inc., Minneapolis, MN, USA) in selection buffer. After 20 min of incubation, unbound streptavidin was removed by washing and 100 μL of HRP reagent substrate (R&D Systems, Inc., Minneapolis, MN, USA) was added. The reaction was stopped by the addition of 50 μL of 2N H2SO4. Absorbance at 450 and 570 nm (correction of optical plate imperfections) was determined using an Infinite 200 PRO multimode reader (Tecan Group Ltd., Männedorf, Switzerland).
4.5. Immobilization Test
Biotinylated aptamers (4 μM; 2c2s or negative control) were immobilized on Streptavidin Mag Sepharose (GE Healthcare, Chicago, IL, USA) by incubation in PBS for 20 min in RT. After washing with PBS, the beads were incubated with 0,2% BSA (BioShop Canada Inc., Burlington, VT, Canada) in the SELEX buffer for 30 min at RT to block unspecific sites and washed with SELEX buffer. Next, the beads were suspended in SELEX buffer containing 40 μg/mL tRNA and incubated with PD-L1 (18–239 or 18–134 Cterm Histag; final concentrations: 90, 45, and 22,2 μg/mL) for 20 min at RT while mixing continuously. After incubation, the beads were washed with SELEX buffer and the bound protein was eluted by boiling briefly in the loading buffer (3% SDS, 10% glycerol, 12,5 mM Tris-HCl, 100 mM DTT, 0,05% bromophenol blue, pH 6.8). The recovered proteins were analyzed by SDS/PAGE. Coomassie blue-stained gels were imaged using ChemiDoc (Bio-Rad Laboratories, Inc. Hercules, CA, USA).
4.6. Cell Culture and Binding Assay
To validate the binding of the aptamers to PD-L1 expressed at the cells surface, aAPC/CHO-K1 cells and PD-L1 aAPC/CHO-K1CHO cells stably expressing hPD-L1 protein (Promega), human melanoma cells WM115 and WM266.4 (ATCC CRL-1675 and ATCC CRL-1675), human glioblastoma LN18 (ATCC CRL-2610) and 786-O renal adenocarcinoma (JCRB Cell Bank, JCRB1397) cells were used. Cells were cultured at 37 °C with 5% CO2 in an appropriate culture medium supplemented with 10% FBS (InvivoGen) and 1% penicillin/streptomycin. aAPC/CHO-K1 and PD-L1 aAPC/CHO-K1 cells were grown in DMEM-F12, melanoma cells were cultured in RPMI-1640, LN18 in DMEM (4.5 g/L glucose) and 786-O in RPMI-1640. PD-L1 aAPC/CHO-K1 cells medium was additionally supplemented with 50 µg/ml hygromycin B (Sigma-Aldrich, St. Louis, MO, USA) and 250 µg/ml G 418 disulfate salt (InvivoGen, San Diego, CA, USA). Non-transfected CHO cells were used as a negative control.
4.7. Flow Cytometry Analysis
To assess aptamer binding to PD-L1 at the cell surface flow cytometry was used. The cells (aAPC/CHO-K1, PD-L1 aAPC/CHO-K1, WM115, WM266.4, LN18 and 786-O) were blocked with HBSS/Dextran solution (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% FBS for 30 min at 37 °C and the FITC-conjugated 2c2s aptamer was incubated with 1 × 105 cells for 30 min in the dark with gentle shaking (180 rpm). Cells were then washed three times with HBSS and harvested with accutase (Thermo Fisher Scientific, Waltham, MA, USA). To confirm the surface expression of PD-L1, cells were incubated with an anti-human PD-L1 antibody (MIH1 clone, Thermo Fisher Scientific, Waltham, MA, USA). Samples were analyzed on a FACSCalibur instrument (Becton Dickinson, Franklin Lakes, NJ, USA; adenocarcinoma cells) using CellQuest software or Guava cytometer (Merck Millipore, Darmstadt, Germany; all other analyses in this study). Data were analyzed in FlowJo v10.8.0.
4.8. In Vitro Kinetic Binding Assay
Real-time cell binding assay (RT-CBA) using LigandTracer® Green (Ridgeview Instruments AB, Vänge, Sweden), and a blue (488 nm)-green (535 nm) detector was performed to evaluate the interactions of FAM-labeled 2c2s with PD-L1 protein expressed at the surface of aAPC/CHO-K1 cells. Cells were seeded in each target compartment of MultiDish 2 × 2 (1 × 106 cells). Uncoated free areas of the Petri dish were measured to enable the subtraction of the background signal. The baseline measurement of cells in the absence of labeled aptamer was also performed. Kinetics of binding was analyzed with increasing concentrations (50, 200 or 650 nM) of the labeled aptamer. To evaluate the dissociation process, a fresh cell medium was provided. Affinity calculations (based on the association and dissociation rates) were performed using the TraceDrawer Software (Ridgeview Instruments AB, Uppsala, Sweden).
4.9. Fluorescence Imaging In Vivo
The 12-week-old male BALB/c nude mice (mean weight, 16 ± 5 g) were purchased from Anima Lab. Mice were maintained in a pathogen-free environment, on 12/12 night/day cycle, with food and water supplied ad libitum throughout the experiments. Weight loss >25% or tumor volume >900 mm3 were identified as endpoints for euthanasia. All animal experiments were performed in accordance with institutional guidelines. All experiments were carried out with approval no. 190/2018 of the 2nd Local Institutional Animal Care and Use Committee, Krakow, Poland.
aAPC/CHO-K1 or PD-L1 aAPC/CHO-K1 cells (Promega) suspension in PBS/BD Matrigel Matrix Growth Factor Reduced (1 × 107 cells) was injected subcutaneously into the left flank of each animal. The diagnostics procedures were started when tumors reached more than 0.5 cm in each diameter (which corresponds to tumor volume of about 80–100 mm3; up to 21 days after tumor inoculation). For the fluorescence imaging experiment, 2 mg/kg BW 2c2s-Cy5.5 aptamer was administrated intravenously via tail vein injection and then tumors were imaged via fiber optics, and fluorescence spectra from tumor or skin were registered. The measurements were performed up to 96 h post-injection. The fluorescence spectra were collected using a Perkin Elmer LS spectrofluorometer equipped with tumor-imaging-dedicated fiber optics.
4.10. Immunohistochemistry and Confocal Imaging
The efficacy of tumor detection with the 2c2s-FAM probe was evaluated ex vivo by immunostaining of tumors obtained from BALB/c nude mice in a prior in vivo imaging experiment. aPD-L1 monoclonal antibody (MIH1 clone, APC conjugated, ThermoFisher Scientific, Waltham, MA, USA) was used as a positive control. aAPC/CHO-K1 and PD-L1 aAPC/CHO-K1 tumors were weighed, snap-frozen and stored at −80 °C. Harvested tumors were cut into 5-μm-thick sections using a cryostat (Leica). Sections were placed on microscope slides for staining. Frozen slides were air dried (30 min) and fixed with ice-cold acetone for 10 min. After acetone evaporation (30 min) slides were washed two times with PBS and then incubated for 10 min in 0.3% H2O2 in MeOH/PBS. The slides were washed two times (5 min) with PBS and blocked for 1 h in blocking buffer (1× TBS, 0.3% Triton X-100, 5% FBS). The blocking solution was removed and slides were incubated with 2c2s-FAM aptamer or aPD-L1 primary antibody overnight at 4 °C. After incubation, slides were washed three times (5 min) with TBS-0.01% Tween 20, stained with Hoechst33342 for 10 min, rinsed with PBS and mounted with prolong gold mounting medium. Tissues were imaged with fluorescence confocal microscope LSM880 (Carl Zeiss) and the images were analyzed using ZEN software (Carl Zeiss). Five complete and non-overlapping regions of interest (ROI) were randomly selected from each prepared slide. All microscopic images were adjusted with the same parameters.
4.11. Orthotopic Kidney Cancer Imaging
Female, 8-week-old Athymic Nude mice were purchased from Janvier Labs, France. The animals were housed and maintained in individually ventilated cages under a 50–60% humidity, 12/12 h light/dark cycle and at 22 ± 2 °C, in SPF conditions at the Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University in Krakow, Poland. All animal experiments were reviewed and approved by the 1st Regional Ethics Committee on Animal Experimentation, Krakow, Poland (approval no: 264/2020).
JCRB1397-786-Luc tumor cells (1 × 105/50µL/kidney) were implanted under the capsule of the kidney using a 1 mL syringe with a 30-gauge needle under anesthesia. Control mice were administrated with PBS into the kidney. Tumor growth monitoring began 4 weeks after cell implantation and continued every 10 days by bioluminescence using an In Vivo Imaging System (IVIS) Lumina (PerkinElmer, Waltham, MA, USA). For this purpose, 0.2 mL (15 mg/ml) D-luciferin (XenoLight D-luciferin potassium salt, PerkinElmer, USA) was injected into the peritoneum of tested mice and visualization was performed after 10 min. The mice were under inhalation anesthesia with 3–4% Isoflurane (Aerrane, Baxter, Poland) throughout the experiment. The 2 mg/kg body weight Cy5.5-2c2s or unspecific sequence (control) labeled with Cyanine 5.5 (Sigma Aldrich, Darmstadt, Germany) was administered systemically by intraperitoneal injection and whole-body imaging using the IVIS 200 Imaging System was performed immediately following and up to 90 min after the injection.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








