
Design and Anticancer Properties of New Water-Soluble Ruthenium–Cyclopentadienyl Complexes

 ,
,  , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. NMR, FT-IR Analysis, and Mass Spectral Analysis
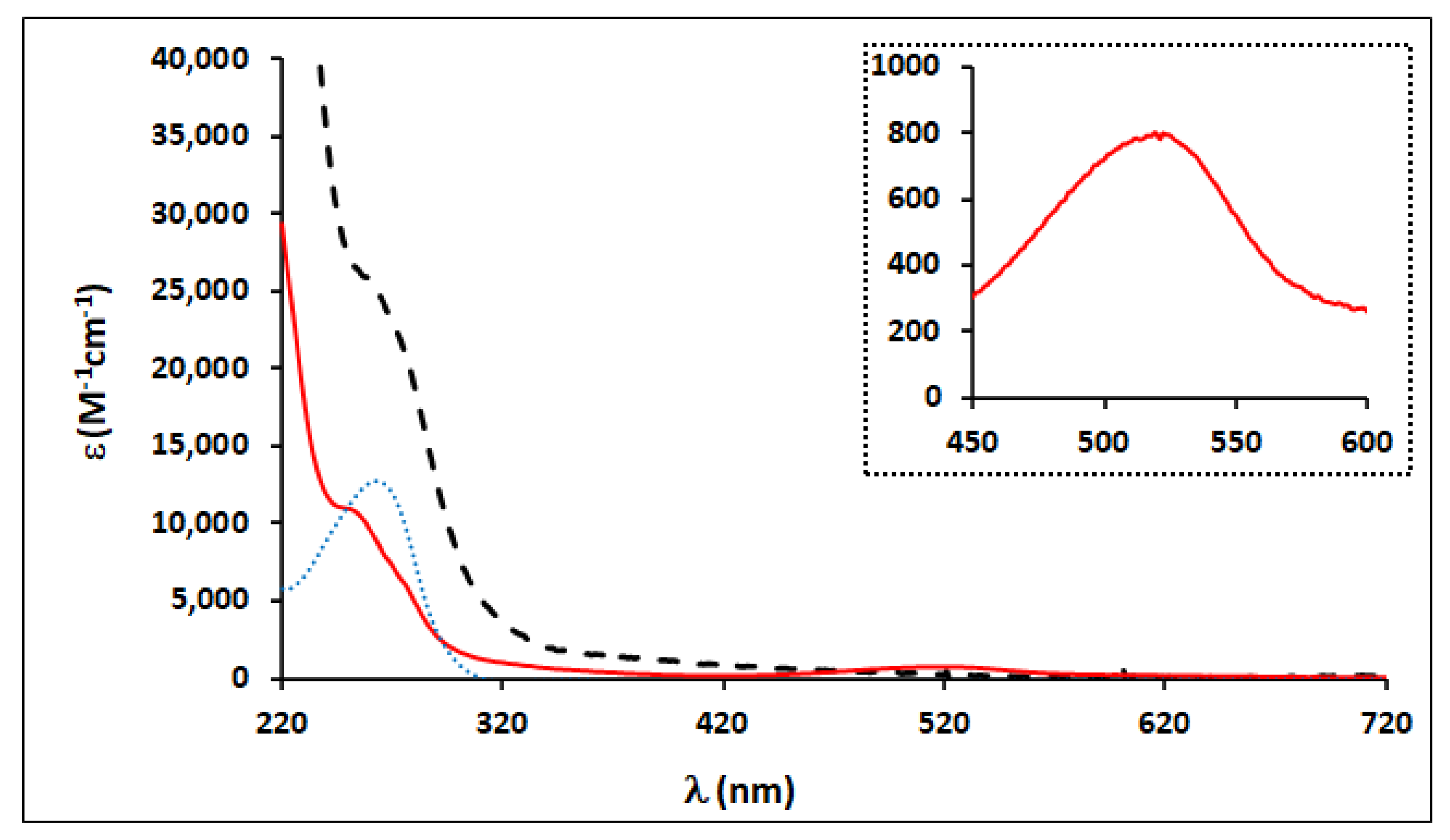
2.2. Electronic Absorption Spectroscopy
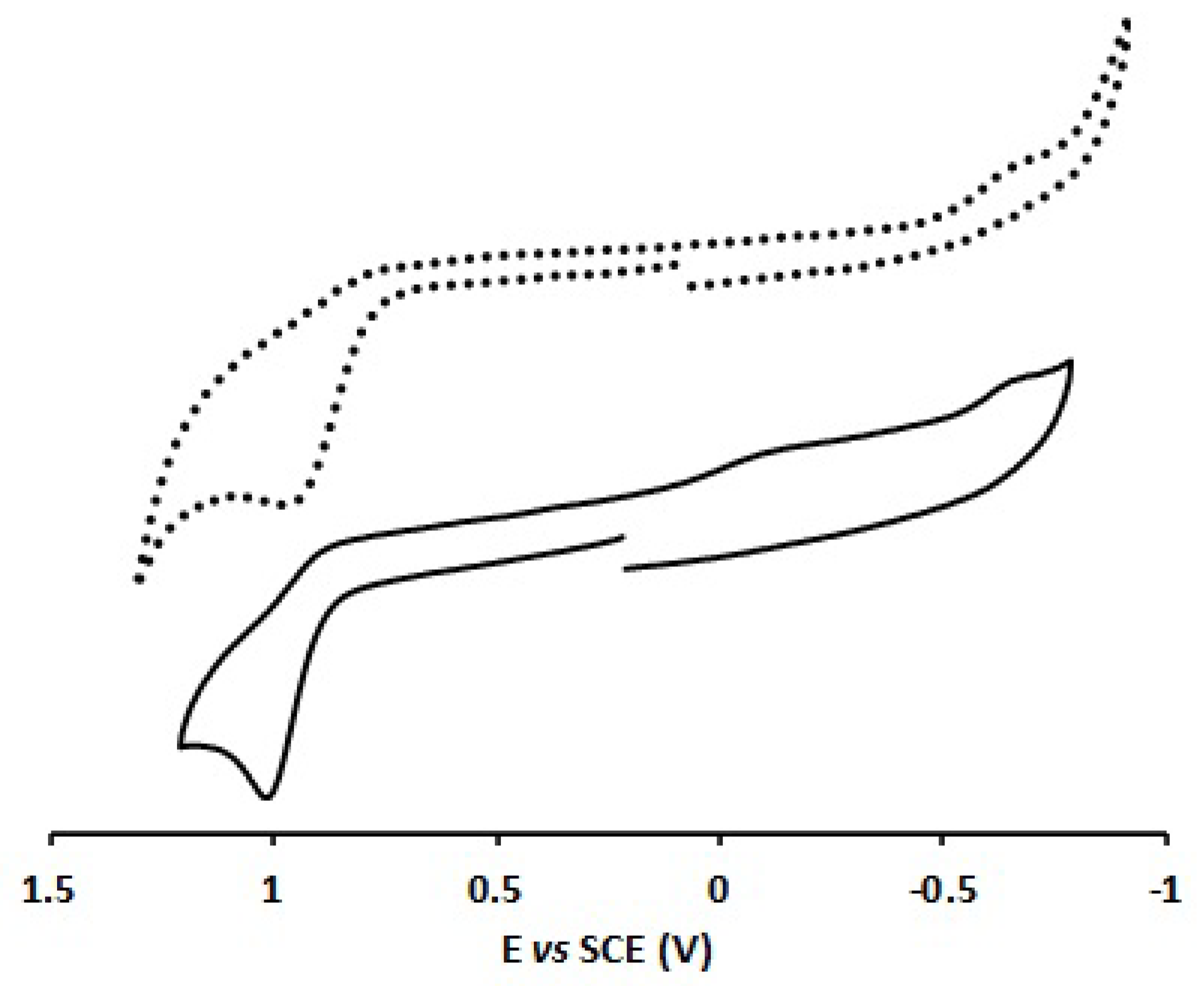
2.3. Electrochemical Characterization of Complexes
2.4. Complex Solubility and Stability in Aqueous Solutions and Reactivity towards O2 and Estimation of Lipophilicity
2.5. Cytotoxicity in Human Cell Lines
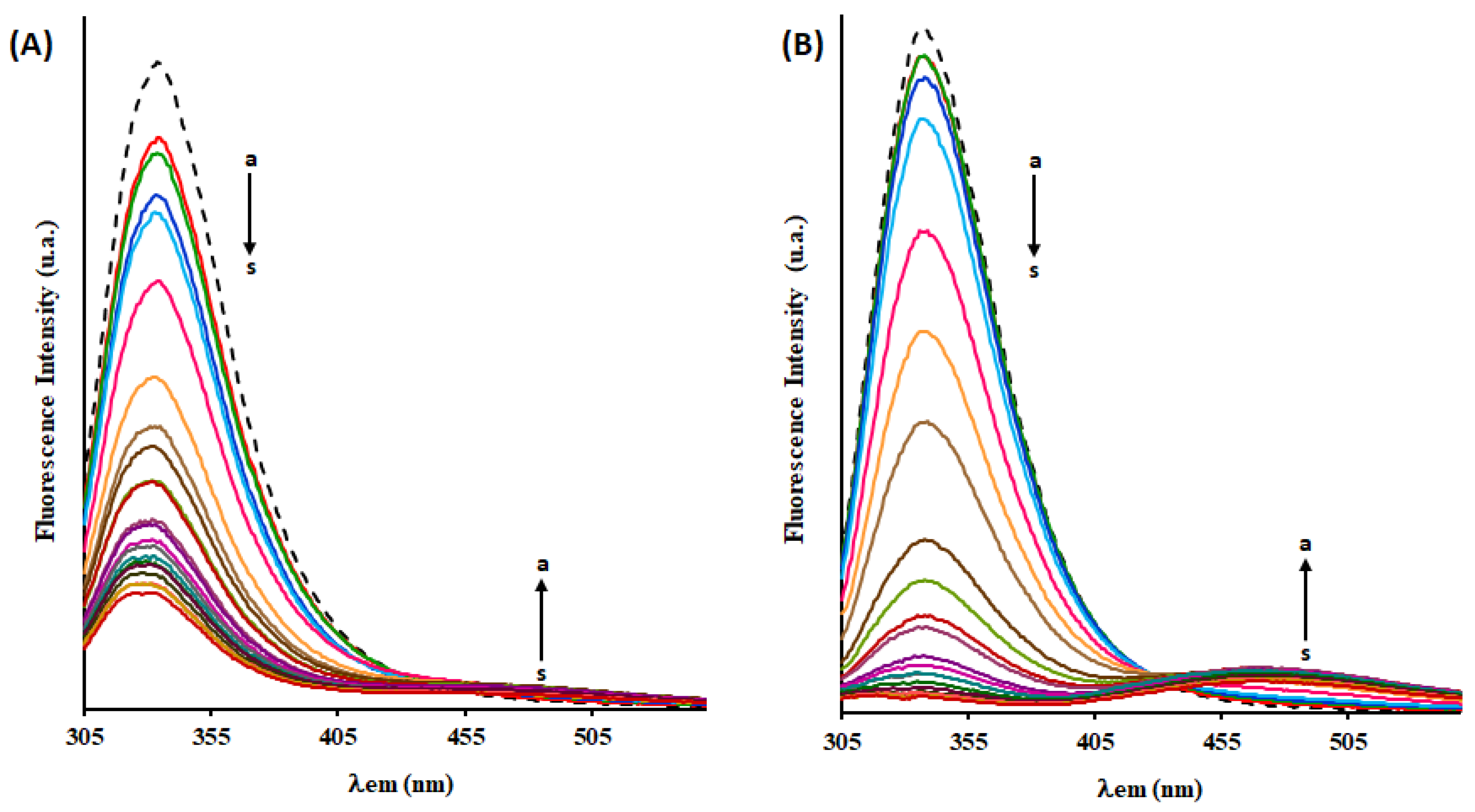
2.6. Fluorescence Quenching of HSA by Complex [Ru(η5-C5H5)(mTPPMS)(bopy)][CF3SO3] (6)
3. Materials and Methods
3.1. Materials and General Procedures
3.2. Chemical Synthesis
3.2.1. General Procedure for the Synthesis of [RuCp(mTPPMSNa)2L][CF3SO3] Complexes (1–5)
3.2.2. General Procedure for the Synthesis of [RuCp(mTPPMSNa)L][CF3SO3] Complexes (6–8)
3.3. Cyclic Voltammetry
3.4. Stability in Aqueous Medium and Air
3.5. Octanol-Water Partition Coefficients
3.6. In Vitro Anticancer Activity
3.7. Preparations of the Stock Solutions for Fluorescence Spectroscopic Measurements
3.8. Fluorescence Spectroscopic Measurements
3.9. Site-Marker Competitive Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Gust, R. Metal N-Heterocyclic Carbene Complexes as Potential Antitumor Metallodrugs. Chem. Soc. Rev. 2012, 42, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic Anticancer Compounds. J. Med. Chem. 2010, 54, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Fong, T.T.-H.; Lok, C.-N.; Chung, C.Y.-S.; Fung, Y.-M.E.; Chow, P.-K.; Wan, P.-K.; Che, C.-M. Cyclometalated Palladium(II) N-Heterocyclic Carbene Complexes: Anticancer Agents for Potent In Vitro Cytotoxicity and In Vivo Tumor Growth Suppression. Angew. Chem. 2016, 128, 12114–12118. [Google Scholar] [CrossRef]
- Zeng, L.; Gupta, P.; Chen, Y.; Wang, E.; Ji, L.; Chao, H.; Chen, Z.S. The Development of Anticancer Ruthenium(II) Complexes: From Single Molecule Compounds to Nanomaterials. Chem. Soc. Rev. 2017, 46, 5771–5804. [Google Scholar] [CrossRef]
- Liu, Z.; Romero-Canelõn, I.; Qamar, B.; Hearn, J.M.; Habtemariam, A.; Barry, N.P.E.; Pizarro, A.M.; Clarkson, G.J.; Sadler, P.J. The Potent Oxidant Anticancer Activity of Organoiridium Catalysts. Angew. Chem. Int. Ed. 2014, 53, 3941–3946. [Google Scholar] [CrossRef] [Green Version]
- Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.; Marzano, C. Advances in Copper Complexes as Anticancer Agents. Chem. Rev. 2013, 114, 815–862. [Google Scholar] [CrossRef]
- Dyson, P.J.; Sava, G. Metal-Based Antitumour Drugs in the Post Genomic Era. Dalton Trans. 2006, 16, 1929–1933. [Google Scholar] [CrossRef]
- Jakupec, M.A.; Galanski, M.; Arion, V.B.; Hartinger, C.G.; Keppler, B.K. Antitumour Metal Compounds: More than Theme and Variations. Dalton Trans. 2007, 2, 183–194. [Google Scholar] [CrossRef]
- Liu, J.; Lai, H.; Xiong, Z.; Chen, B.; Chen, T. Functionalization and Cancer-Targeting Design of Ruthenium Complexes for Precise Cancer Therapy. Chem. Commun. 2019, 55, 9904–9914. [Google Scholar] [CrossRef]
- Thota, S.; Rodrigues, D.A.; Crans, D.C.; Barreiro, E.J. Ru(II) Compounds: Next-Generation Anticancer Metallotherapeutics? J. Med. Chem. 2018, 61, 5805–5821. [Google Scholar] [CrossRef]
- Broomfield, L.M.; Alonso-Moreno, C.; Martin, E.; Shafir, A.; Posadas, I.; Ceña, V.; Castro-Osma, J.A. Aminophosphine Ligands as a Privileged Platform for Development of Antitumoral Ruthenium(II) Arene Complexes. Dalton Trans. 2017, 46, 16113–16125. [Google Scholar] [CrossRef]
- Meier-Menches, S.M.; Gerner, C.; Berger, W.; Hartinger, C.G.; Keppler, B.K. Structure–Activity Relationships for Ruthenium and Osmium Anticancer Agents—Towards Clinical Development. Chem. Soc. Rev. 2018, 47, 909–928. [Google Scholar] [CrossRef]
- Sudhindra, P.; Ajay Sharma, S.; Roy, N.; Moharana, P.; Paira, P. Recent Advances in Cytotoxicity, Cellular Uptake and Mechanism of Action of Ruthenium Metallodrugs: A Review. Polyhedron 2020, 192, 114827. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Emadi, A. Ruthenium-Based Chemotherapeutics: Are They Ready for Prime Time? Cancer Chemother. Pharmacol. 2010, 66, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Rademaker-Lakhai, J.M.; Van Den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H.M. A Phase I and Pharmacological Study with Imidazolium-Trans-DMSO-Imidazole-Tetrachlororuthenate, a Novel Ruthenium Anticancer Agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef] [Green Version]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; Van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H.M. Phase I/II Study with Ruthenium Compound NAMI-A and Gemcitabine in Patients with Non-Small Cell Lung Cancer after First Line Therapy. Investig. New Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef] [Green Version]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, A New Redox-Active Anticancer Agent—Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Zorbas-Seifried, S.; Jakupec, M.A.; Kynast, B.; Zorbas, H.; Keppler, B.K. From Bench to Bedside—Preclinical and Early Clinical Development of the Anticancer Agent Indazolium Trans-[Tetrachlorobis(1H-Indazole)Ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891–904. [Google Scholar] [CrossRef]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the First Ruthenium-Based Anticancer Drug on the Edge to Clinical Application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef] [Green Version]
- Alessio, E.; Messori, L. NAMI-A and KP1019/1339, Two Iconic Ruthenium Anticancer Drug Candidates Face-to-Face: A Case Story in Medicinal Inorganic Chemistry. Molecules 2019, 24, 1995. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, P.S.; Pichler, V.; Roller, A.; Hejl, M.; Jakupec, M.A.; Kandioller, W.; Keppler, B.K. Improved Reaction Conditions for the Synthesis of New NKP-1339 Derivatives and Preliminary Investigations on Their Anticancer Potential. Dalton Trans. 2014, 44, 659–668. [Google Scholar] [CrossRef]
- Fong, J.; Kasimova, K.; Arenas, Y.; Kaspler, P.; Lazic, S.; Mandel, A.; Lilge, L. A Novel Class of Ruthenium-Based Photosensitizers Effectively Kills in Vitro Cancer Cells and in Vivo Tumors. Photochem. Photobiol. Sci. 2015, 14, 2014–2023. [Google Scholar] [CrossRef]
- Chen, Q.; Ramu, V.; Aydar, Y.; Groenewoud, A.; Zhou, X.Q.; Jager, M.J.; Cole, H.; Cameron, C.G.; McFarland, S.A.; Bonnet, S.; et al. TLD1433 Photosensitizer Inhibits Conjunctival Melanoma Cells in Zebrafish Ectopic and Orthotopic Tumour Models. Cancers 2020, 12, 587. [Google Scholar] [CrossRef] [Green Version]
- Kar, B.; Roy, N.; Pete, S.; Moharana, P.; Paira, P. Ruthenium and Iridium Based Mononuclear and Multinuclear Complexes: A Breakthrough of Next-Generation Anticancer Metallopharmaceuticals. Inorg. Chim. Acta 2020, 512, 119858. [Google Scholar] [CrossRef]
- Fan, C.; Wu, Q.; Chen, T.; Zhang, Y.; Zheng, W.; Wang, Q.; Mei, W. Arene Ruthenium(II) Complexes Induce S-Phase Arrest in MG-63 Cells through Stabilization of c-Myc G-Quadruplex DNA. MedChemComm 2014, 5, 597–602. [Google Scholar] [CrossRef]
- Vock, C.A.; Ang, W.H.; Scolaro, C.; Phillips, A.D.; Lagopoulos, L.; Juillerat-Jeanneret, L.; Sava, G.; Scopelliti, R.; Dyson, P.J. Development of Ruthenium Antitumor Drugs That Overcome Multidrug Resistance Mechanisms. J. Med. Chem. 2007, 50, 2166–2175. [Google Scholar] [CrossRef]
- Lv, G.; Qiu, L.; Li, K.; Liu, Q.; Li, X.; Peng, Y.; Wang, S.; Lin, J. Enhancement of Therapeutic Effect in Breast Cancer with a Steroid-Conjugated Ruthenium Complex. New J. Chem. 2019, 43, 3419–3427. [Google Scholar] [CrossRef]
- Ruiz, J.; Rodríguez, V.; Cutillas, N.; Espinosa, A.; Hannon, M.J. A Potent Ruthenium(II) Antitumor Complex Bearing a Lipophilic Levonorgestrel Group. Inorg. Chem. 2011, 50, 9164–9171. [Google Scholar] [CrossRef]
- Teixeira, R.G.; Belisario, D.C.; Fontrodona, X.; Romero, I.; Tomaz, A.I.; Garcia, M.H.; Riganti, C.; Valente, A. Unprecedented Collateral Sensitivity for Cisplatin-Resistant Lung Cancer Cells Presented by New Ruthenium Organometallic Compounds. Inorg. Chem. Front. 2021, 8, 1983–1996. [Google Scholar] [CrossRef]
- Biancalana, L.; Zacchini, S.; Ferri, N.; Lupo, M.G.; Pampaloni, G.; Marchetti, F. Tuning the Cytotoxicity of Ruthenium(II) Para-Cymene Complexes by Mono-Substitution at a Triphenylphosphine/Phenoxydiphenylphosphine Ligand. Dalton Trans. 2017, 46, 16589–16604. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Berndsen, R.H.; Dubois, M.; Müller, C.; Schibli, R.; Griffioen, A.W.; Dyson, P.J.; Nowak-Sliwinska, P. In Vivo Anti-Tumor Activity of the Organometallic Ruthenium(II)-Arene Complex [Ru(H6-p-Cymene)Cl2(Pta)] (RAPTA-C) in Human Ovarian and Colorectal Carcinomas. Chem. Sci. 2014, 5, 4742–4748. [Google Scholar] [CrossRef] [Green Version]
- Dyson, P.J. Systematic Design of a Targeted Organometallic Antitumour Drug in Pre-Clinical Development. CHIMIA Int. J. Chem. 2007, 61, 698–703. [Google Scholar] [CrossRef]
- Côrte-Real, L.; Mendes, F.; Coimbra, J.; Morais, T.S.; Tomaz, A.I.; Valente, A.; Garcia, M.H.; Santos, I.; Bicho, M.; Marques, F. Anticancer Activity of Structurally Related Ruthenium(II) Cyclopentadienyl Complexes. J. Biol. Inorg. Chem. 2014, 19, 853–867. [Google Scholar] [CrossRef]
- Côrte-Real, L.; Matos, A.P.; Alho, I.; Morais, T.S.; Tomaz, A.I.; Garcia, M.H.; Santos, I.; Bicho, M.P.; Marques, F. Cellular Uptake Mechanisms of an Antitumor Ruthenium Compound: The Endosomal/Lysosomal System as a Target for Anticancer Metal-Based Drugs. Microsc. Microanal. 2013, 19, 1122–1130. [Google Scholar] [CrossRef]
- Morais, T.S.; Valente, A.; Tomaz, A.I.; Marques, F.; Garcia, M.H. Tracking Antitumor Metallodrugs: Promising Agents with the Ru(II)- and Fe(II)-Cyclopentadienyl Scaffolds. Future Med. Chem. 2016, 8, 527–544. [Google Scholar] [CrossRef]
- Morais, T.S.; Silva, T.J.L.; Marques, F.; Robalo, M.P.; Avecilla, F.; Madeira, P.J.A.; Mendes, P.J.G.; Santos, I.; Garcia, M.H. Synthesis of Organometallic Ruthenium(II) Complexes with Strong Activity against Several Human Cancer Cell Lines. J. Inorg. Biochem. 2012, 114, 65–74. [Google Scholar] [CrossRef]
- Gano, L.; Pinheiro, T.; Matos, A.P.; Tortosa, F.; Jorge, T.F.; Gonçalves, M.S.; Martins, M.; Morais, T.S.; Valente, A.; Tomaz, A.I.; et al. Antitumour and Toxicity Evaluation of a Ru(II)-Cyclopentadienyl Complex in a Prostate Cancer Model by Imaging Tools. Anti-Cancer Agents Med. Chem. 2019, 19, 1262–1275. [Google Scholar] [CrossRef]
- Mendes, N.; Tortosa, F.; Valente, A.; Marques, F.; Matos, A.; Morais, T.; Tomaz, A.; Gärtner, F.; Garcia, M. In Vivo Performance of a Ruthenium-Cyclopentadienyl Compound in an Orthotopic 1 Triple Negative Breast Cancer Model. Anti-Cancer Agents Med. Chem. 2016, 16, 126–136. [Google Scholar] [CrossRef]
- Morais, T.S.; Santos, F.C.; Jorge, T.F.; Côrte-Real, L.; Madeira, P.J.A.; Marques, F.; Robalo, M.P.; Matos, A.; Santos, I.; Garcia, M.H. New Water-Soluble Ruthenium(II) Cytotoxic Complex: Biological Activity and Cellular Distribution. J. Inorg. Biochem. 2014, 130, 1–14. [Google Scholar] [CrossRef]
- Mishra, H.; Mukherjee, R. Half-Sandwich H6-Benzene Ru(II) Complexes of Pyridylpyrazole and Pyridylimidazole Ligands: Synthesis, Spectra, and Structure. J. Organomet. Chem. 2006, 691, 3545–3555. [Google Scholar] [CrossRef]
- Hollósy, F.; Lóránd, T.; Örfi, L.; Erös, D.; Kéri, G.; Idei, M. Relationship between Lipophilicity and Antitumor Activity of Molecule Library of Mannich Ketones Determined by High-Performance Liquid Chromatography, ClogP Calculation and Cytotoxicity Test. J. Chromatogr. B 2002, 768, 361–368. [Google Scholar] [CrossRef]
- Gama, S.; Santos, I.; Mendes, F.; Marques, F.; Santos, I.C.; Carvalho, M.F.; Correia, I.; Pessoa, J.C.; Paulo, A. Copper(II) Complexes with Tridentate Pyrazole-Based Ligands: Synthesis, Characterization, DNA Cleavage Activity and Cytotoxicity. J. Inorg. Biochem. 2011, 105, 637–644. [Google Scholar] [CrossRef]
- Pope, A.J.; Bruce, C.; Kysela, B.; Hannon, M.J. Issues Surrounding Standard Cytotoxicity Testing for Assessing Activity of Non-Covalent DNA-Binding Metallo-Drugs. Dalton Trans. 2010, 39, 2772–2774. [Google Scholar] [CrossRef]
- Butsch, K.; Gust, R.; Klein, A.; Ott, I.; Romanski, M. Tuning the Electronic Properties of Dppz-Ligands and Their Palladium(II) Complexes. Dalton Trans. 2010, 39, 4331–4340. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: Boston, MA, USA, 2006. [Google Scholar]
- Sarzehi, S.; Chamani, J. Investigation on the Interaction between Tamoxifen and Human Holo-Transferrin: Determination of the Binding Mechanism by Fluorescence Quenching, Resonance Light Scattering and Circular Dichroism Methods. Int. J. Biol. Macromol. 2010, 47, 558–569. [Google Scholar] [CrossRef]
- Valeur, B.; Berberan-Santos, M.N. Molecular Fluorescence: Principles and Applications; Wiley-VCH: Weinheim, Germany, 2013; ISBN 978-3-527-32837-6. [Google Scholar]
- Morais, T.S.; Santos, F.C.; Corte-Real, L.; Garcia, M.H. Exploring the Effect of the Ligand Design on the Interactions between [Ru(H5-C5H5)(PPh3)(N,O)][CF3SO3] Complexes and Human Serum Albumin. J. Inorg. Biochem. 2013, 129, 94–101. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Zhang, H.M.; Cao, J. Binding of Hydroxylated Single-Walled Carbon Nanotubes to Two Hemoproteins, Hemoglobin and Myoglobin. J. Photochem. Photobiol. B Biol. 2014, 141, 26–35. [Google Scholar] [CrossRef]
- Romerosa, A.; Saoud, M.; Campos-Malpartida, T.; Lidrissi, C.; Serrano-Ruiz, M.; Peruzzini, M.; Garrido, J.A.; García-Maroto, F. DNA Interactions Mediated by Cyclopentadienidoruthenium(II) Complexes Containing Water-Soluble Phosphanes. Eur. J. Inorg. Chem. 2007, 2007, 2803–2812. [Google Scholar] [CrossRef]
- Berthod, A.; Carda-Broch, S. Determination of Liquid-Liquid Partition Coefficients by Separation Methods. J. Chromatogr. A 2004, 1037, 3–14. [Google Scholar] [CrossRef]
- Fotakis, G.; Timbrell, J.A. In Vitro Cytotoxicity Assays: Comparison of LDH, Neutral Red, MTT and Protein Assay in Hepatoma Cell Lines Following Exposure to Cadmium Chloride. Toxicol. Lett. 2006, 160, 171–177. [Google Scholar] [CrossRef]
- Beaven, G.H.; Chen, S.-H.; D’albis, A.; Gratzer, W.B. A Spectroscopic Study of the Haemin–Human-Serum-Albumin System. Eur. J. Biochem. 1974, 41, 539–546. [Google Scholar] [CrossRef]
- Kubista, M.; Sjöback, R.; Eriksson, S.; Albinsson, B. Experimental Correction for the Inner-Filter Effect in Fluorescence Spectra. Analyst 1994, 119, 417–419. [Google Scholar] [CrossRef]
- Coutinho, A.; Prieto, M. Ribonuclease T1 and Alcohol Dehydrogenase Fluorescence Quenching by Acrylamide: A Laboratory Experiment for Undergraduate Students. J. Chem. Educ. 1993, 70, 425. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
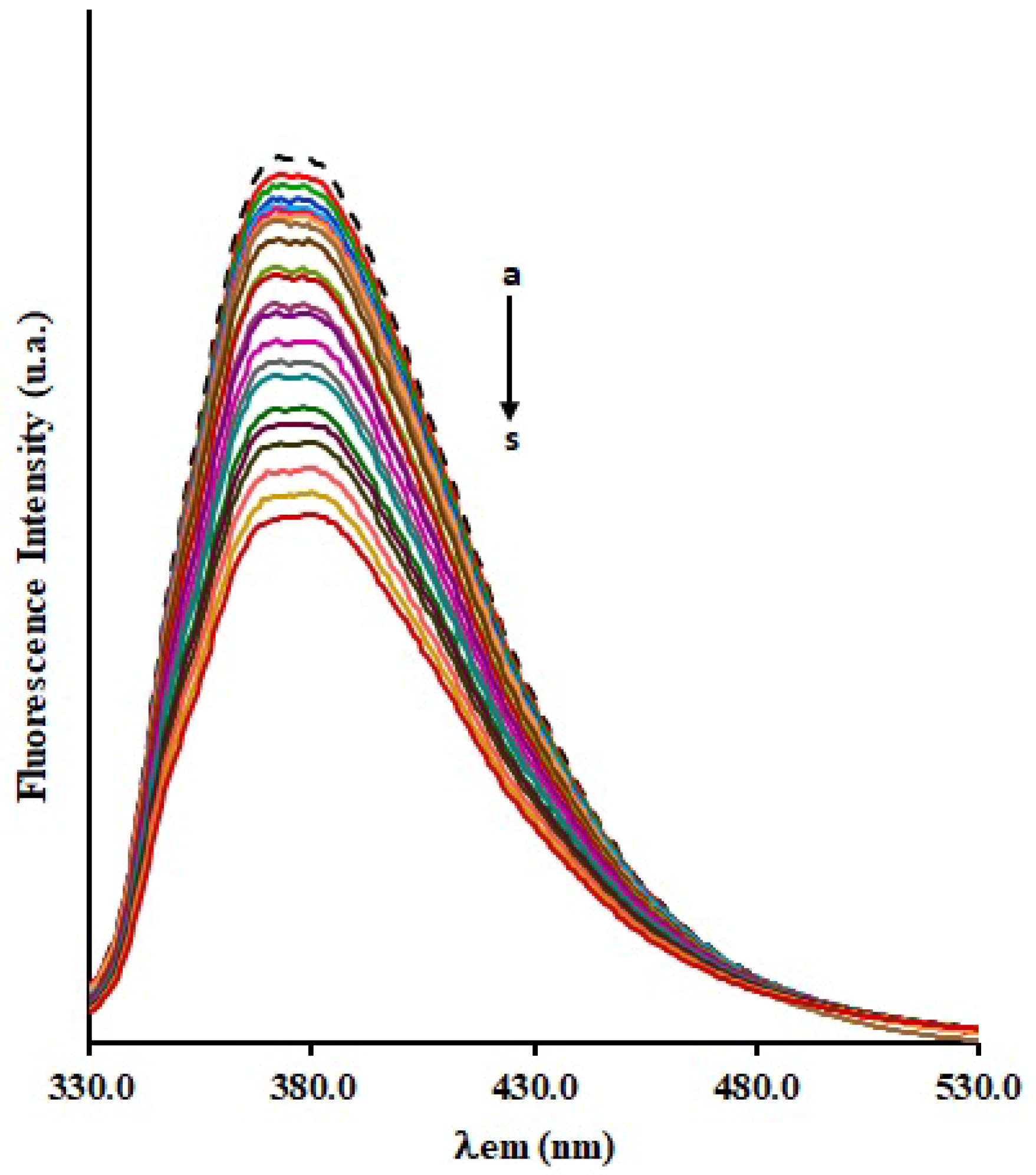{kind=link}
{kind=link}
| Compound | λmax (nm) (εM−1 cm−1) | Compound | λmax (nm) (εM−1 cm−1) | ||
|---|---|---|---|---|---|
| MeOH | Water | MeOH | Water | ||
| 1 L = ImH, n = 2 | 238 (34,600) | 245 (20,300) | 5 L = 4-MpIm, n = 2 | 248 (44,600) | 245 (29,700) |
| 281 (sh) | 283 (sh) | 282 (sh) | 277 (sh) | ||
| 383 (1780) | 379 (2010) | 383 (2950) | 379 (1900) | ||
| 2 L = 1-BI, n = 2 | 238 (64,500) | 218 (87,000) | 6 L = bopy, n = 1 | 259 (9830) | 260 (8520) |
| 256 (sh) | − | 283 (sh) | − | ||
| 395 (2290) | 377 (2720) | 530 (759) | 536 (526) | ||
| 3 L = 1-BuIm, n = 2 | 234 (39,500) | 234 (14,200) | 7 L = dpk, n = 1 | 237 (42,100) | − |
| 267 (sh) | 281 (sh) | 279 (25,800) | 288 (12,200) | ||
| 346 (sh) | 333 (sh) | ||||
| 379 (186) | 386 (827) | 515 (3380) | 510 (1590) | ||
| 4 L = 3-ApIm, n = 2 | 239(29,400) | 238 (26,800) | 8 L = pbt, n = 1 | 259 (27,800) | 261 (4900) |
| 278 (sh) | − | ||||
| 271 (sh) | 264 (sh) | 318 (15,900) | 318 (2670) | ||
| 381 (1910) | 383 (2020) | 331 (sh) | 332 (sh) | ||
| 446 (716) | 428 (228) | ||||
| Compound | Epa (V) vs. SCE | |
|---|---|---|
| Dichoromethane | HEPES Buffer (7.4) | |
| 1 L = ImH; n = 2 | -- | 0.86 |
| 2 L = 1-BI; n = 2 | -- | 0.92 |
| 3 L = 1-BuIm; n = 2 | -- | 0.93 |
| 4 L = 3-ApIm; n = 2 | -- | 0.90 |
| 5 L = 4-MpIm; n = 2 | -- | 0.92 |
| 6 L = bopy; n = 1 | 1.10 | 0.77 |
| 7 L = dpk; n = 1 | 1.02 | 0.83 |
| 8 L = pbt; n = 1 | 1.17 | 1.09 |
| Compound | S(H2O)/mg mL−1 | Time (Days) | Log Po/w |
|---|---|---|---|
| 1 L = ImH, n = 2 | 21.4 (2.1 × 10−2 M) | − [a] | − |
| 2 L = 1-BI, n = 2 | 38.6 (3.9 × 10−2 M) | 0.25 | − |
| 3 L = 1-BuIm, n = 2 | 42.8 (4.3 × 10−2 M) | 0.12 | − |
| 4 L = 3-ApIm, n = 2 | 48.6 (4.9 × 10−2 M) | 1 | 4.43 ± 0.03 |
| 5 L = 4-MpIm, n = 2 | 28.8 (2.9 × 10−2 M) | 0.12 | − |
| 6 L = bopy, n = 1 | 15.3 (1.5 × 10−2 M) | >4 [b] | 5.68 ± 0.05 |
| 7 L = dpk, n = 1 | 19.3 (1.9 × 10−2 M) | >4 [b] | 5.95 ± 0.07 |
| 8 L = pbt, n = 1 | 20.0 (2.0 × 10−2 M) | >4 [b,c] | − |
| Compound | IC50 (μM) | ||
|---|---|---|---|
| A2780 | MDAMB231 | HT29 | |
| 6 | 0.37 ± 0.20 | 13.4 ± 0.4 | 72.4 ± 50 |
| 7 | 0.45 ± 0.30 | >100 | >100 |
| 8 | 0.20 ± 0.08 | 25.4 ± 5.0 | 11.3 ± 2.4 |
| CisPt [a] | 1.90 ± 0.10 | 39 ± 5.0 | 7.0 ± 2.0 |
| System | T (K) | KSV (L mol−1) | Kq (L mol−1 s−1) | R2 |
|---|---|---|---|---|
| HSA-Ru | 293.15 | (7.91 ± 0.10) × 104 | (7.91 ± 0.10) × 1012 | 0.9959 |
| 298.15 | (8.73 ± 0.12) × 104 | (8.73 ± 0.12) × 1012 | 0.9953 | |
| 310.15 | (10.4 ± 0.15) × 104 | (10.4 ± 0.15) × 1012 | 0.9922 | |
| HSAfaf-Ru | 293.15 | (28.7 ± 0.49) × 104 | (28.7 ± 0.49) × 1012 | 0.9906 |
| 298.15 | (40.7 ± 0.75) × 104 | (40.7 ± 0.75) × 1012 | 0.9917 | |
| 310.15 | (72.6 ± 1.02) × 104 | (72.6 ± 1.02) × 1012 | 0.9921 |
| System | T (K) | Ka (L mol−1) | n | R2 | ΔH (kJ mol−1) | ΔG (kJ mol−1) | ΔS (J mol−1 K−1) |
|---|---|---|---|---|---|---|---|
| HSA-Ru | 293.15 | (38.806 ± 1.26) × 104 | 1.136 | 0.9940 | 11.99 (R2 = 0.9963) | −72.37 | 287.92 |
| 298.15 | (19.467 ± 1.14) × 104 | 1.053 | 0.9978 | −73.81 | |||
| 310.15 | (13.948 ± 1.15) × 104 | 0.994 | 0.9970 | −77.26 | |||
| HSAfaf-Ru | 293.15 | (57.293 ± 1.27) × 104 | 1.048 | 0.9923 | 40.51 (R2 = 0.9938) | −156.59 | 396.16 |
| 298.15 | (66.764 ± 1.29) × 104 | 1.023 | 0.9909 | −158.57 | |||
| 310.15 | (80.668 ± 1.17) × 104 | 1.006 | 0.9963 | −163.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morais, T.S.; Marques, F.; Madeira, P.J.A.; Robalo, M.P.; Garcia, M.H. Design and Anticancer Properties of New Water-Soluble Ruthenium–Cyclopentadienyl Complexes. Pharmaceuticals 2022, 15, 862. https://doi.org/10.3390/ph15070862
Morais TS, Marques F, Madeira PJA, Robalo MP, Garcia MH. Design and Anticancer Properties of New Water-Soluble Ruthenium–Cyclopentadienyl Complexes. Pharmaceuticals. 2022; 15(7):862. https://doi.org/10.3390/ph15070862
Chicago/Turabian StyleMorais, Tânia S., Fernanda Marques, Paulo J. Amorim Madeira, Maria Paula Robalo, and Maria Helena Garcia. 2022. "Design and Anticancer Properties of New Water-Soluble Ruthenium–Cyclopentadienyl Complexes" Pharmaceuticals 15, no. 7: 862. https://doi.org/10.3390/ph15070862
APA StyleMorais, T. S., Marques, F., Madeira, P. J. A., Robalo, M. P., & Garcia, M. H. (2022). Design and Anticancer Properties of New Water-Soluble Ruthenium–Cyclopentadienyl Complexes. Pharmaceuticals, 15(7), 862. https://doi.org/10.3390/ph15070862