
Novel Therapies to Address Unmet Needs in ITP

 , ,
, ,  , ,
, ,
Abstract
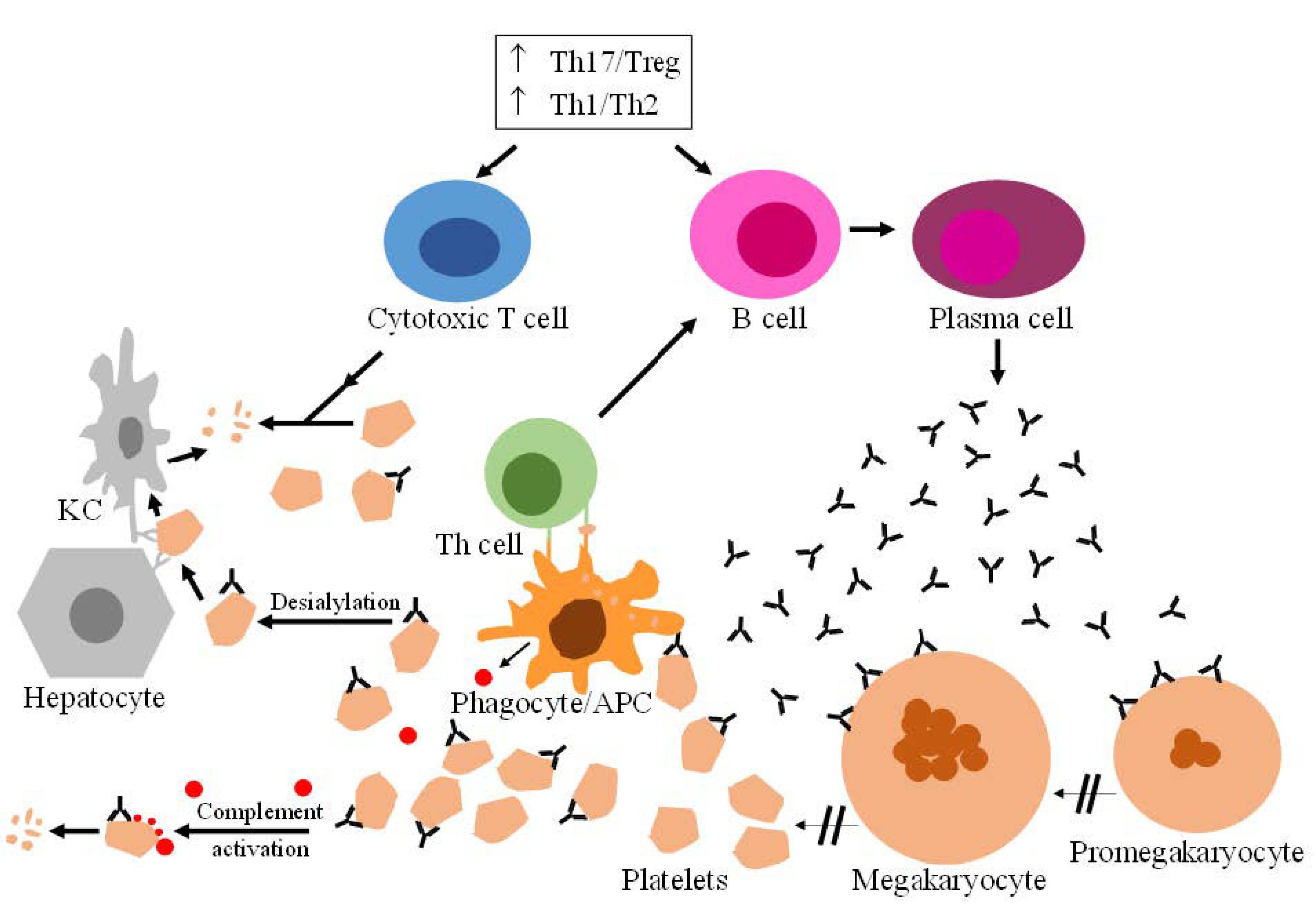
:1. Introduction
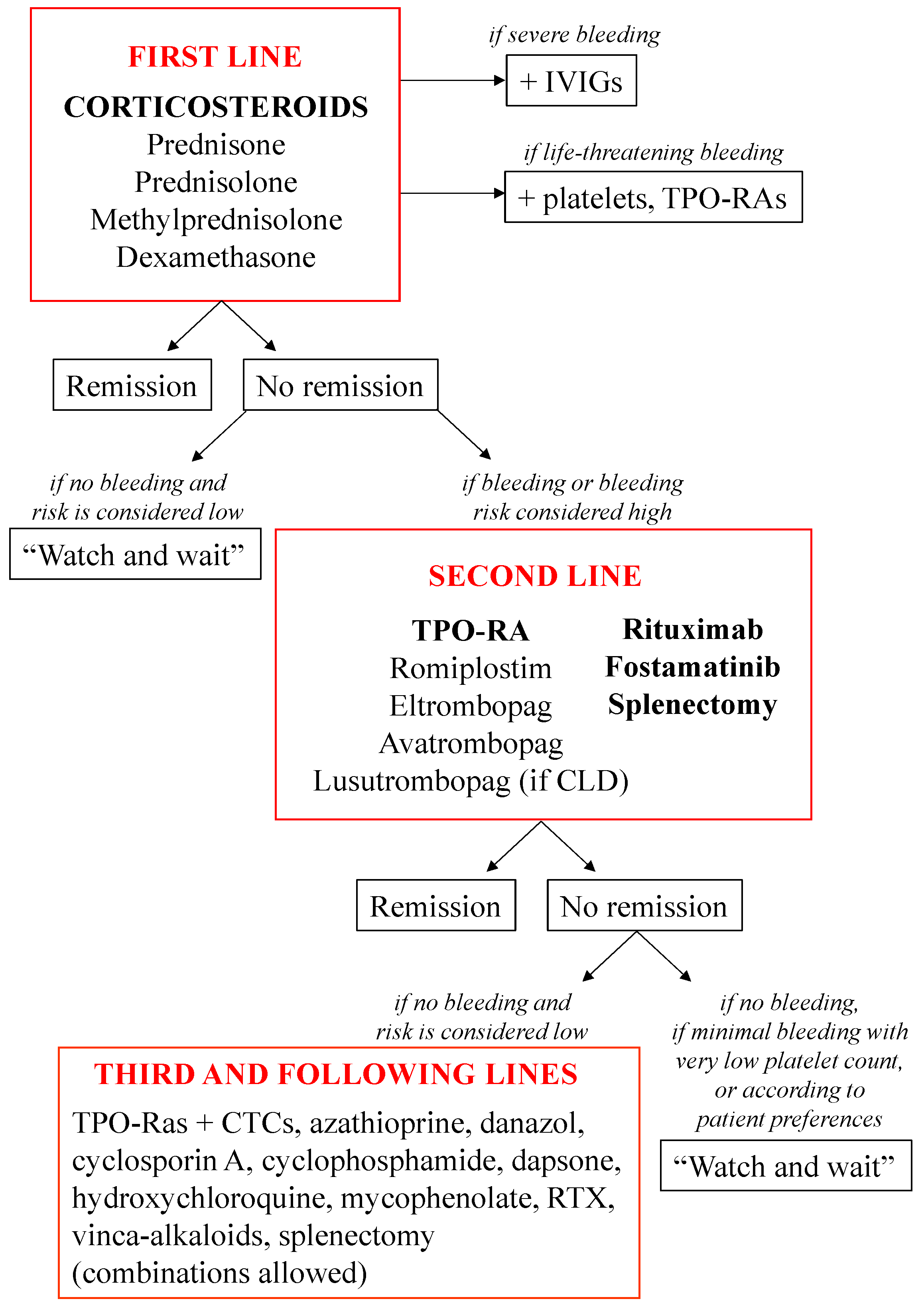
2. Currently Approved Treatments to Manage ITP
2.1. First-Line Treatments
2.2. Second-Line Treatments
2.2.1. TPO-RAs
2.2.2. Rituximab
2.2.3. Fostamatinib
2.2.4. Splenectomy
2.3. Third-Line Treatments
3. Novel Drugs and Therapies to Treat ITP
3.1. Other TPO-RAs: Hetrombopag Olamine
3.2. Inhibitors of Kinases Involved in Phagocyte Cytoskeleton Rearrangements
3.2.1. Syk Inhibitors
HMPL-523
SKI-O-703 (Cevidoplenib)
3.2.2. Bruton’s Tyrosine Kinase Inhibitors
Rilzabrutinib
Orelabrutinib
3.3. Inhibitors of Neonatal Fc Receptors
Efgartigimod
Rozanolixizumab
Other FcRn blockers
3.4. Staphylococcal Protein A
PRTX-100
3.5. Immunoglobulin-Based Therapies
3.5.1. Hyper-Sialylated Immunoglobulin G
M254
3.5.2. Other Immunoglobulin-Derived Drugs
3.6. Complement Inhibitors
Sutimlimab
3.7. Platelet Desialylation Inhibitors
Oseltamivir
3.8. Anti-CD38 Antibody
Daratumumab
Mezagitamab
3.9. Proteasome Inhibitors
Bortezomib
Other Proteasome Inhibitors
3.10. Therapies Targeting T or B Cells
3.11. Other Future Options
3.11.1. VPAC1 Inhibitors
3.11.2. Amifostine
3.11.3. Atorvastatin
3.11.4. Epigenetic Modulation
3.11.5. Low-Level Laser Light
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Terrell, D.R.; Beebe, L.A.; Vesely, S.K.; Neas, B.R.; Segal, J.B.; George, J.N. The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am. J. Hematol. 2010, 85, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, D.; Zhu, X.; Qu, X.; Ji, C.; Hou, M. Elevated profile of Th17, Th1 and Tc1 cells in patients with immune thrombocytopenic purpura. Haematologica 2009, 94, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, W.; Zhang, W.; Lee-Sundlov, M.M.; Casari, C.; Berndt, M.C.; Lanza, F.; Bergmeier, W.; Hoffmeister, K.M.; Zhang, X.F.; et al. Desialylation of O-glycans on glycoprotein Ibα drives receptor signaling and platelet clearance. Haematologica 2021, 106, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Fu, J.; Ling, Y.; Yago, T.; McDaniel, J.M.; Song, J.; Bai, X.; Kondo, Y.; Qin, Y.; Hoover, C.; et al. Sialylation on O-glycans protects platelets from clearance by liver Kupffer cells. Proc. Natl. Acad. Sci. USA 2017, 114, 8360–8365. [Google Scholar] [CrossRef] [Green Version]
- Cooper, N.; Bussel, J. The pathogenesis of immune thrombocytopaenic purpura. Br. J. Haematol. 2006, 133, 364–374. [Google Scholar] [CrossRef]
- Neunert, C.; Noroozi, N.; Norman, G.; Buchanan, G.R.; Goy, J.; Nazi, I.; Kelton, J.G.; Arnold, D.M. Severe bleeding events in adults and children with primary immune thrombocytopenia: A systematic review. J. Thromb. Haemost. 2015, 13, 457–464. [Google Scholar] [CrossRef]
- Singh, A.; Uzun, G.; Bakchoul, T. Primary Immune Thrombocytopenia: Novel Insights into Pathophysiology and Disease Management. J. Clin. Med. 2021, 10, 789. [Google Scholar] [CrossRef]
- Lozano, M.L.; Sanz, M.A.; Vicente, V.; Grupo Español de PTI (GEPTI). Guidelines of the Spanish ITP Group for the diagnosis, treatment and follow-up of patients with immune thrombopenia. Med. Clin. 2021, 157, 191–198. [Google Scholar] [CrossRef]
- Stasi, R. Immune Thrombocytopenia: Pathophysiologic and Clinical Update. Semin. Thromb. Hemost. 2012, 38, 454–462. [Google Scholar] [CrossRef]
- Provan, D.; Arnold, D.M.; Bussel, J.B.; Chong, B.H.; Cooper, N.; Gernsheimer, T.; Ghanima, W.; Godeau, B.; González-López, T.J.; Grainger, J.; et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019, 3, 3780–3817. [Google Scholar] [CrossRef] [Green Version]
- Neunert, C.; Terrell, D.R.; Arnold, D.M.; Buchanan, G.; Cines, D.B.; Cooper, N.; Cuker, A.; Despotovic, J.M.; George, J.N.; Grace, R.F.; et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019, 3, 3829–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solar, G.P.; Kerr, W.G.; Zeigler, F.C.; Hess, D.; Donahue, C.; de Sauvage, F.J.; Eaton, D.L. Role of c-mpl in Early Hematopoiesis. Blood 1998, 92, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Kuter, D.J. New drugs for familiar therapeutic targets: Thrombopoietin receptor agonists and immune thrombocytopenic purpura. Eur. J. Haematol. 2008, 80 (Suppl. 69), 9–18. [Google Scholar] [CrossRef] [PubMed]
- Tortolani, P.J.; Johnston, J.A.; Bacon, C.M.; McVicar, D.W.; Shimosaka, A.; Linnekin, D.; Longo, D.L.; O’Shea, J.J. Thrombo-poietin induces tyrosine phosphorylation and activation of the Janus kinase, JAK2. Blood 1995, 85, 3444–3451. [Google Scholar] [CrossRef] [Green Version]
- Stasi, R.; Bosworth, J.; Rhodes, E.; Shannon, M.S.; Willis, F.; Gordon-Smith, E.C. Thrombopoietic agents. Blood Rev. 2010, 24, 179–190. [Google Scholar] [CrossRef]
- Neunert, C.E. Thrombopoietin Receptor Agonist Use for Immune Thrombocytopaenia. Hamostaseologie 2019, 39, 272–278. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Kuter, D.J. Immune Thrombocytopenia in Adults: Modern Approaches to Diagnosis and Treatment. Semin. Thromb. Hemost. 2020, 46, 275–288. [Google Scholar] [CrossRef]
- Kuter, D.J. Novel therapies for immune thrombocytopenia. Br. J. Haematol. 2021, 196, 1311–1328. [Google Scholar] [CrossRef]
- Lozano, M.L.; Godeau, B.; Grainger, J.; Matzdorff, A.; Rodeghiero, F.; Hippenmeyer, J.; Kuter, D.J. Romiplostim in adults with newly diagnosed or persistent immune thrombocytopenia. Expert Rev. Hematol. 2020, 13, 1319–1332. [Google Scholar] [CrossRef]
- González-Porras, J.R.; Mingot-Castellano, M.E.; Andrade, M.M.; Alonso, R.; Caparrós, I.; Arratibel, M.C.; Fernández-Fuertes, F.; Cortti, M.J.; Pascual, C.; Sánchez-González, B.; et al. Use of eltrombopag after romiplostim in primary immune thrombocytopenia. Br. J. Haematol. 2015, 169, 111–116. [Google Scholar] [CrossRef]
- Stasi, R.; Evangelista, M.L.; Amadori, S. Novel thrombopoietic agents: A review of their use in idiopathic thrombocytopenic purpura. Drugs 2008, 68, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Saleh, M.N.; Marcher, C.; Vasey, S.; Mayer, B.; Aivado, M.; Arning, M.; Stone, N.L.; Bussel, J.B. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): A 6-month, randomised, phase 3 study. Lancet 2011, 377, 393–402. [Google Scholar] [CrossRef]
- Kuter, D.J.; Bussel, J.B.; Lyons, R.M.; Pullarkat, V.; Gernsheimer, T.B.; Senecal, F.M.; Aledort, L.M.; George, J.N.; Kessler, C.M.; Sanz, M.A.; et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: A double-blind randomised controlled trial. Lancet 2008, 371, 395–403. [Google Scholar] [CrossRef]
- Bussel, J.B.; Provan, D.; Shamsi, T.; Cheng, G.; Psaila, B.; Kovaleva, L.; Salama, A.; Jenkins, J.M.; Roychowdhury, D.; Mayer, B.; et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: A randomised, double-blind, placebo-controlled trial. Lancet 2009, 373, 641–648. [Google Scholar] [CrossRef]
- Newland, A.; Godeau, B.; Priego, V.; Viallard, J.-F.; Fernández, M.F.L.; Orejudos, A.; Eisen, M. Remission and platelet responses with romiplostim in primary immune thrombocytopenia: Final results from a phase 2 study. Br. J. Haematol. 2016, 172, 262–273. [Google Scholar] [CrossRef]
- González-López, T.J.; Pascual, C.; Álvarez-Román, M.T.; Fernández-Fuertes, F.; Sánchez-González, B.; Caparrós, I.; Jarque, I.; Castellano, M.E.M.; Hernandez-Rivas, J.-A.; Martín-Salces, M.; et al. Successful discontinuation of eltrombopag after complete remission in patients with primary immune thrombocytopenia. Am. J. Hematol. 2015, 90, E40–E43. [Google Scholar] [CrossRef]
- Janssens, A.; Rodeghiero, F.; Anderson, D.; Chong, B.H.; Boda, Z.; Pabinger, I.; Červinek, L.; Terrell, D.R.; Wang, X.; Franklin, J. Changes in bone marrow morphology in adults receiving romiplostim for the treatment of thrombocytopenia associated with primary immune thrombocytopenia. Ann. Hematol. 2016, 95, 1077–1087. [Google Scholar] [CrossRef] [Green Version]
- Mingot-Castellano, M.E.; Caparrós, I.S.; Fernández, F.; Perera-Alvarez, M.D.M.; Jimenez-Bárcenas, R.; García, A.C.; González-Silva, M.; Yera-Cobo, M.; Nieto-Hernandez, M.M.; Rodríguez-Fernandez, M.J.; et al. Treatment characteristics, efficacy and safety of thrombopoietin analogues in routine management of primary immune thrombocytopenia. Blood Coagul. Fibrinolysis 2018, 29, 374–380. [Google Scholar] [CrossRef]
- Lozano, M.L.; Mingot-Castellano, M.E.; Perera, M.M.; Jarque, I.; Campos-Alvarez, R.M.; González-López, T.J.; Carreño-Tarragona, G.; Bermejo, N.; Lopez-Fernandez, M.F.; De Andrés, A.; et al. Deciphering predictive factors for choice of thrombopoietin receptor agonist, treatment free responses, and thrombotic events in immune thrombocytopenia. Sci. Rep. 2019, 9, 16680. [Google Scholar] [CrossRef] [Green Version]
- Kuter, D.J.; Rummel, M.; Boccia, R.; Macik, B.G.; Pabinger, I.; Selleslag, D.; Rodeghiero, F.; Chong, B.H.; Wang, X.; Berger, D.P. Romiplostim or Standard of Care in Patients with Immune Thrombocytopenia. N. Engl. J. Med. 2010, 363, 1889–1899. [Google Scholar] [CrossRef] [Green Version]
- Jurczak, W.; Chojnowski, K.; Mayer, J.; Krawczyk, K.; Jamieson, B.D.; Tian, W.; Allen, L.F. Phase 3 randomised study of avatrombopag, a novel thrombopoietin receptor agonist for the treatment of chronic immune thrombocytopenia. Br. J. Haematol. 2018, 183, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GlaxoSmithKline. Promacta (Eltrombopag) [Prescribing Information]; GlaxoSmithKline: Research Triangle Park, NC, USA, 2017. [Google Scholar]
- Psaila, B.; Bussel, J.B.; Linden, M.D.; Babula, B.; Li, Y.; Barnard, M.R.; Tate, C.; Mathur, K.; Frelinger, A.L.; Michelson, A.D. In vivo effects of eltrombopag on platelet function in immune thrombocytopenia: No evidence of platelet activation. Blood 2012, 119, 4066–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Samkari, H.; Van Cott, E.M.; Kuter, D.J. Platelet aggregation response in immune thrombocytopenia patients treated with romiplostim. Ann. Hematol. 2019, 98, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Bussel, J.B.; Kuter, D.J.; Pullarkat, V.; Lyons, R.M.; Guo, M.; Nichol, J.L. Safety and efficacy of long-term treatment with romi-plostim in thrombocytopenic patients with chronic ITP. Blood 2009, 113, 2161–2171. [Google Scholar] [CrossRef]
- Cines, D.B.; Gernsheimer, T.; Wasser, J.; Godeau, B.; Provan, D.; Lyons, R.; Altomare, I.; Wang, X.; Lopez, A. Integrated analysis of long-term safety in patients with chronic immune thrombocytopaenia (ITP) treated with the thrombopoietin (TPO) receptor agonist romiplostim. Int. J. Hematol. 2015, 102, 259–270. [Google Scholar] [CrossRef]
- Kuter, D.J.; Mufti, G.J.; Bain, B.J.; Hasserjian, R.P.; Davis, W.; Rutstein, M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood 2009, 114, 3748–3756. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, M.; Du, X.; Cheng, Y.; Cheng, G. Safety and efficacy of eltrombopag plus pulsed dexamethasone as first-line therapy for immune thrombocytopenia. Br. J. Haematol. 2020, 189, 369–378. [Google Scholar] [CrossRef]
- Roll, P.; Palanichamy, A.; Kneitz, C.; Dorner, T.; Tony, H.P. Regeneration of B cell subsets after transient B cell depletion using anti-CD20 an-tibodies in rheumatoid arthritis. Arthritis Rheum. 2006, 54, 2377–2386. [Google Scholar] [CrossRef]
- Lucchini, E.; Zaja, F.; Bussel, J. Rituximab in the treatment of immune thrombocytopenia: What is the role of this agent in 2019? Haematologica 2019, 104, 1124–1135. [Google Scholar] [CrossRef]
- Thai, L.-H.; Le Gallou, S.; Robbins, A.; Crickx, E.; Fadeev, T.; Zhou, Z.; Cagnard, N.; Mégret, J.; Bole, C.; Weill, J.-C.; et al. BAFF and CD4(+) T cells are major survival factors for long-lived splenic plasma cells in a B-cell–depletion context. Blood 2018, 131, 1545–1555. [Google Scholar] [CrossRef]
- Audia, S.; Samson, M.; Mahévas, M.; Ferrand, C.; Trad, M.; Ciudad, M.; Gautheron, A.; Seaphanh, F.; Leguy, V.; Berthier, S.; et al. Preferential splenic CD8(+) T-cell activation in rituximab-nonresponder patients with immune thrombocytopenia. Blood 2013, 122, 2477–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussel, J.; Arnold, D.M.; Grossbard, E.; Mayer, J.; Treliński, J.; Homenda, W.; Hellmann, A.; Windyga, J.; Sivcheva, L.; Khalafallah, A.; et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. Am. J. Hematol. 2018, 93, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Bussel, J.B.; Arnold, D.M.; Boxer, M.A.; Cooper, N.; Mayer, J.; Zayed, H.; Tong, S.; Duliege, A. Long-term fostamatinib treatment of adults with immune thrombocytopenia during the phase 3 clinical trial program. Am. J. Hematol. 2019, 94, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Boccia, R.; Cooper, N.; Ghanima, W.; Boxer, M.A.; Hill, Q.A.; Sholzberg, M.; Tarantino, M.D.; Todd, L.K.; Tong, S.; Bussel, J.B.; et al. Fostamatinib is an effective second-line therapy in patients with immune thrombocytopenia. Br. J. Haematol. 2020, 190, 933–938. [Google Scholar] [CrossRef]
- Kojouri, K.; Vesely, S.; Terrell, D.; George, J.N. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: A systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004, 104, 2623–2634. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowska, A.; Radkowiak, D.; Wysocki, M.; Torbicz, G.; Gajewska, N.; Lasek, A.; Kulawik, J.; Budzyński, A.; Pędziwiatr, M. Prognostic Factors for Immune Thrombocytopenic Purpura Remission after Laparoscopic Splenectomy: A Cohort Study. Medicina 2019, 55, 112. [Google Scholar] [CrossRef] [Green Version]
- Dou, X.; Yang, R. Current and emerging treatments for immune thrombocytopenia. Expert Rev. Hematol. 2019, 12, 723–732. [Google Scholar] [CrossRef]
- Zheng, L.; Liang, M.-Z.; Zeng, X.-L.; Li, C.-Z.; Zhang, Y.-F.; Chen, X.-Y.; Zhu, X.; Xiang, A.-B. Safety, Pharmacokinetics and Pharmacodynamics of Hetrombopag Olamine, a Novel TPO-R Agonist, in Healthy Individuals. Basic Clin. Pharmacol. Toxicol. 2017, 121, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Huang, R.; Yang, S.; Zhang, X.; Yang, X.; Chen, H.; Huang, Z.; Guo, C.; Pei, Q.; Tai, Y.; et al. Effect of postdose fasting duration on hetrombopag olamine pharmacokinetics and pharmacodynamics in healthy volunteers. Br. J. Clin. Pharmacol. 2020, 86, 1528–1536. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, X.; Li, A.; Chen, L.; Wang, Y.; Zheng, L. Effect of Food on the Pharmacokinetic and Pharmacodynamic Profiles of Hetrombopag in Healthy Volunteers. Clin. Ther. 2020, 42, 2280–2288. [Google Scholar] [CrossRef]
- Xie, C.; Zhao, H.; Bao, X.; Fu, H.; Lou, L. Pharmacological characterization of hetrombopag, a novel orally active human thrombopoietin receptor agonist. J. Cell. Mol. Med. 2018, 22, 5367–5377. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Liu, X.; Li, Y.; Zhou, H.; Feng, Y.; Gao, G.; Cheng, P.; Huang, R.; Yang, L.; Hu, J.; et al. Dose tapering to withdrawal stage and long-term efficacy and safety of hetrombopag for the treatment of immune thrombocytopenia: Results from an open-label extension study. J. Thromb. Haemost. 2022, 20, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Lickliter, J.; Wu, Y.; Hua, Y.; Yuan, I.; Dai, G.; Li, X.; Wang, J.; Sai, Y.; Sun, Z.; Pan, A.; et al. A Phase I, Randomized, Double Blind, Placebo-Controlled, Dose Escalating Study of the Safety, Tolerability and Pharmacokinetics and Pharmacodynamics of Single and Multiple Doses of Hmpl 523 in Australian Male Healthy Subjects. In Proceedings of the 2016 ACR/ARHP Annual Meeting, Washington, DC, USA, 11–16 November 2016; Abstract Number 1621. [Google Scholar]
- Yang, R.; Zhou, H.; Hu, Y.; Yin, J.; Li, J.; Chen, W.; Huang, R.; Gong, Y.; Luo, C.; Xiaofan, L.; et al. Safety, Pharmacokinetics and Preliminary Efficacy of HMPL-523 in Adult Patients with Primary Immune Thrombocytopenia: A Randomized, Double-Blind and Placebo-Controlled Phase 1b Study. Blood 2021, 138 (Suppl. 1), 16. [Google Scholar] [CrossRef]
- Neys, S.F.H.; Hendriks, R.W.; Corneth, O.B.J. Targeting Bruton’s Tyrosine Kinase in Inflammatory and Autoimmune Pathologies. Front. Cell Dev. Biol. 2021, 9, 668131. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.F.; Krishnarajah, J.; Nunn, P.A.; Hill, R.J.; Karr, D.; Tam, D.; Masjedizadeh, M.; Funk, J.O.; Gourlay, S.G. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br. J. Clin. Pharmacol. 2017, 83, 2367–2376. [Google Scholar] [CrossRef]
- Kuter, D.J.; Tzvetkov, N.; Efraim, M.; Kaplan, Z.; Mayer, J.; Choi, P.; Jansen, A.G.; McDonald, V.; Baker, R.; Bird, R.J.; et al. Updated Phase I/II Safety and Efficacy Results for Oral Bruton Tyrosine Kinase Inhibitor Rilzabrutinib in Patients with Relapsed/Refractory Immune Thrombocytopenia. Blood 2021, 138 (Suppl. 1), 14. [Google Scholar] [CrossRef]
- Kuter, D.J.; Efraim, M.; Mayer, J.; Trněný, M.; McDonald, V.; Bird, R.; Regenbogen, T.; Garg, M.; Kaplan, Z.; Tzvetkov, N.; et al. Rilzabrutinib, an Oral BTK Inhibitor, in Immune Thrombocytopenia. N. Engl. J. Med. 2022, 386, 1421–1431. [Google Scholar] [CrossRef]
- Kuter, D.J.; Bussel, J.B.; Cooper, N.; Gernsheimer, T.; Lambert, M.P.; Liebman, H.; Tarantino, M.D.; Bandman, O.; Arora, P.; Neale, A.; et al. LUNA3 Phase III Multicenter, Double-Blind, Randomized, Placebo-Controlled Trial of the Oral BTK Inhibitor Rilzabrutinib in Adults and Adolescents with Persistent or Chronic Immune Thrombocytopenia. Blood 2021, 138 (Suppl. 1), 1010. [Google Scholar] [CrossRef]
- Zhang, B.; Zhao, R.; Liang, R.; Gao, Y.; Liu, R.; Chen, X.; Lu, Z.; Wang, Z.; Yu, L.; Shakib, S.; et al. Orelabrutinib, a potent and selective Bruton’s tyrosine kinase inhibitor with superior safety profile and excellent PK/PD properties. Cancer Res. 2020, 80 (Suppl. 16), CT132. [Google Scholar] [CrossRef]
- Yu, T.; Wang, L.; Ni, X.; Hou, Y.; Liu, X.; Hou, M. Orelabrutinib, a Selective Bruton’s Tyrosine Kinase (BTK) Inhibitor in the Treatment of Primary Immune Thrombocytopenia (ITP). Blood 2021, 138 (Suppl. 1), 3172. [Google Scholar] [CrossRef]
- Lamamy, J.; Boulard, P.; Brachet, G.; Tourlet, S.; Gouilleux-Gruart, V.; Ramdani, Y. Ways in which the neonatal Fc-receptor is involved in autoimmunity. J. Transl. Autoimmun. 2021, 4, 100122. [Google Scholar] [CrossRef] [PubMed]
- Newland, A.C.; Sánchez-González, B.; Rejtő, L.; Egyed, M.; Romanyuk, N.; Godar, M.; Verschueren, K.; Gandini, D.; Ulrichts, P.; Beauchamp, J.; et al. Phase 2 study of efgartigimod, a novel FcRn antagonist, in adult patients with primary immune thrombocytopenia. Am. J. Hematol. 2020, 95, 178–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiessling, P.; Lledo-Garcia, R.; Watanabe, S.; Langdon, G.; Tran, D.; Bari, M.; Christodoulou, L.; Jones, E.; Price, G.; Smith, B.; et al. The FcRn inhibitor rozanolixizumab reduces human serum IgG concentration: A randomized phase 1 study. Sci. Transl. Med. 2017, 9, 414. [Google Scholar] [CrossRef] [PubMed]
- Zuercher, A.W.; Spirig, R.; Morelli, A.B.; Käsermann, F. IVIG in autoimmune disease—Potential next generation biologics. Autoimmun. Rev. 2016, 15, 781–785. [Google Scholar] [CrossRef]
- Sondermann, P. The FcgammaR/IgG Interaction as target for the treatment of autoimmune diseases. J. Clin. Immunol. 2016, 36 (Suppl. 1), 95–99. [Google Scholar] [CrossRef]
- Lambris, J.D.; Ricklin, D.; Geisbrecht, B.V. Complement evasion by human pathogens. Nat. Rev. Genet. 2008, 6, 132–142. [Google Scholar] [CrossRef]
- Goodyear, C.S.; Silverman, G.J. Death by a B cell superantigen: In vivo VH-targeted apoptotic supraclonal B cell deletion by a Staphylococcal Toxin. J. Exp. Med. 2003, 197, 1125–1139. [Google Scholar] [CrossRef]
- Kapur, R.; Catalina, M.; Aslam, R.; Speck, E.R.; Francovitch, R.F.; Semple, J.W. A highly purified form of staphylococcal protein A alleviates murine immune thrombocytopenia (ITP). Br. J. Haematol. 2018, 183, 501–503. [Google Scholar] [CrossRef] [Green Version]
- Bethesda (MD): National Library of Medicine (US). 27 March 2015. Identifier NCT02401061. PRTX-100-202 Open-Label, Dose Escalation Study in Adult Patients with ITP. 5 July 2019. Available online: https://clinicaltrials.gov/ (accessed on 26 November 2021).
- Kaneko, Y.; Nimmerjahn, F.; Ravetch, J.V. Anti-Inflammatory Activity of Immunoglobulin G Resulting from Fc Sialylation. Science 2006, 313, 670–673. [Google Scholar] [CrossRef] [Green Version]
- Bagnoli, F.; Bertholet, S.; Grandi, G. Inferring Reasons for the Failure of Staphylococcus aureus Vaccines in Clinical Trials. Front. Cell. Infect. Microbiol. 2012, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Lou, Y.; Zhang, Y.; Liu, S.; Li, J.; Tao, J. Sialylated immunoglobulin G: A promising diagnostic and therapeutic strategy for autoimmune diseases. Theranostics 2021, 11, 5430–5446. [Google Scholar] [CrossRef] [PubMed]
- Washburn, N.; Schwab, I.; Ortiz, D.; Bhatnagar, N.; Lansing, J.C.; Medeiros, A.; Tyler, S.; Mekala, D.; Cochran, E.; Sarvaiya, H.; et al. Controlled tetra-Fc sialylation of IVIg results in a drug candidate with consistent enhanced anti-inflammatory activity. Proc. Natl. Acad. Sci. USA 2015, 112, E1297–E1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debré, M.; Griscelli, C.; Bonnet, M.C.; Carosella, E.; Philippe, N.; Reinert, P.; Vilmer, E.; Kaplan, C.; Fridman, W.H.; Teillaud, J.L. Infusion of Fc gamma fragments for treatment of children with acute immune thrombocytopenic purpura. Lancet 1993, 42, 945–949. [Google Scholar] [CrossRef]
- Augener, W.; Friedmann, B.; Brittinger, G. Are aggregates of IgG the effective part of high-dose immunoglobulin therapy in adult idiopathic thrombocytopenic purpura (ITP)? Blut 1985, 50, 249–252. [Google Scholar] [CrossRef]
- Zhou, H.; Olsen, H.; So, E.; Mérigeon, E.; Rybin, D.; Owens, J.; LaRosa, G.; Block, D.S.; Strome, S.E.; Zhang, X. A fully recombinant human IgG1 Fc multimer (GL-2045) inhibits complement-mediated cytotoxicity and induces iC3b. Blood Adv. 2017, 1, 504–515. [Google Scholar] [CrossRef]
- Bethesda (MD): National Library of Medicine (US). 24 June 2020. Identifier NCT03375606. Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Subcutaneous CSL730 in Healthy Adult Subjects. 30 September 2021. Available online: https://clinicaltrials.gov/ (accessed on 26 November 2021).
- Najaoui, A.; Bakchoul, T.; Stoy, J.; Bein, G.; Rummel, M.J.; Santoso, S.; Sachs, U.J. Autoantibody-mediated complement activation on platelets is a common finding in patients with immune thrombocytopenic purpura (ITP). Eur. J. Haematol. 2012, 88, 167–174. [Google Scholar] [CrossRef]
- Castelli, R.; Delilliers, G.L.; Gidaro, A.; Cicardi, M.; Bergamaschini, L. Complement activation in patients with immune thrombocytopenic purpura according to phases of disease course. Clin. Exp. Immunol. 2020, 201, 258–265. [Google Scholar] [CrossRef]
- Peerschke, E.I.; Panicker, S.; Bussel, J. Classical complement pathway activation in immune thrombocytopenia purpura: Inhibition by a novel C1s inhibitor. Br. J. Haematol. 2016, 173, 942–945. [Google Scholar] [CrossRef] [PubMed]
- Roesch, E.E.; Broome, C. Complement Blockade with C1 Esterase Inhibitor in Refractory Immune Thrombocytopenia. Blood 2015, 126, 2253. [Google Scholar] [CrossRef]
- Röth, A.; Barcellini, W.; D’Sa, S.; Miyakawa, Y.; Broome, C.M.; Michel, M.; Kuter, D.J.; Jilma, B.; Tvedt, T.H.A.; Fruebis, J.; et al. Sutimlimab in Cold Agglutinin Disease. N. Engl. J. Med. 2021, 384, 1323–1334. [Google Scholar] [CrossRef]
- Broome, C.M.; Röth, A.; Kuter, D.J.; Scully, M.; Smith, R.; Wang, J.; Jiang, X.; Reuter, C.; Daak, A.; Hobbs, W. Long-Term Safety and Efficacy of Sutimlimab in Patients with Chronic Immune Thrombocytopenia. Blood 2020, 136 (Suppl. 1), 14–15. [Google Scholar] [CrossRef]
- Rumjantseva, V.; Grewal, P.K.; Wandall, H.H.; Josefsson, E.C.; Sørensen, A.L.; Larson, G.; Marth, J.D.; Hartwig, J.H.; Hoffmeister, K.M. Dual roles for hepatic lectin receptors in the clearance of chilled platelets. Nat. Med. 2009, 15, 1273–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmeister, K.M.; Falet, H. Platelet clearance by the hepatic Ashwell-Morrell receptor: Mechanisms and biological significance. Thromb. Res. 2016, 141 (Suppl. 2), S68–S72. [Google Scholar] [CrossRef] [Green Version]
- Grozovsky, R.; Begonja, A.J.; Liu, K.; Visner, G.; Hartwig, J.H.; Falet, H.; Hoffmeister, K.M. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat. Med. 2015, 21, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; van der Wal, D.E.; Zhu, G.; Xu, M.; Yougbare, I.; Ma, L.; Vadasz, B.; Carrim, N.; Grozovsky, R.; Ruan, M.; et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat. Commun. 2015, 6, 7737. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Liu, X.; Li, X.; Zhang, X.; Han, P.; Zhou, H.; Shao, L.; Hou, Y.; Min, Y.; Kong, Z.; et al. CD8(+) T cells induce platelet clearance in the liver via platelet desialylation in immune thrombocytopenia. Sci. Rep. 2016, 6, 27445. [Google Scholar] [CrossRef] [Green Version]
- Bigot, P.; Auffret, M.; Gautier, S.; Weinborn, M.; Ettahar, N.; Coupé, P. Unexpected platelets elevation in a patient with idiopathic thrombocytopenia treated with oseltamivir for influenza infection. Fundam. Clin. Pharmacol. 2016, 30, 483–485. [Google Scholar] [CrossRef]
- Shao, L.; Wu, Y.; Zhou, H.; Qin, P.; Ni, H.; Peng, J.; Hou, M. Successful treatment with oseltamivir phosphate in a patient with chronic immune thrombocytopenia positive for anti-GPIb/IX autoantibody. Platelets 2015, 26, 495–497. [Google Scholar] [CrossRef]
- Shaim, H.; Mccaffrey, P.; Trieu, J.A.; DeAnda, A.; Yates, S.G. Evaluating the effects of oseltamivir phosphate on platelet counts: A retrospective review. Platelets 2020, 31, 1080–1084. [Google Scholar] [CrossRef]
- Sun, L.; Wang, J.; Shao, L.; Yuan, C.; Zhao, H.; Li, D.; Wang, Z.; Han, P.; Yu, Y.; Xu, M.; et al. Dexamethasone plus oseltamivir versus dexamethasone in treatment-naive primary immune thrombocytopenia: A multicentre, randomised, open-label, phase 2 trial. Lancet Haematol. 2021, 8, e289–e298. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Burrows, P.D.; Wang, J.-Y. B Cell Development and Maturation. Adv. Exp. Med. Biol. 2020, 1254, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Mittelbrunn, M.; de la Fuente, H.; Pérez-Martínez, M.; García-Pérez, A.; Ariza-Veguillas, A.; Malavasi, F.; Zubiaur, M.; Sánchez-Madrid, F.; Sancho, J. Antigen-induced clustering of surface CD38 and recruitment of intracellular CD38 to the immunologic synapse. Blood 2008, 111, 3653–3664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Weers, M.; Tai, Y.-T.; Van Der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.H.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, F.; Kwa, F.A.A.; Burchall, G.; Jackson, D.E. New generation drugs for treatment of multiple myeloma. Drug Discov. Today 2019, 25, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Tsykunova, G.; Holme, P.A.; Tran, H.T.T.; Tvedt, T.H.A.; Munthe, L.A.; Michel, M.; Frederiksen, H.; Bussel, J.B.; Kuter, D.J.; Ghanima, W. Daratumumab as a Treatment for Adult Immune Thrombocytopenia: A Phase II Study with Safety Run-in (the DART Study). Blood 2021, 138 (Suppl. 1), 2088. [Google Scholar] [CrossRef]
- Crickx, E.; Audia, S.; Robbins, A.; Boutboul, D.; Comont, T.; Cheminant, M.; Oksenhendler, E.; Godeau, B.; Michel, M.; Mahevas, M. Daratumumab, an original approach for treating multi-refractory autoimmune cytopenia. Haematologica 2021, 106, 3198–3201. [Google Scholar] [CrossRef]
- Feist, E.; Burmester, G.-R.; Krüger, E. The proteasome—Victim or culprit in autoimmunity. Clin. Immunol. 2016, 172, 83–89. [Google Scholar] [CrossRef]
- Li, G.; Wang, S.; Li, N.; Liu, Y.; Feng, Q.; Zuo, X.; Li, X.; Hou, Y.; Shao, L.; Ma, C.; et al. Proteasome Inhibition with Bortezomib Induces Apoptosis of Long-Lived Plasma Cells in Steroid-Resistant or Relapsed Immune Thrombocytopaenia. Thromb. Haemost. 2018, 118, 1752–1764. [Google Scholar] [CrossRef]
- Mazepa, M.A.; Raval, J.; Moll, S.; Ma, A.; Park, Y.A. Bortezomib induces clinical remission and reduction of ADAMTS13 inhibitory antibodies in relapsed refractory idiopathic thrombotic thrombocytopenic purpura. Br. J. Haematol. 2014, 164, 900–902. [Google Scholar] [CrossRef] [Green Version]
- Fadlallah, J.; Michel, M.; Crickx, E.; Limal, N.; Costedoat, N.; Malphettes, M.; Fieschi, C.; Galicier, L.; Oksenhendler, E.; Godeau, B.; et al. Bortezomib and dexamethasone, an original approach for treating multi-refractory warm autoimmune haemolytic anaemia. Br. J. Haematol. 2019, 187, 124–128. [Google Scholar] [CrossRef]
- Beckman, J.; Rollins-Raval, M.; Raval, J.; Park, Y.A.; Mazepa, M.; Ma, A. Bortezomib for Refractory Immune-Mediated Thrombocytopenia Purpura. Am. J. Ther. 2018, 25, e270–e272. [Google Scholar] [CrossRef] [PubMed]
- Conti, F.; Gottardi, F.; Moratti, M.; Belotti, T.; Ferrari, S.; Selva, P.; Bassi, M.; Zama, D.; Pession, A. Refractory immune thrombocytopenia successfully treated with bortezomib in a child with 22q11.2 deletion syndrome, complicated by Evans syndrome and hypogammaglobulinemia. Platelets 2022, 33, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.W.B.; Lowe, E.; Anderl, J.L.; Fan, A.; Muchamuel, T.; Bowers, S.; Moebius, D.C.; Kirk, C.; McMINN, D.L. Required Immunoproteasome Subunit Inhibition Profile for Anti-Inflammatory Efficacy and Clinical Candidate KZR-616 ((2S,3R)-N-((S)-3-(Cyclopent-1-en-1-yl)-1-((R)-2-methyloxiran-2-yl)-1-oxopropan-2-yl)-3-hydroxy-3-(4-methoxyphenyl)-2-((S)-2-(2-morpholinoacetamido)propanamido)propenamide). J. Med. Chem. 2018, 61, 11127–11143. [Google Scholar] [PubMed]
- Patel, V.L.; Schwartz, J.; Bussel, J.B. The effect of anti-CD40 ligand in immune thrombocytopenic purpura. Br. J. Haematol. 2008, 141, 545–548. [Google Scholar] [CrossRef]
- Kuwana, M.; Nomura, S.; Fujimura, K.; Nagasawa, T.; Muto, Y.; Kurata, Y.; Tanaka, S.; Ikeda, Y. Effect of a single injection of humanized anti-CD154 monoclonal antibody on the platelet-specific autoimmune response in patients with immune thrombocytopenic purpura. Blood 2004, 103, 1229–1236. [Google Scholar] [CrossRef] [Green Version]
- Spolski, R.; Leonard, W.J. Interleukin-21: A double-edged sword with therapeutic potential. Nat. Rev. Drug Discov. 2014, 13, 379–395. [Google Scholar] [CrossRef]
- Peeters, K.; Loyen, S.; Van Kerckhoven, S.; Stoffels, K.; Hoylaerts, M.F.; van Geet, C.; Freson, K. Thrombopoietic effect of VPAC1 inhibition during megakaryopoiesis. Br. J. Haematol. 2010, 151, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Zhu, H.-L.; Li, S.-X.; Lu, X.-C.; Zhai, B.; Guo, B.; Yao, S.-Q.; Liu, Y. Efficacy of Amifostine in Treating Patients with Idiopathic Thrombocytopenia Purpura. Cell Biophys. 2011, 59, 7–12. [Google Scholar] [CrossRef]
- Podolanczuk, A.; Lazarus, A.H.; Crow, A.R.; Grossbard, E.; Bussel, J.B. Of mice and men: An open-label pilot study for treatment of immune thrombocytopenic purpura by an inhibitor of Syk. Blood 2009, 113, 3154–3160. [Google Scholar] [CrossRef] [Green Version]
- Han, P.; Hou, Y.; Zhao, Y.; Liu, Y.; Yu, T.; Sun, Y.; Wang, H.; Xu, P.; Li, G.; Sun, T.; et al. Low-dose decitabine modulates T-cell homeostasis and restores immune tolerance in immune thrombocytopenia. Blood 2021, 138, 674–688. [Google Scholar] [CrossRef]
- Zhao, H.-Y.; Ma, Y.-H.; Li, D.-Q.; Sun, T.; Li, L.-Z.; Li, P.; Liu, X.-G.; Zhou, H.; Hou, Y.; Liu, Y.; et al. Low-dose chidamide restores immune tolerance in ITP in mice and humans. Blood 2019, 133, 730–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.R.; Hartwig, J.H.; Italiano, J.E. The biogenesis of platelets from megakaryocyte proplatelets. J. Clin. Investig. 2005, 115, 3348–3354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, J.L.; Shivdasani, R.A.; Boers, C.; Hartwig, J.H.; Italiano, J.E. Mechanisms of organelle transport and capture along proplatelets during platelet production. Blood 2005, 106, 4066–4075. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Dong, T.; Li, P.; Wu, M.X. Noninvasive low-level laser therapy for thrombocytopenia. Sci. Transl. Med. 2016, 8, 349ra101. [Google Scholar] [CrossRef] [Green Version]
- Kuffler, D.P. Photobiomodulation in promoting wound healing: A review. Regen. Med. 2016, 11, 107–122. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | Absorption | Vd | PB | Elimination | t1/2 | Clearance |
|---|---|---|---|---|---|---|
| Prednisone | Cmax: n.a. Tmax: 2 h | 29.3 L (0.15 mg/kg dose); 44.2 L (0.30 mg/kg dose) | <50% | Urine | 2–3 h | 0.066 ± 0.12 L/h/kg (5.5 µg/h/kg dose) |
| Prednisolone | Cmax: 113–1343 ng/mL Tmax: 1.0–2.6 h Bioavailability: 70% | 29.3 L (0.15 mg/kg dose); 44.2 L (0.30 mg/kg dose) | 65–91% | Urine | 2–3 h | 0.09 L/kg/h (0.15 mg/kg dose) 0.12 L/kg/h (0.30 mg/kg dose) |
| Methylprednisolone | Bioavailability: 89.9% | 1.38 L/kg | 76.8% | Urine | 2.3 h | 336 mL/h/kg |
| Dexamethasone | Cmax: 13.9 ± 6.8 ng/mL Tmax: 2.0 ± 0.5 h (1.5 mg oral dose) Bioavailability: 70–78% | 51.0 L (1.5 mg dose) | 77% | <10% urine | 6.6 h | 15.6 ± 4.9 L/h (1.5 mg dose) |
| Romiplostim | Tmax: median 14 h (range: 7–50 h) | 10 µg/kg (48.2 mL/kg) | n.a. | Renal (high doses), c-Mpl receptors (low doses) | median 3.5 d (range 1–34 d) | n.a. |
| Eltrombopag | Total absorption: ≥52% (75 mg oral dose) Tmax: 2–6 h | In blood cells, concentrations are 50–79% of those in plasma | >99% | Feces (59%), urine (31%) | 26–35 h | n.a. |
| Avatrombopag | Cmax: 166 ng/mL (40 mg dose) Tmax: 5–8 h | 180 L (CV 25%) | >96% | Feces (88%) urine (6%) | 19 h | 6.9 L/h (CV 29%) |
| Rituximab | Cmax: 157 ± 46 and 183 ± 55 ng/mL after 1st and 2nd infusion of a 500 mg dose (RA) | 3.1 L (RA) | n.a. | Reticuloendothelial system | median 22 d (range 16–28 d) | 0.335 L/d (RA) |
| Fostamatinib | Proportional exposure up to doses of 200 mg BD Tmax: 1.5 h Bioavailability: 55% | 400 L | 98.3% | Feces (80%), urine (20%) | 15 h | 300 mL/min |
| Drug | Pharmacodynamics |
|---|---|
| Prednisone | Inhibits pro-inflammatory signals and promotes anti-inflammatory signals by binding to the glucocorticoid receptor. Its duration of action is short, in agreement with its half-life of 2–3 h |
| Prednisolone | Inhibits pro-inflammatory signals and promotes anti-inflammatory signals by binding to the glucocorticoid receptor. Its duration of action is short, in agreement with its half-life of 2–3 h |
| Methylprednisolone | Inhibits pro-inflammatory signals and promotes anti-inflammatory signals by binding to the glucocorticoid receptor. Its duration of action is short, in agreement with its half-life of 2.3 h |
| Dexamethasone | Inhibits pro-inflammatory signals and promotes anti-inflammatory signals by binding to the glucocorticoid receptor. Its duration of action is somewhat longer than that of other glucocorticoids, in agreement with its longer half-life |
| TPOa (Romiplostim, Eltrombopag, Avatrombopag) | Drug dose-dependency has been reported for platelet increase. The extent of the effect may vary among patients, which means that dose individualization is required. |
| Rituximab | Binding to the CD20 antigen on mature B cell surfaces induces the selective killing of B cells. More details on pharmacodynamics are available for other autoimmune conditions. In RA, the near-complete depletion of peripheral B cells is achieved within 2 weeks after the first dose, which may be sustained for ≥6 months |
| Fostamatinib | The active metabolite R406 inhibits signal transduction by Fcγ receptors involved in the antibody-mediated destruction of platelets by immune cells, thus increasing platelet counts. R406 inhibits T and B lymphocyte activation by T-cell and B-cell receptors. The inhibition of Fc receptor signaling suppresses dendritic cell maturation and antigen presentation. Production of the major inflammatory mediators and cytokines is also reduced |
| Drug | Dose | Onset of Action/ Peak Response (d) | Overall Response | Side Effects |
|---|---|---|---|---|
| Prednisone or prednisolone | 0.5–2 mg/kg/d p.o. for 2–3 wk, gradually tapered next 6–8 wk. Rapid 2 wk tapering in case of no response | 4–14/7–28 | In the short term: 60–80%. Sustained after drug discontinuation: 30–50%. | Weight gain, cushingoid phenotype, infection, hypertension, hyperglycemia, hirsutism, cataracts, mood disorders |
| Methylprednisolone | 1 g/d i.v. for 3 d (or 15 mg/kg/d) | 2–14/4–28 | Similar to those of prednisone or prednisolone | Similar to those of prednisone or prednisolone |
| Dexamethasone | 40 mg p.o. or i.v. from D1 to D4, up to 3–4 cycles, each cycle after 2–4 wk | 2–14/4–28 | More rapid response than prednisone, but similar in the long term | Weight gain and cushingoid phenotype more attenuated, and infection rate lower than that observed with prednisone |
| IVIG a | 1 g/kg/d i.v. for 1–2 d or 400 mg/kg/d i.v. for 5 d | 1–3/2–7 | n.a. | Headache, anaphylaxis |
| Romiplostim | 1–10 μg/kg/wk s.c., initially with the minimum dose. Titration must be according to platelet response | 7–14/16–60 | 70–80% (maintenance therapy required)Sustained after discontinuation: 10–30% | Pain at injection site, body ache, headache, thrombosis, bone marrow fibrosis, reticulin increase |
| Eltrombopag | 25–75 mg/d p.o. (2 h before or 4 h after food or polyanion (Ca, Fe)-containing products) | 7–14/16–90 | Similar to romiplostim | Transaminitis, gastrointestinal discomfort, thrombosis, bone-marrow fibrosis |
| Avatrombopag | 5–40 mg/d p.o. | 3–5/10–13 | 65% on D8 of treatment | Thrombosis (rarely), arthralgia, headache |
| Rituximab | 375 or 100 mg/m2/wk i.v. for 4 wk | 7–56/14–180 | At short term: 60–80%. At long term (2–5 yr): 20–30% | Infusion related reactions: fever, chills, rigor. At long-term: hypogammaglobulinemia, infection, reactivation of hepatitis B, PML |
| Fostamatinib | 50–150 mg p.o. BD | 7–14/n.a. | 18–43% | Diarrhea, hypertension, infection |
| Gaps to Fill Regarding ITP Treatment |
|---|
| Increase in the efficacy of new drugs, redirecting research toward targets where modulation results in a more durable improvement |
| Increased sustained response to first-line treatment |
| New immune system modulation-based treatments may shed light on the mechanisms underlying distorted immune tolerance, thus allowing the use (or future design) of more specific drugs |
| Analyzing the efficacy of new drugs targeting specific mechanisms should shed light on their relevance, unveiling those pathways on which therapies should focus |
| Type of Approach | Mechanism of Action | Drug-Development Stage (Finished/Ongoing Trials) |
|---|---|---|
| TPO-RAs | Increase in megakaryocytes and subsequent platelet production | Hetrombopag
|
| Syk inhibition | Inhibition of macrophage phagocytosis and the subsequent decrease in platelet destruction | HMPL-523
|
| BTK inhibition | Inhibition of macrophage phagocytosis and the subsequent decrease in platelet destruction | Rilzabrutinib
|
| FcRn blockers | Increase in antiplatelet autoantibody clearance, thus decreasing the peripheral platelet destruction and immune response against megakaryocytes | Efgartigimod
|
| Staphylococcal protein A | Inhibition of macrophage phagocytosis by preventing FcγRIII (CD16) participation | PRTX-100
|
| Immunoglobulin-based drugs | Decrease in platelet destruction by splenic macrophages and increase in antiplatelet autoantibody clearance by FcRn saturation | M254
|
| Complement inhibition | Decrease in complement-dependent cytotoxicity | Sutimlimab
|
| Neuraminidase inhibition | Inhibition of platelet desialylation prevents liver platelet destruction | Oseltamivir
|
| Anti-CD38 inhibition | Reduction of autoantibody production and immune imbalance by the inhibition of CD38 on the surface of plasma cells and other immune cells | Daratumumab
|
| Proteasome inhibition | Decreased autoantibody production by preventing long-lived plasma cells | Bortezomib
|
| Interference with the interaction between CD40 and CD154 | Partial resolution of the imbalance between cellular and humoral adaptive immunity processes | Letolizumab
|
| Drug | Absorption | Vd | PB | Elimination | t1/2 | Clearance |
|---|---|---|---|---|---|---|
| Hetrombopag | Cmax: 24 ng/mL (5 mg oral dose) Tmax: 8 h | n.a. | n.a. | Feces (62.5) | 11.9–40 h | 15.6 L/h |
| HMPL-523 | Cmax: dose-dependent until 800 mg dose Tmax: 3–6 h | n.a. | n.a. | n.a. | 9.8–13.5 h (dose range tested: 100–800 mg) | n.a. |
| Rilzabrutinib | Cmax: 91 ng/mL (300 mg dose) Tmax: 1.5 h | 4910 L (300 mg dose) | n.a. | n.a. | 1.3–3.9 h (dose range tested: 50–1200 mg) | 1.6%/h (occupancydecay rate) |
| Efgartigimod | Exposure increases proportionally up to 50 mg/kg | 15–20 L | n.a. | Proteolytic enzymes (urine < 0.1%) | 80–120 h | n.a. |
| Rozanolixizumab | (i.v.) Cmax: 89–154 µg/mL (4–7 mg/kg dose) Tmax: 1–2.5 h (s.c.) Cmax: 12 µg/mL (7 mg/kg dose) Tmax: 48 h | n.a. | n.a. | Predominantly by reticuloendothelial macrophages | n.a. | n.a. |
| Sutimlimab | Exposure increases proportionately with increasing dosage. Steady-state achieved by 7th week | 5.8 L | n.a. | Predominantly by reticuloendothelial macrophages | 21 d (6.5–7.5 g i.v. dose) | 0.14 L/d |
| Oseltamivir | Cmax: 65 ng/mL (oral 75 mg BD) | 23–26 L | 42% | Renal excretion (>90%), feces (<20%) | 1–3 h (oral 75 mg BD) | 18.8 L/h |
| Daratumumab | Cmax: 592 µg/mL (1800 mg s.c. dose) | 5.2 L (central compartment), 3.8 L (peripheral compartment) | n.a. | Predominantly by reticuloendothelial macrophages | 20 d | 119 mL/d |
| Bortezomib | Cmax: 57–112 ng/mL (1–1.3 mg/m2 i.v. dose) | 498–1884 L/m2 (1–1.3 mg/m2 i.v. dose) | 83% | Renal and hepatic routes | 40–193 h (1 mg/m2 i.v. dose) | 102–112 L/h (1–1.3 mg/m2 i.v. dose) |
| Drug | Pharmacodynamics |
|---|---|
| Hetrombopag | Dynamic changes in blood platelets are best characterized by four-transit compartment models.Single dose (5–40 mg): a consistent increase in platelet counts is observed after D4, the maximal effect being reported on D10. The thrombopoietic activity is dose-dependent, with platelet counts increasing twofold with a dose of 40 mg.Repeated daily doses (2.5–7.5 mg) for 10 d: an increase in platelet counts starts after 6 d, with the peak at 12–14 d. Eighteen days after the last dose, platelet counts are still 18.8% and 32.2% above baseline in those patients administered 5 and 7.5 mg doses, respectively |
| HMPL-523 | Inhibits anti-IgE-induced basophil (CD63+) dose-dependently, with an EC50 of 47.70 ng/mL |
| Rilzabrutinib | Occupancy of BTK occurs rapidly and dose-dependently, with doses ≥ 150 mg and the maximum occupancy (>90%) within the first 4 h. A 30–35% reduction in occupancy is observed with all doses between hours 4 and 24. IC50 = 1.3 nM |
| Orelabrutinib | Near-complete BTK occupancy is achieved at doses ≥ 50 mg (Cmax to achieve EC99: 300 ng/mL), the effect being sustained for 24 h post-dosing, which is consistent with the covalent binding mode of action. The IC50 value is 1.6 nM |
| Efgartigimod | The pharmacologic effect is exerted by reducing the circulating levels of autoantibodies. Because efgartigimod also reduces the level of the rest of the IgGs, patients may be at greater risk of infection. Treatment should not be initiated in patients with an active infection. Accordingly, efgartigimod discontinuation should be considered in patients who develop a serious infection during therapy |
| Rozanolixizumab | A reduction in total serum IgG concentration over time is observed with both i.v. and s.c. administration, in a dose-dependent manner. Similar maximum reductions are observed via i.v. or s.c. The greatest IgG reduction is reported to be seen by days 7 to 10, the baseline level being restored by day 57. Reductions in the serum IgG on day 10 are 14.5, 33.4, and 47.6%, with 1, 4, and 7 mg/kg doses in the case of i.v. administration, and 16.8, 25.9, and 43.4% when s.c. administration is used |
| Sutimlimab | After a single i.v. injection, > 90% inhibition of the complement pathway is observed, which is sustained for concentrations of sutimlimab ≥100 µg/mL. The impairment in complement-mediated immune response makes necessary appropriate vaccination against encapsulated bacteria at least 2 weeks prior to treatment initiation. Since patients are at a higher risk of serious infections, they have to be closely monitored throughout therapy |
| Oseltamivir | Once hydrolyzed to its active metabolite oseltamivir carboxylate, the drug exerts neuraminidase inhibitor activity, via competitive inhibition of the activity of neuraminidase upon sialic acid, which is found on glycoproteins on the surface of platelets. By blocking the activity of the enzyme, platelet destruction in the liver may be prevented |
| Daratumumab | Apoptosis is induced in CD38 highly expressing cells. The long duration of action allows dosing on a weekly basis. It is advisable to counsel patients regarding the risk of neutropenia, thrombocytopenia, embryo-fetal toxicity, hypersensitivity, and interference with cross-matching and red blood cell antibody screening |
| Bortezomib | The target is the ubiquitin-proteasome pathway, which regulates intracellular concentrations of proteins and promotes protein degradation, and may be dysregulated in pathological conditions. By reversibly inhibiting proteasome, proteasome-mediated proteolysis is prevented. Inhibition occurs in a dose-dependent manner |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mingot-Castellano, M.E.; Bastida, J.M.; Caballero-Navarro, G.; Entrena Ureña, L.; González-López, T.J.; González-Porras, J.R.; Butta, N.; Canaro, M.; Jiménez-Bárcenas, R.; Gómez del Castillo Solano, M.d.C.; et al. Novel Therapies to Address Unmet Needs in ITP. Pharmaceuticals 2022, 15, 779. https://doi.org/10.3390/ph15070779
Mingot-Castellano ME, Bastida JM, Caballero-Navarro G, Entrena Ureña L, González-López TJ, González-Porras JR, Butta N, Canaro M, Jiménez-Bárcenas R, Gómez del Castillo Solano MdC, et al. Novel Therapies to Address Unmet Needs in ITP. Pharmaceuticals. 2022; 15(7):779. https://doi.org/10.3390/ph15070779
Chicago/Turabian StyleMingot-Castellano, María Eva, José María Bastida, Gonzalo Caballero-Navarro, Laura Entrena Ureña, Tomás José González-López, José Ramón González-Porras, Nora Butta, Mariana Canaro, Reyes Jiménez-Bárcenas, María del Carmen Gómez del Castillo Solano, and et al. 2022. "Novel Therapies to Address Unmet Needs in ITP" Pharmaceuticals 15, no. 7: 779. https://doi.org/10.3390/ph15070779
APA StyleMingot-Castellano, M. E., Bastida, J. M., Caballero-Navarro, G., Entrena Ureña, L., González-López, T. J., González-Porras, J. R., Butta, N., Canaro, M., Jiménez-Bárcenas, R., Gómez del Castillo Solano, M. d. C., Sánchez-González, B., Pascual-Izquierdo, C., & on behalf of the GEPTI. (2022). Novel Therapies to Address Unmet Needs in ITP. Pharmaceuticals, 15(7), 779. https://doi.org/10.3390/ph15070779