
Design and Synthesis of Hepatitis B Virus (HBV) Capsid Assembly Modulators and Evaluation of Their Activity in Mammalian Cell Model


 ,
,  , , , ,
, , , ,  , and
, and
Abstract
:
1. Introduction
2. Results
2.1. Chemistry
- (1)
- Changes at position 5 of the dihydropyrimidine core led to compounds containing carboxylic acid moiety (comp. 1b), ester moiety (comp. 1c-1g and 1j-1k, including alkoxyalkyl esters 1d-1g), and amide moiety (including anilide 1h and pyridylamide 1i).
- (2)
- Variations at position 4 of the dihydropyrimidine core led to the formation of compounds with phenyl and substituted phenyl (including 4-fluoro, 2-difluoromethoxy, and 2-chloro-4-fluoro substituents) groups.
- (3)
- Derivatization at position 2 of the dihydropyrimidine core led to 3,5-difluoropyridyl-2 (comp. 1a-1i) or thiazolyl-2 (comp. 1j-1k) group containing analogues.
- (4)
- Insertion of bromine atoms or cation-substituted ammonium groups at position 6 of the dihydropyrimidine core led to compounds 3a-3d.
- (5)
- Dihydropyridine 6 (or deaza-HAP) was synthesized via the cyclocondensation reaction of the aminovinylcarbonyl and arylidene carbonyl compounds.
- (6)
- Heteroarylpyrimidines 4a and 4b—pyrimidine derivatives as oxidized HAP representatives were obtained by aromatization of comp. 1j and 1k.
- (7)
- Additionally, derivative at positions 5 + 6 (condensed furanone cyclic comp. 5) was obtained by heating of 2-bromomethyl-HAPs compound.
2.2. Evaluation of the Compound Effects on HBV Capsid Assembly in HBc-Producing BHK-21 Cells
- (i)
- By prolongation of the methyl ester group at position 5 of the Bay 41-4109: 5-ethoxyethoxycarbonyl 1d and 5-propoxyethoxycarbonyl 1f, that is, in the case of n-alkoxyalkylcarbonyl substituents. In the case of small deviations in the structures of comp. 1d and 1f, comp. 1e, containing an i-propoxyethoxycarbonyl moiety at position 5, did not lead to HBc accumulation; in contrast, a dose-dependent capsid signal decrease was observed. Comp. 1g, a close analogue of comp. 1f, comprising an o-difluoromethoxyphenyl group in position 4 of the dihydropyrimidine molecule, showed some increase in capsid band signal (Figure 5);
- (ii)
- In the case of lactone derivative 5 of the acid 1b;
- (iii)
- In the case of 2-(thiazolyl-2)-5-ethoxycarbonyl-6-methyl-1,4-dihydropyrimidones containing 4-phenyl and 4-(4-fluoro)-phenyl substituents (comp. 1j and 1k).
2.3. Cytotoxicity Estimation of Selected Compounds in BHK-21 Cells with the MTT Viability Assay
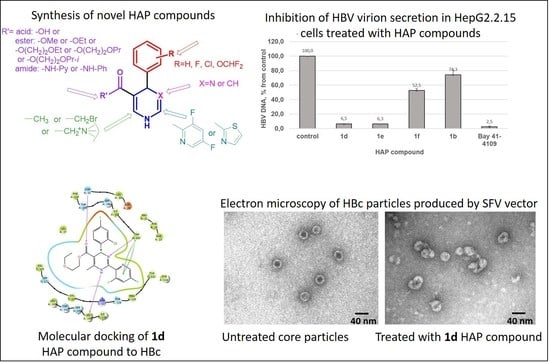
2.4. Antiviral Effects Study in HBV-Producing HepG2.2.15 Cells
2.5. Evaluation of the HBc Polymer Structures Induced by Compound 1d in BHK-21 Cells
2.6. Molecular Modeling of HAP Compound Interaction with HBc Protein
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Molecular Modeling
4.3. Cell Cultures
4.4. HBc Protein Production in BHK-21 Cells
4.5. Evaluation of Compound Toxicity
4.6. Evaluation of Compound Effects on HBV Capsid Assembly in BHK-21 Cells
4.7. HBV Virion DNA Isolation from the HepG2.2.15 Cell Culture Supernatant
4.8. HBV Virion DNA Quantification by Real Time PCR
4.9. HBc Protein Detection in BHK-21 Cells by Immunostaining
4.10. HBV Capsid Isolation from BHK-21 Cells and Evaluation by Electron Microscopy
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akbar, S.M.F.; Al-Mahtab, M.; Khan, S.I. Nature of Host Immunity during Hepatitis B Virus Infection and designing Immune Therapy. Euroasian J. Hepato-Gastroenterol. 2018, 8, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Revill, P.A.; Chisari, F.V.; Block, J.M.; Dandri, M.; Gehring, A.J.; Guo, H.; Hu, J.; Kramvis, A.; Lampertico, P.; Janssen, H.L.A.; et al. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol. Hepatol. 2019, 4, 545–558. [Google Scholar] [CrossRef]
- Gish, R.; Given, B.; Lai, C.; Locarnini, S.; Lewis, D.; Schleup, T. Chronic hepatitis B: Virology, natural history, current management and glimpse at future opportunities. Antivir. Res. 2015, 121, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Glebe, D.; Goldmann, N.; Lauber, C.; Seitz, S. HBV evolution and genetic variability: Impact on prevention, treatment and development of antivirals. Antivir. Res. 2021, 186, 104973. [Google Scholar] [CrossRef] [PubMed]
- Phyo, W.; Soh, A.; Lim, S.; Lee, G. Search for a cure for chronic hepatitis B infection: How close are we? World J. Hepatol. 2015, 7, 1272–1281. [Google Scholar] [CrossRef]
- Mueller, H.; Wildum, S.; Luangsay, S.; Walther, J.; Lopez, A.; Tropberger, P.; Ottaviani, G.; Lu, W.; Parrott, N.J.; Zhang, J.D.; et al. A novel orally available small molecule that inhibits hepatitis B virus expression. J. Hepatol. 2018, 68, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, J.; Gelson, W.; Rushbrook, M.S. Therapeutic advances in the management of chronic hepatitis B infection. Ther. Adv. Chronic Dis. 2013, 4, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.P.; Gutfreund, K.S.; Tyrrell, D.L. Lamivudine resistance in hepatitis B: Mechanisms and clinical implications. Drug Resist. Update 2004, 4, 118–128. [Google Scholar] [CrossRef]
- Liaw, Y.F.; Chu, C.M. Hepatitis B virus infection. Lancet 2009, 373, 582–592. [Google Scholar] [CrossRef]
- Liu, C.; Fan, G.; Wang, Z.; Chen, H.S.; Yin, C.C. Allosteric conformational changes of human HBV core protein transform its assembly. Sci. Rep. 2017, 7, 1404. [Google Scholar] [CrossRef]
- Deres, K.; Schroder, C.H.; Paessens, A.; Goldmann, S.; Hacker, H.J.; Weber, O.; Kramer, T.; Niewohner, U.; Pleiss, U.; Stoltefuss, J.; et al. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 2003, 299, 893–896. [Google Scholar] [CrossRef] [PubMed]
- Bakail, M.; Ochsenbein, F. Targeting protein—Protein interactions, a wide open field for drug design. Comptes Rendus Chim. 2016, 19, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Voter, A.F.; Keck, J.L. Development of Protein-Protein Interaction Inhibitors for the Treatment of Infectious Diseases. Adv. Protein Chem. Struct. Biol. 2018, 111, 197–222. [Google Scholar] [CrossRef] [PubMed]
- Nijampatnam, B.; Liotta, D.C. Recent advances in the development of HBV capsid assembly modulators. Curr. Opin. Chem. Biol. 2019, 50, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Schlicksup, C.J.; Zlotnick, A. Viral structural proteins as targets for antivirals. Curr. Opin. Virol. 2020, 45, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Schinazi, R.F.; Ehteshami, M.; Bassit, L.; Asselah, T. Towards HBV Curative Therapies. Liver Int. 2018, 38, 102–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stray, S.; Bourne, R.C.; Punna, S.; Lewis, G.W.; Finn, M.G.; Zlotnick, A. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc. Natl. Acad. Sci. USA 2005, 102, 8138–8143. [Google Scholar] [CrossRef] [Green Version]
- Prifti, G.-M.; Moianos, D.; Giannokopoulou, E.; Pardali, V.; Tavis, J.E.; Zoidis, G. Recent Advances in Hepatitis B Treatment. Pharmaceuticals 2021, 14, 417. [Google Scholar] [CrossRef]
- Wu, G.; Liu, B.; Zhang, Y.; Li, J.; Arzumanyan, A.; Clayton, M.M.; Schinazi, R.F.; Wang, Z.; Goldmann, S.; Ren, Q.; et al. Preclinical characterization of GLS4, an inhibitor of Hepatitis B virus core particle assembly. Antimicrob. Agents Chemother. 2013, 57, 5344–5354. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, F.; Zhu, X.; Chen, Y.; Chen, H.; Li, X.; Wu, M.; Li, C.; Liu, J.; Zhang, Y.; et al. Antiviral activity and pharmacokinetics of the hepatitis B virus (HBV) capsid assembly modulator GLS4 in patients with chronic HBV infection. Clin. Infect. Dis. 2021, 73, 175–182. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, H.; Zhang, Y.; Jiang, D.; Li, J.; Goldmann, S.; Ren, Q.; Fei, R.; Wang, X.; Wei, L. Influences on viral replication and sensitivity to GLS4, a HAP compound, of naturally occurring T109/V124 mutations in hepatitis B virus core protein. J. Med. Virol. 2017, 89, 1804–1810. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ko, C.; Lee, J.-Y.; Kim, M. Current progress in the development of hepatitis B virus capsid assembly modulators: Chemical structure, mode-of-action and efficacy. Molecules 2021, 26, 7420. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhao, S.; Ren, Y.; Cherukupalli, S.; Li, Q.; Woodson, M.E.; Bradley, D.P.; Tavis, J.E.; Liu, X.; Zhan, P. Design, synthesis and evaluation of heteroaryldihydropyrimidine analogues bearing spiro ring as hepatitis B virus capsid protein inhibitors. Eur. J. Med. Chem. 2021, 225, 113780. [Google Scholar] [CrossRef] [PubMed]
- Lahlali, T.; Berke, J.M.; Vergauwen, K.; Foca, A.; Vandyck, K.; Pauwels, F.; Zoulim, F.; Durantel, D. Novel potent capsid assembly modulators regulate multiple steps of the hepatitis B virus life cycle. Antimicrob. Agents Chemother. 2018, 62, e00835-18. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Liu, F.; Tong, X.; Hoffmann, D.; Zuo, J.; Lu, M. Treatment of chronic hepatitis B virus infection using small molecule modulators of nucleocapsid assembly: Recent advances and perspectives. ACS Infect. Dis. 2019, 5, 713–724. [Google Scholar] [CrossRef]
- Wang, S.; Fogeron, M.L.; Schledorn, M.; Dujardin, M.; Penzel, S.; Burdette, D.; Berke, J.M.; Nassal, M.; Lecoq, L.; Meier, B.H.; et al. Combining cell-free protein synthesis and NMR into a tool to study capsid assembly modulation. Front. Mol. Biosci. 2019, 6, 67. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Hu, T.; Zhou, X.; Wildum, S.; Garcia-Alcalde, F.; Xu, Z.; Wu, D.; Mao, Y.; Tian, X.; Zhou, Y.; et al. Heteroaryldihydropyrimidine (HAP) and sulfamoylbenzamide (SBA) inhibit Hepatitis B virus replication by different molecular mechanisms. Sci. Rep. 2017, 13, 42374. [Google Scholar] [CrossRef]
- Zlotnick, A.; Venkatakrishnan, B.; Tan, Z.; Lewellyn, E.; Turner, W.; Francis, S. Core protein: A pleiotropic keystone in the HBV lifecycle. Antivir. Res. 2015, 121, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Böttcher, B.; Nassal, M. Structure of mutant Hepatitis B core protein capsids with premature secretion phenotype. J. Mol. Biol. 2018, 430, 4941–4954. [Google Scholar] [CrossRef]
- Zajakina, A.; Bruvere, R.; Kozlovska, T. A Semliki Forest virus expression system as a model for investigating the nuclear import and export of hepatitis B virus nucleocapsid protein. Acta Virol. 2014, 58, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Kurena, B.; Vežāne, A.; Skrastiņa, D.; Trofimova, O.; Zajakina, A. Magnetic nanoparticles for efficient cell transduction with Semliki Forest virus. J. Virol. Methods 2017, 245, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Korba, B. Hepatitis B virus cell culture assays for antiviral activity. Methods Mol. Med. 2000, 24, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Lin, X.; Zhou, M.; Liu, Y.; Zhu, W.; Chen, W.; Zhang, W.; Guo, L.; Liu, H.; Wu, G.; et al. Design and synthesis of orally bioavailable 4-methyl heteroaryldihydropyrimidine based Hepatitis B virus (HBV) capsid inhibitors. J. Med. Chem. 2016, 59, 7651–7666. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Liu, X.; Luo, Z.; Li, Y.; Wang, Z.; Goldmann, S.; Zhang, J.; Zhang, Y. Discovery of hepatitis B virus capsid assembly inhibitors leading to a heteroaryldihydropyrimidine based clinical candidate (GLS4). Bioorg. Med. Chem. 2017, 25, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.W.; Seo, J.P.; Cho, Y.; Ko, E.K.; Kim, Y.J.; Jung, G. Cetylpyridinium chloride interaction with the hepatitis B virus core protein inhibits capsid assembly. Virus Res. 2019, 263, 102–111. [Google Scholar] [CrossRef]
- Sells, M.A.; Chen, M.L.; Acs, G. Production of hepatitis B virus particles in HepG2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. USA 1987, 84, 1005–1009. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Luo, Y.; Viswanathan, U.; Kulp, J.; Cheng, J.; Hu, Z.; Xu, Q.; Zhou, Y.; Gong, G.Z.; Chang, J.; et al. CpAMs induce assembly of HBV capsids with altered electrophoresis mobility: Implications for mechanism of inhibiting pgRNA packaging. Antivir. Res. 2018, 159, 1–12. [Google Scholar] [CrossRef]
- Cheng, X.; Guan, W.; Sun, S.; Li, B.; Li, H.; Kang, F.; Kang, J.; Yang, D.; Nassal, M.; Sun, D. Stable human hepatoma cell lines for efficient regulated expression of nucleoside/nucleotide analog resistant and vaccine escape Hepatitis B virus variants and woolly monkey Hepatitis B virus. PLoS ONE 2015, 10, e0145746. [Google Scholar] [CrossRef] [Green Version]
- Weigand, K.; Knaust, A.; Schaller, H. Assembly and export determine the intracellular distribution of hepatitis B virus core protein subunits. J. Gen. Virol. 2010, 91, 59–67. [Google Scholar] [CrossRef]
- Nair, S.; Zlotnick, A. HBV Core Protein Is in Flux between Cytoplasmic, Nuclear, and Nucleolar Compartments. Virology 2021, 12, e03514-20. [Google Scholar] [CrossRef]
- Stray, S.J.; Zlotnick, A. BAY 41-4109 has multiple effects on Hepatitis B virus capsid assembly. J. Mol. Recognit. 2006, 19, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Bourne, C.R.; Finn, M.G.; Zlotnick, A. Global structural changes in hepatitis B virus capsids induced by the assembly effector HAP1. J. Virol. 2006, 80, 11055–11061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Zhao, Q.; Zhang, P.; Kulp, J.; Hu, L.; Hwang, N.; Zhang, J.; Block, T.M.; Xu, X.; Du, Y.; et al. Discovery and mechanistic study of benzamide derivatives that modulate hepatitis B virus capsid assembly. J. Virol. 2017, 91, e00519-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campagna, M.R.; Liu, F.; Mao, R.; Mills, C.; Cai, D.; Guo, F.; Zhao, X.; Ye, H.; Cuconati, A.; Guo, H.; et al. Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. J. Virol. 2013, 87, 6931–6942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Wu, C.Q.; Cao, A.M.; Sheng, H.Z.; Yan, X.Z.; Liao, M.Y. NMR-spectroscopy-based metabonomic approach to the analysis of Bay41-4109, a novel anti-HBV compound, induced hepatotoxicity in rats. Toxicol. Lett. 2007, 173, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Lavanya, P.; Maddila, S.; Jonnalagadda, S.B.; Venkata Rao, C. Synthesis and biological evaluation of novel thio-1,4-dihydropyrimidine-5-carboxylate derivatives. Asian J. Chem. 2013, 25, 385–389. [Google Scholar] [CrossRef]
- Kumar, B.R.P.; Sankar, G.; Nasir Baig, R.B.; Chandrashekuran, S. Novel Biginelli dihydropyrimidines with potential anticancer activity: A parallel synthesis and COMSIA study. Eur. J. Med. Chem. 2009, 44, 4192–4198. [Google Scholar] [CrossRef]
- Timm, M.; Saaby, L.; Moesby, L.; Hansen, E.W. Considerations regarding use of solvents in in vitro cell based assays. Cytotechnology 2013, 65, 887–894. [Google Scholar] [CrossRef] [Green Version]
- Guan, H.; Zhao, G.; Chen, W.; Wu, G.; Liu, H.; Jiang, X.; Li, S.; Wang, L.L. The novel compound Z060228 inhibits assembly of the HBV capsid. Life Sci. 2015, 133, 1–7. [Google Scholar] [CrossRef]
- Berke, J.M.; Dehertogh, P.; Vergauwen, K.; Van Damme, E.; Mostmans, W.; Vandyck, K.; Pauwels, F. Capsid assembly modulators have a dual mechanism of action in primary human hepatocytes infected with Hepatitis B virus. Antimicrob. Agents Chemother. 2017, 61, e00560-17. [Google Scholar] [CrossRef] [Green Version]
- Klumpp, K.; Lam, A.M.; Lukacs, C.; Vogel, R.; Ren, S.; Espiritu, C.; Baydo, R.; Atkins, K.; Abendroth, J.; Liao, G.; et al. High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein. Proc. Natl. Acad. Sci. USA 2015, 112, 15196–15201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berke, J.M.; Tan, Y.; Verbinnen, T.; Dehertogh, P.; Vergauwen, K.; Vos, A.; Lenz, O.; Pauwels, F. Antiviral profiling of the capsid assembly modulator BAY41-4109 on full-length HBV genotype A-H clinical isolates and core site-directed mutants in vitro. Antivir. Res. 2017, 144, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Zajakina, A.; Aleksejeva, J.; Sharipo, A.; Bruvere, R.; Ose, V.; Pumpens, P.; Garoff, H.; Meisel, H.; Kozlovska, T. Proteasomal degradation of core protein variants from chronic hepatitis B patients. J. Med. Virol. 2007, 79, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Bichko, V.; Schödel, F.; Nassal, M.; Gren, E.; Berzinsh, I.; Borisova, G.; Miska, S.; Peterson, D.L.; Gren, E.; Pushko, P.; et al. Epitopes recognized by antibodies to denatured core protein of hepatitis B virus. Mol. Immunol. 1993, 30, 221–231. [Google Scholar] [CrossRef]
- Rat, V.; Seigneuret, F.; Burlaud-Gaillard, J.; Lemoine, R.; Hourioux, C.; Zoulim, F.; Testoni, B.; Meunier, J.C.; Tauber, C.; Roingeard, P.; et al. BAY 41-4109-mediated aggregation of assembled and misassembled HBV capsids in cells revealed by electron microscopy. Antivir. Res. 2019, 169, 104557. [Google Scholar] [CrossRef] [PubMed]
- Brezillon, N.; Brunelle, M.N.; Massinet, H.; Giang, E.; Lamant, C.; DaSilva, L.; Berissi, S.; Belghiti, J.; Hannoun, L.; Puerstinger, G.; et al. Antiviral activity of Bay 41-4109 on hepatitis B virus in humanized Alb-uPA/SCID mice. PLoS ONE 2011, 6, e25096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Kim, N.D.; Seong, B.L. Discovery and development of anti-HBV agents and their resistance. Molecules 2010, 15, 5878–5908. [Google Scholar] [CrossRef]
- Thenin-Houssier, S.; Valente, S.T. HIV capsid inhibitors as antiretroviral agents. Curr. Hiv. Res. 2016, 14, 270–282. [Google Scholar] [CrossRef] [Green Version]
- Baines, J.D. Herpes simplex virus capsid assembly and DNA packaging: A present and future antiviral drug target. Trends Microbiol. 2011, 19, 606–613. [Google Scholar] [CrossRef]
- Boucle, S.; Lu, X.; Bassit, L.; Ozturk, T.; Ollinger Russell, O.; Amblard, F.; Coats, S.J.; Schinazi, R.F. Synthesis and antiviral evaluation of novel heteroarylpyrimidines analogs as HBV capsid effectors. Bioorg. Med. Chem. Lett. 2017, 27, 904–910. [Google Scholar] [CrossRef] [Green Version]
- Stolting, J.; Stoltefuss, J.; Goldmann, S.; Kramer, T.; Schlemmer, K.H.; Niewohner, U.; Paessens, A.; Graef, E.; Lottmann, S.; Deres, K.; et al. Dihydropyrimidines and Their Use in the Treatment of Hepatitis B. Patent WO0058302A1, 5 October 2000. [Google Scholar]
- Kappe, C.O. 100 Years of the Biginelli Dihydropyrimidine Synthesis. Tetrahedron 1993, 49, 6937–6963. [Google Scholar] [CrossRef]
- Suresh, A.; Sandhu, J.S. Past, present and future of the Biginelli reaction: A critical perspective. ARKIVOC 2012, 1, 66–133. [Google Scholar] [CrossRef] [Green Version]
- Kaur, R.; Chaudhary, S.; Kumar, K.; Gupta, M.; Rawal, R.K. Recent synthetic and medicinal perspectives of dihydropyrimidinones: A review. Eur. J. Med. Chem. 2017, 132, 108–134. [Google Scholar] [CrossRef] [PubMed]
- Mingzhe, J.; Hua, B.; Lifei, L.; Yongxiang, G.; Yi, L.; Qixiong, C.; Yongxian, D.; Jian, C.; Hongying, L. Dihydropyrimidines, Preparation Method and Use Thereof. Patent WO2018045911 A1, 15 March 2018. [Google Scholar]
- Sharma, V.K.; Singh, S.K. Synthesis, utility, and medicinal importance of 1,2- and 1,4-dihydropyridines. RSC Adv. 2017, 7, 2682–2732. [Google Scholar] [CrossRef] [Green Version]
- Dubur, G.Y.; Dobretsov, G.E.; Deme, A.K.; Dubure, R.R.; Lapshin, E.N.; Spirin, M.M. Fluorescent probes based on styrylpyridinium derivatives: Optical properties and membrane binding. J. Biochem. Biophys. Methods 1984, 10, 123–134. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository—New features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2018-4: Schrödinger Suite 2018-4 Induced Fit Docking Protocol; Glide, Schrödinger, LLC: New York, NY, USA; Prime, Schrödinger, LLC: New York, NY, USA, 2018.
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an Induced Fit Receptor Structure in Virtual Screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef]
- Liljeström, P.; Garoff, H. A new generation of animal cell expression vectors based on the Semliki Forest virus replicon. Biotechnology 1991, 9, 1356–1361. [Google Scholar] [CrossRef]
- Zajakina, A.; Vasilevska, J.; Zhulenkovs, D.; Skrastina, D.; Spaks, A. High efficiency of alphaviral gene transfer in combination with 5-fluorouracil in a mouse mammary tumor model. BMC cancer 2014, 14, 460. [Google Scholar] [CrossRef]
- Zajakina, A.; Kozlovska, T.; Bruvere, R.; Aleksejeva, J.; Pumpens, P.; Garoff, H. Translation of hepatitis B virus (HBV) surface proteins from the HBV pregenome and precore RNAs in Semliki Forest virus-driven expression. J. Gen. Virol. 2004, 85, 3343–3351. [Google Scholar] [CrossRef]
- Hutornojs, V.; Niedre-Otomere, B.; Kozlovska, T.; Zajakina, A. Comparison of ultracentrifugation methods for concentration of recombinant alphaviruses: Sucrose and iodixanol cushions. Environ. Exp. Biol. 2012, 10, 117–123. [Google Scholar]
- Heger-Stevic, J.; Kolb, P.; Walker, A.; Nassal, M. Displaying Whole-Chain Proteins on Hepatitis B Virus Capsid-Like Particles. Methods Mol. Biol. 2018, 1776, 503–531. [Google Scholar] [CrossRef]
- Changotra, H.; Sehajpal, P.K. An improved method for the isolation of hepatitis B virus DNA from human serum. Indian J. Virol. 2013, 24, 174–179. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | ||||||
 | ||||||
| Nr. | R1 | R2 | N+ | The Highest Non-Toxic Concentration, μM | Effect on Capsid Assembly, Starting Concentration | |
| Bay 41-4109 (1a) | 2-Cl, 4-F | Me | 100 | Dose dependent capsid signal decrease, 1 μM | ||
| 1b | 2-Cl, 4-F | H | 50 | Dose dependent capsid signal decrease, 10 μM | ||
| 1c | 4-F | Et | <25 | Capsid signal decrease, 5 μM | ||
| 1d | 2-Cl, 4-F | CH2CH2OEt | 200 | HBc accumulation, aggregates/or incorrect capsid structures, 5 μM | ||
| 1e | 2-Cl, 4-F | CH2CH2O-Pr-i | 100 | Dose dependent capsid signal decrease, 10 μM | ||
| 1f | 2-Cl, 4-F | CH2CH2OPr | 100 | HBc accumulation, aggregates/or incorrect capsid structures, 10 μM | ||
| 1g | 2-OCHF2 | CH2CH2OPr | 50 | Insignificant capsid amount increases | ||
| 1h | 2-Cl, 4-F | 2-pyridyl | 150 | Dose dependent capsid signal decrease, 20 μM | ||
| 1i | 2-Cl, 4-F | o-methoxyphenyl | 50 | Dose dependent capsid signal decrease, 20 μM | ||
| 1j | 4-F | Et | <25 | HBc accumulation, aggregates/or incorrect capsid structures, 5 μM | ||
| 1k | H | Et | 25 | Dose dependent HBc accumulation, aggregates/or incorrect capsid structures, 1 μM | ||
| 3a |  | 200 | No effect | |||
| 3b |  | 200 | No effect | |||
| 3c |  | 150 | No effect | |||
| 3d |  | 100 | Capsid signal decrease, possible changes in NS packaging | |||
| (B) | ||||||
 | ||||||
| The highest non-toxic concentration, μM | 200 | 25 | 200 | 50 | <25 | 150 |
| Effect to capsid assembly, starting concentration, μM | HBc accumulation, aggregates/or incorrect capsid structures, 50 μM | No effect | No effect | No effect | No effect | No effect |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spunde, K.; Vigante, B.; Dubova, U.N.; Sipola, A.; Timofejeva, I.; Zajakina, A.; Jansons, J.; Plotniece, A.; Pajuste, K.; Sobolev, A.; et al. Design and Synthesis of Hepatitis B Virus (HBV) Capsid Assembly Modulators and Evaluation of Their Activity in Mammalian Cell Model. Pharmaceuticals 2022, 15, 773. https://doi.org/10.3390/ph15070773
Spunde K, Vigante B, Dubova UN, Sipola A, Timofejeva I, Zajakina A, Jansons J, Plotniece A, Pajuste K, Sobolev A, et al. Design and Synthesis of Hepatitis B Virus (HBV) Capsid Assembly Modulators and Evaluation of Their Activity in Mammalian Cell Model. Pharmaceuticals. 2022; 15(7):773. https://doi.org/10.3390/ph15070773
Chicago/Turabian StyleSpunde, Karina, Brigita Vigante, Unda Nelda Dubova, Anda Sipola, Irena Timofejeva, Anna Zajakina, Juris Jansons, Aiva Plotniece, Karlis Pajuste, Arkadij Sobolev, and et al. 2022. "Design and Synthesis of Hepatitis B Virus (HBV) Capsid Assembly Modulators and Evaluation of Their Activity in Mammalian Cell Model" Pharmaceuticals 15, no. 7: 773. https://doi.org/10.3390/ph15070773
APA StyleSpunde, K., Vigante, B., Dubova, U. N., Sipola, A., Timofejeva, I., Zajakina, A., Jansons, J., Plotniece, A., Pajuste, K., Sobolev, A., Muhamadejev, R., Jaudzems, K., Duburs, G., & Kozlovska, T. (2022). Design and Synthesis of Hepatitis B Virus (HBV) Capsid Assembly Modulators and Evaluation of Their Activity in Mammalian Cell Model. Pharmaceuticals, 15(7), 773. https://doi.org/10.3390/ph15070773