
Matrix Metalloproteinase 2 as a Pharmacological Target in Heart Failure



Abstract
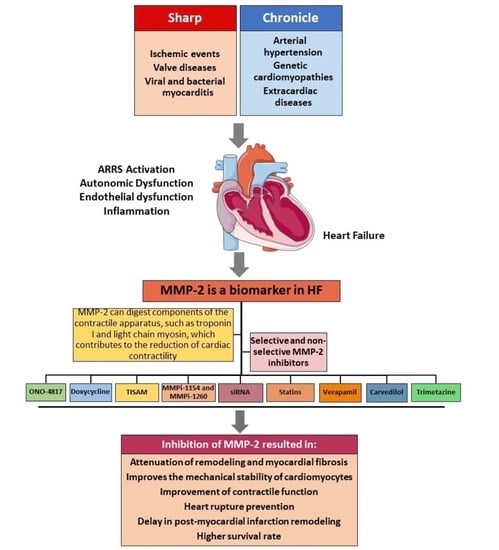
:
1. Introduction
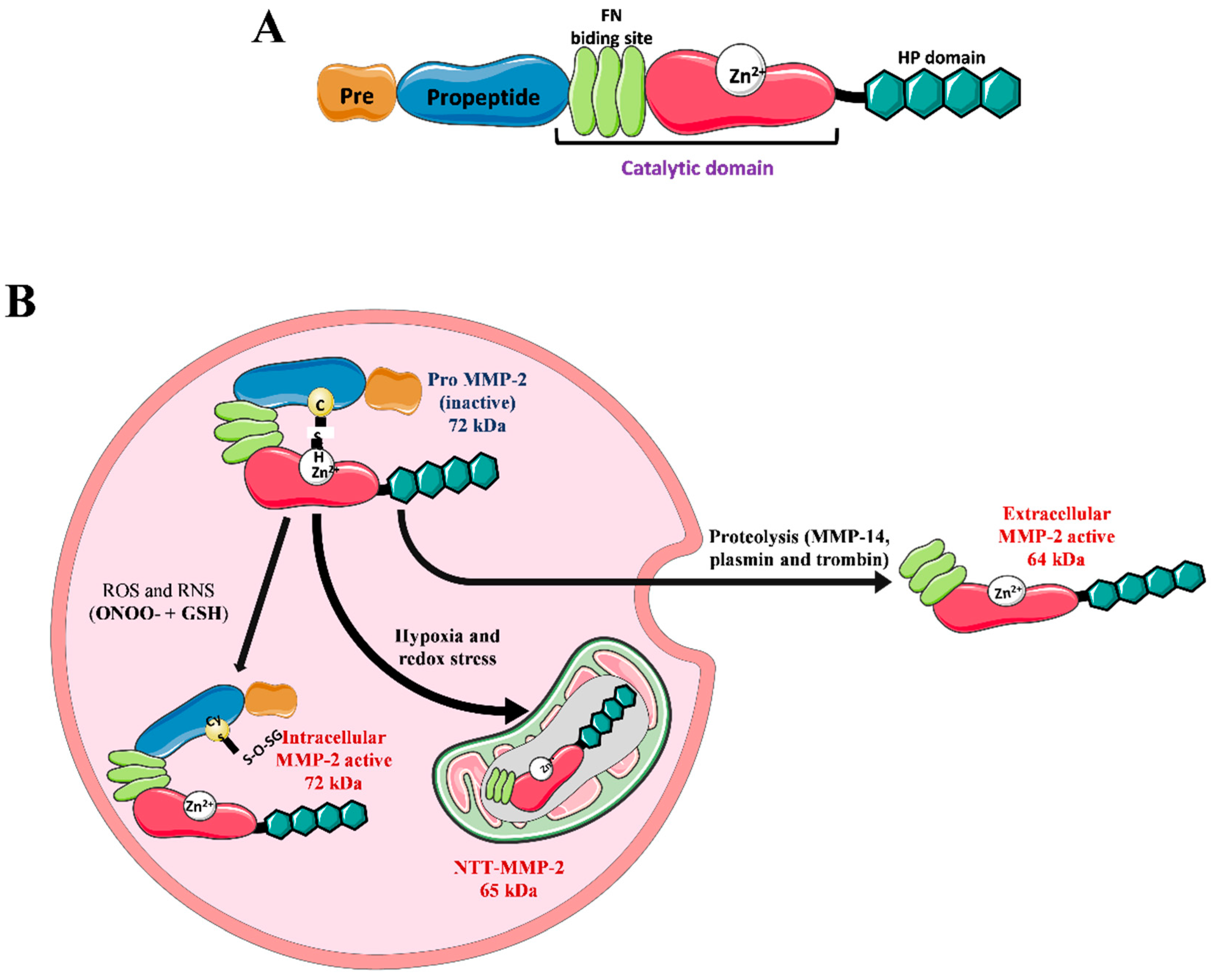
2. Matrix Metalloproteinase 2 (MMP-2)
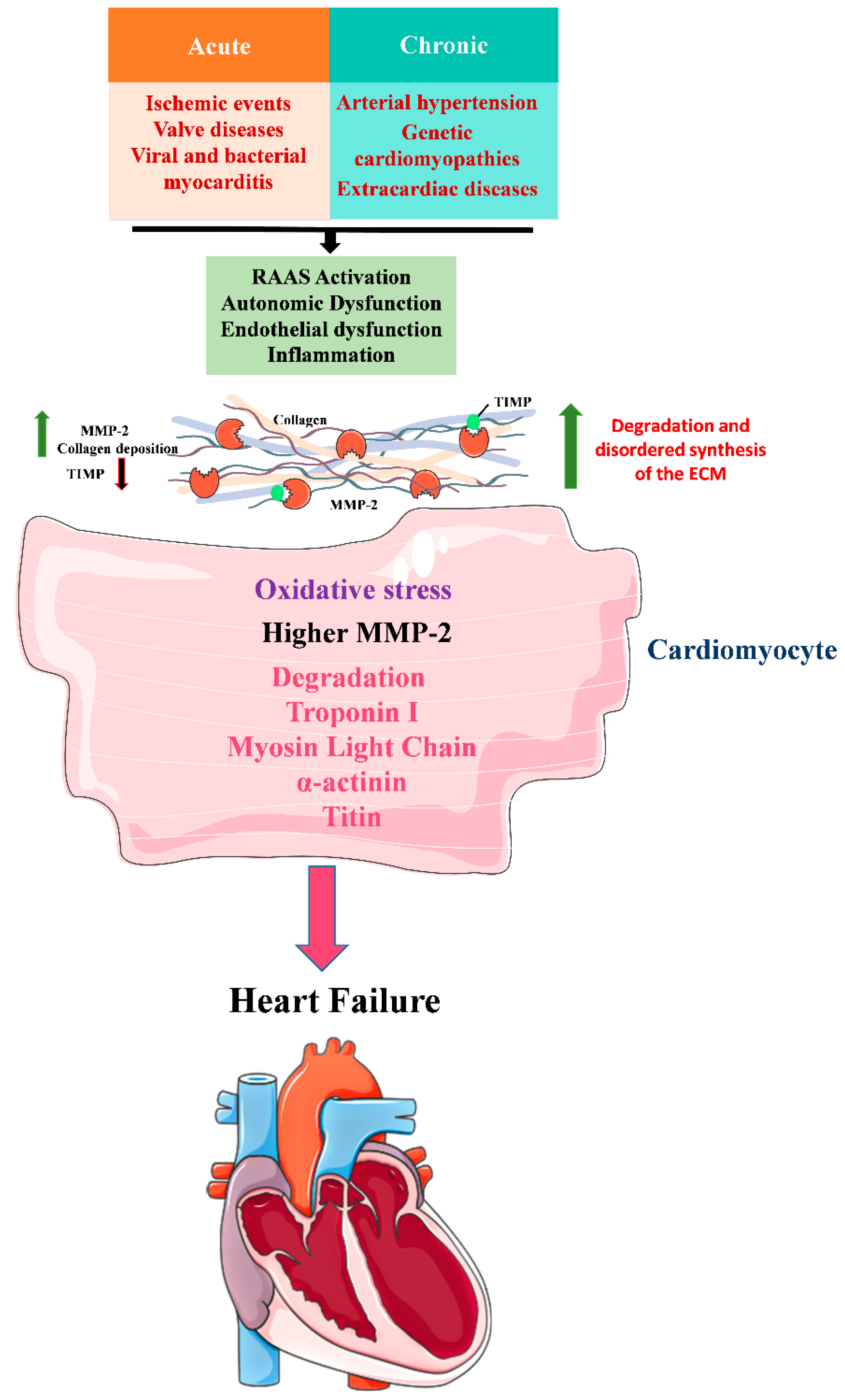
3. Role of MMP-2 in HF
4. The Development of Inhibitors for MMPs and Their Use as a Pharmacological Tool in Disease
5. Use of Nonspecific and Specific Inhibitors for MMP-2 in HF
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Savarese, G.; Lund, L.H. Global Public Health Burden of Heart Failure. Card. Fail Rev. 2017, 3, 7–11. [Google Scholar] [CrossRef]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Seferovic, P.M.; Vardas, P.; Jankowska, E.A.; Maggioni, A.P.; Timmis, A.; Milinkovic, I.; Polovina, M.; Gale, C.P.; Lund, L.H.; Lopatin, Y.; et al. The Heart Failure Association Atlas: Heart Failure Epidemiology and Management Statistics 2019. Eur. J. Heart Fail. 2021, 23, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Allen, L.A.; Whellan, D.J. Economic burden of heart failure in the elderly. PharmacoEconomics 2008, 26, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.; Cole, G.; Asaria, P.; Jabbour, R.; Francis, D.P. The annual global economic burden of heart failure. Int. J. Cardiol. 2014, 171, 368–376. [Google Scholar] [CrossRef]
- Malik, A.; Brito, D.; Vaqar, S.; Chhabra, L. Congestive Heart Failure; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Mazurek, J.A.; Jessup, M. Understanding Heart Failure. Heart Fail Clin. 2017, 13, 1–19. [Google Scholar] [CrossRef]
- Snipelisky, D.; Chaudhry, S.P.; Stewart, G.C. The Many Faces of Heart Failure. Card Electrophysiol. Clin. 2019, 11, 11–20. [Google Scholar] [CrossRef]
- Pagliaro, B.R.; Cannata, F.; Stefanini, G.G.; Bolognese, L. Myocardial ischemia and coronary disease in heart failure. Heart Fail. Rev. 2020, 25, 53–65. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013, 128, 1810–1852. [Google Scholar] [CrossRef]
- Lalande, S.; Johnson, B.D. Diastolic dysfunction: A link between hypertension and heart failure. Drugs Today 2008, 44, 503–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Marteles, M.; Rubio Gracia, J.; Gimenez Lopez, I. Pathophysiology of acute heart failure: A world to know. Rev. Clin. Esp 2016, 216, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, K.R.; Stewart, J.A., Jr.; Lucchesi, P.A. Extracellular matrix remodeling during the progression of volume overload-induced heart failure. J. Mol. Cell. Cardiol. 2010, 48, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Segura, A.M.; Frazier, O.H.; Buja, L.M. Fibrosis and heart failure. Heart Fail. Rev. 2014, 19, 173–185. [Google Scholar] [CrossRef]
- Kobusiak-Prokopowicz, M.; Krzysztofik, J.; Kaaz, K.; Jolda-Mydlowska, B.; Mysiak, A. MMP-2 and TIMP-2 in Patients with Heart Failure and Chronic Kidney Disease. Open Med. 2018, 13, 237–246. [Google Scholar] [CrossRef]
- Piccoli, M.T.; Gupta, S.K.; Viereck, J.; Foinquinos, A.; Samolovac, S.; Kramer, F.L.; Garg, A.; Remke, J.; Zimmer, K.; Batkai, S.; et al. Inhibition of the Cardiac Fibroblast-Enriched lncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ. Res. 2017, 121, 575–583. [Google Scholar] [CrossRef]
- Hughes, B.G.; Schulz, R. Targeting MMP-2 to treat ischemic heart injury. Basic Res. Cardiol. 2014, 109, 424. [Google Scholar] [CrossRef]
- Massaro, M.; Scoditti, E.; Carluccio, M.A.; De Caterina, R. Oxidative stress and vascular stiffness in hypertension: A renewed interest for antioxidant therapies? Vasc. Pharmacol. 2019, 116, 45–50. [Google Scholar] [CrossRef]
- Wang, C.; Qian, X.; Sun, X.; Chang, Q. Angiotensin II increases matrix metalloproteinase 2 expression in human aortic smooth muscle cells via AT1R and ERK1/2. Exp. Biol. Med. 2015, 240, 1564–1571. [Google Scholar] [CrossRef] [Green Version]
- White, D.A.; Su, Y.; Kanellakis, P.; Kiriazis, H.; Morand, E.F.; Bucala, R.; Dart, A.M.; Gao, X.M.; Du, X.J. Differential roles of cardiac and leukocyte derived macrophage migration inhibitory factor in inflammatory responses and cardiac remodelling post myocardial infarction. J. Mol. Cell. Cardiol. 2014, 69, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendry, R.G.; Bilawchuk, L.M.; Marchant, D.J. Targeting matrix metalloproteinase activity and expression for the treatment of viral myocarditis. J. Cardiovasc. Transl. Res. 2014, 7, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Squire, I.B.; Evans, J.; Ng, L.L.; Loftus, I.M.; Thompson, M.M. Plasma MMP-9 and MMP-2 following acute myocardial infarction in man: Correlation with echocardiographic and neurohumoral parameters of left ventricular dysfunction. J. Card. Fail. 2004, 10, 328–333. [Google Scholar] [CrossRef]
- Dostal, D.; Glaser, S.; Baudino, T.A. Cardiac fibroblast physiology and pathology. Compr. Physiol. 2015, 5, 887–909. [Google Scholar] [CrossRef]
- Turner, N.A.; Porter, K.E. Regulation of myocardial matrix metalloproteinase expression and activity by cardiac fibroblasts. IUBMB Life 2012, 64, 143–150. [Google Scholar] [CrossRef]
- Pytliak, M.; Vaník, V.; Bojčík, P. Heart Remodelation: Role of MMPs. In The Role of Matrix Metalloproteinase in Human Body Pathologies; Travascio, F., Ed.; IntechOpen: London, UK, 2017. [Google Scholar]
- Gouveia, M.R.A.; Ascencao, R.; Fiorentino, F.; Costa, J.; Broeiro-Goncalves, P.M.; Fonseca, M.; Borges, M. Current costs of heart failure in Portugal and expected increases due to population aging. Rev. Port. Cardiol. Engl. Ed. 2020, 39, 3–11. [Google Scholar] [CrossRef]
- Nagase, H.; Woessner, J.F., Jr. Matrix metalloproteinases. J. Biol. Chem. 1999, 274, 21491–21494. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Khalil, R.A. Matrix Metalloproteinase Inhibitors as Investigational and Therapeutic Tools in Unrestrained Tissue Remodeling and Pathological Disorders. Prog. Mol. Biol. Transl. Sci. 2017, 148, 355–420. [Google Scholar] [CrossRef] [Green Version]
- Huhtala, P.; Chow, L.T.; Tryggvason, K. Structure of the human type IV collagenase gene. J. Biol. Chem. 1990, 265, 11077–11082. [Google Scholar] [CrossRef]
- Collier, I.E.; Wilhelm, S.M.; Eisen, A.Z.; Marmer, B.L.; Grant, G.A.; Seltzer, J.L.; Kronberger, A.; He, C.S.; Bauer, E.A.; Goldberg, G.I. H-ras oncogene-transformed human bronchial epithelial cells (TBE-1) secrete a single metalloprotease capable of degrading basement membrane collagen. J. Biol. Chem. 1988, 263, 6579–6587. [Google Scholar] [CrossRef]
- Lovett, D.H.; Mahimkar, R.; Raffai, R.L.; Cape, L.; Maklashina, E.; Cecchini, G.; Karliner, J.S. A novel intracellular isoform of matrix metalloproteinase-2 induced by oxidative stress activates innate immunity. PLoS ONE 2012, 7, e34177. [Google Scholar] [CrossRef] [PubMed]
- Van Wart, H.E.; Birkedal-Hansen, H. The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA 1990, 87, 5578–5582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Viappiani, S.; Nicolescu, A.C.; Holt, A.; Sawicki, G.; Crawford, B.D.; Leon, H.; van Mulligen, T.; Schulz, R. Activation and modulation of 72kDa matrix metalloproteinase-2 by peroxynitrite and glutathione. Biochem. Pharmacol. 2009, 77, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, A.; Prado, A.F.; Antonio, R.C.; Issa, J.P.; Gerlach, R.F. Matrix metalloproteinases are involved in cardiovascular diseases. Basic Clin. Pharmacol. Toxicol. 2014, 115, 301–314. [Google Scholar] [CrossRef]
- Prado, A.F.; Batista, R.I.M.; Tanus-Santos, J.E.; Gerlach, R.F. Matrix Metalloproteinases and Arterial Hypertension: Role of Oxidative Stress and Nitric Oxide in Vascular Functional and Structural Alterations. Biomolecules 2021, 11, 585. [Google Scholar] [CrossRef]
- Willerson, J.T. The Medical and Device-Related Treatment of Heart Failure. Circ. Res. 2019, 124, 1519. [Google Scholar] [CrossRef]
- Yamazaki, T.; Lee, J.D.; Shimizu, H.; Uzui, H.; Ueda, T. Circulating matrix metalloproteinase-2 is elevated in patients with congestive heart failure. Eur. J. Heart Fail. 2004, 6, 41–45. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Patal, S.; Wexler, D.; Roth, A.; Sheps, D.; Keren, G. Circulating matrix metalloproteinase-2 but not matrix metalloproteinase-3, matrix metalloproteinase-9, or tissue inhibitor of metalloproteinase-1 predicts outcome in patients with congestive heart failure. Am. Heart J. 2005, 150, 484–487. [Google Scholar] [CrossRef]
- Biolo, A.; Fisch, M.; Balog, J.; Chao, T.; Schulze, P.C.; Ooi, H.; Siwik, D.; Colucci, W.S. Episodes of acute heart failure syndrome are associated with increased levels of troponin and extracellular matrix markers. Circulation. Heart Fail. 2010, 3, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Tager, T.; Wiebalck, C.; Frohlich, H.; Corletto, A.; Katus, H.A.; Frankenstein, L. Biological variation of extracellular matrix biomarkers in patients with stable chronic heart failure. Clin. Res. Cardiol. Off. J. Ger. Card. Soc. 2017, 106, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Cogni, A.L.; Farah, E.; Minicucci, M.F.; Azevedo, P.S.; Okoshi, K.; Matsubara, B.B.; Zanati, S.; Haggeman, R.; Paiva, S.A.; Zornoff, L.A. Metalloproteinases-2 and -9 predict left ventricular remodeling after myocardial infarction. Arq. Bras. De Cardiol. 2013, 100, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Zile, M.R.; Desantis, S.M.; Baicu, C.F.; Stroud, R.E.; Thompson, S.B.; McClure, C.D.; Mehurg, S.M.; Spinale, F.G. Plasma biomarkers that reflect determinants of matrix composition identify the presence of left ventricular hypertrophy and diastolic heart failure. Circulation. Heart Fail. 2011, 4, 246–256. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Bai, X.; Lu, J.; Zhang, L.; Yan, X.; Huang, X.; Dai, H.; Wang, Y.; Hou, L.; Wang, S.; et al. Prognostic Value of Multiple Circulating Biomarkers for 2-Year Death in Acute Heart Failure With Preserved Ejection Fraction. Front. Cardiovasc. Med. 2021, 8, 779282. [Google Scholar] [CrossRef] [PubMed]
- Bergman, M.R.; Teerlink, J.R.; Mahimkar, R.; Li, L.; Zhu, B.Q.; Nguyen, A.; Dahi, S.; Karliner, J.S.; Lovett, D.H. Cardiac matrix metalloproteinase-2 expression independently induces marked ventricular remodeling and systolic dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1847–H1860. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.Z.; Ma, X.; Gray, M.O.; Zhu, B.Q.; Nguyen, A.P.; Baker, A.J.; Simonis, U.; Cecchini, G.; Lovett, D.H.; Karliner, J.S. Transgenic MMP-2 expression induces latent cardiac mitochondrial dysfunction. Biochem. Biophys. Res. Commun. 2007, 358, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Jacob-Ferreira, A.L.; Schulz, R. Activation of intracellular matrix metalloproteinase-2 by reactive oxygen-nitrogen species: Consequences and therapeutic strategies in the heart. Arch. Biochem. Biophys. 2013, 540, 82–93. [Google Scholar] [CrossRef]
- Ali, M.A.; Cho, W.J.; Hudson, B.; Kassiri, Z.; Granzier, H.; Schulz, R. Titin is a target of matrix metalloproteinase-2: Implications in myocardial ischemia/reperfusion injury. Circulation 2010, 122, 2039–2047. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Schulze, C.J.; Suarez-Pinzon, W.L.; Dyck, J.R.; Sawicki, G.; Schulz, R. Intracellular action of matrix metalloproteinase-2 accounts for acute myocardial ischemia and reperfusion injury. Circulation 2002, 106, 1543–1549. [Google Scholar] [CrossRef] [Green Version]
- Cadete, V.J.; Sawicka, J.; Jaswal, J.S.; Lopaschuk, G.D.; Schulz, R.; Szczesna-Cordary, D.; Sawicki, G. Ischemia/reperfusion-induced myosin light chain 1 phosphorylation increases its degradation by matrix metalloproteinase 2. FEBS J. 2012, 279, 2444–2454. [Google Scholar] [CrossRef] [Green Version]
- Sawicki, G.; Leon, H.; Sawicka, J.; Sariahmetoglu, M.; Schulze, C.J.; Scott, P.G.; Szczesna-Cordary, D.; Schulz, R. Degradation of myosin light chain in isolated rat hearts subjected to ischemia-reperfusion injury: A new intracellular target for matrix metalloproteinase-2. Circulation 2005, 112, 544–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, M.M.; Schulz, C.G.; Wang, W.; Sawicki, G.; Bautista-Lopez, N.L.; Schulz, R. Matrix metalloproteinase-2 degrades the cytoskeletal protein alpha-actinin in peroxynitrite mediated myocardial injury. J. Mol. Cell. Cardiol. 2007, 43, 429–436. [Google Scholar] [CrossRef]
- Lovett, D.H.; Mahimkar, R.; Raffai, R.L.; Cape, L.; Zhu, B.Q.; Jin, Z.Q.; Baker, A.J.; Karliner, J.S. N-terminal truncated intracellular matrix metalloproteinase-2 induces cardiomyocyte hypertrophy, inflammation and systolic heart failure. PLoS ONE 2013, 8, e68154. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Iwanaga, S.; Mochizuki, S.; Okamoto, H.; Ogawa, S.; Okada, Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J. Clin. Investig. 2005, 115, 599–609. [Google Scholar] [CrossRef] [Green Version]
- Matsusaka, H.; Ide, T.; Matsushima, S.; Ikeuchi, M.; Kubota, T.; Sunagawa, K.; Kinugawa, S.; Tsutsui, H. Targeted deletion of matrix metalloproteinase 2 ameliorates myocardial remodeling in mice with chronic pressure overload. Hypertension 2006, 47, 711–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, E.; Hardy-Sosa, A.; Fernandez-Patron, C. MMP-2: Is too low as bad as too high in the cardiovascular system? Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1332–H1340. [Google Scholar] [CrossRef]
- Matsusaka, H.; Ikeuchi, M.; Matsushima, S.; Ide, T.; Kubota, T.; Feldman, A.M.; Takeshita, A.; Sunagawa, K.; Tsutsui, H. Selective disruption of MMP-2 gene exacerbates myocardial inflammation and dysfunction in mice with cytokine-induced cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1858–H1864. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Berry, E.; Hernandez-Anzaldo, S.; Takawale, A.; Kassiri, Z.; Fernandez-Patron, C. Matrix metalloproteinase-2 mediates a mechanism of metabolic cardioprotection consisting of negative regulation of the sterol regulatory element-binding protein-2/3-hydroxy-3-methylglutaryl-CoA reductase pathway in the heart. Hypertension 2015, 65, 882–888. [Google Scholar] [CrossRef] [Green Version]
- Martignetti, J.A.; Aqeel, A.A.; Sewairi, W.A.; Boumah, C.E.; Kambouris, M.; Mayouf, S.A.; Sheth, K.V.; Eid, W.A.; Dowling, O.; Harris, J.; et al. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat. Genet. 2001, 28, 261–265. [Google Scholar] [CrossRef]
- Tuysuz, B.; Mosig, R.; Altun, G.; Sancak, S.; Glucksman, M.J.; Martignetti, J.A. A novel matrix metalloproteinase 2 (MMP2) terminal hemopexin domain mutation in a family with multicentric osteolysis with nodulosis and arthritis with cardiac defects. Eur. J. Hum. Genet. EJHG 2009, 17, 565–572. [Google Scholar] [CrossRef] [Green Version]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuna, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, B.; Brown, P.D.; East, N.; Crimmin, M.J.; Balkwill, F.R. A synthetic matrix metalloproteinase inhibitor decreases tumor burden and prolongs survival of mice bearing human ovarian carcinoma xenografts. Cancer Res. 1993, 53, 2087–2091. [Google Scholar] [PubMed]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef]
- Shepherd, F.A.; Giaccone, G.; Seymour, L.; Debruyne, C.; Bezjak, A.; Hirsh, V.; Smylie, M.; Rubin, S.; Martins, H.; Lamont, A.; et al. Prospective, randomized, double-blind, placebo-controlled trial of marimastat after response to first-line chemotherapy in patients with small-cell lung cancer: A trial of the National Cancer Institute of Canada-Clinical Trials Group and the European Organization for Research and Treatment of Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2002, 20, 4434–4439. [Google Scholar] [CrossRef]
- Wada, C.K.; Holms, J.H.; Curtin, M.L.; Dai, Y.; Florjancic, A.S.; Garland, R.B.; Guo, Y.; Heyman, H.R.; Stacey, J.R.; Steinman, D.H.; et al. Phenoxyphenyl sulfone N-formylhydroxylamines (retrohydroxamates) as potent, selective, orally bioavailable matrix metalloproteinase inhibitors. J. Med. Chem. 2002, 45, 219–232. [Google Scholar] [CrossRef]
- Bramhall, S.R.; Hallissey, M.T.; Whiting, J.; Scholefield, J.; Tierney, G.; Stuart, R.C.; Hawkins, R.E.; McCulloch, P.; Maughan, T.; Brown, P.D.; et al. Marimastat as maintenance therapy for patients with advanced gastric cancer: A randomised trial. Br. J. Cancer 2002, 86, 1864–1870. [Google Scholar] [CrossRef]
- Das, S.; Amin, S.A.; Jha, T. Inhibitors of gelatinases (MMP-2 and MMP-9) for the management of hematological malignancies. Eur. J. Med. Chem. 2021, 223, 113623. [Google Scholar] [CrossRef]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef]
- Tan Ide, A.; Ricciardelli, C.; Russell, D.L. The metalloproteinase ADAMTS1: A comprehensive review of its role in tumorigenic and metastatic pathways. Int. J. Cancer. J. Int. Du Cancer 2013, 133, 2263–2276. [Google Scholar] [CrossRef]
- Iwasaki, M.; Nishikawa, A.; Fujimoto, T.; Akutagawa, N.; Manase, K.; Endo, T.; Yoshida, K.; Maekawa, R.; Yoshioka, T.; Kudo, R. Anti-invasive effect of MMI-166, a new selective matrix metalloproteinase inhibitor, in cervical carcinoma cell lines. Gynecol. Oncol. 2002, 85, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Leighl, N.B.; Paz-Ares, L.; Douillard, J.Y.; Peschel, C.; Arnold, A.; Depierre, A.; Santoro, A.; Betticher, D.C.; Gatzemeier, U.; Jassem, J.; et al. Randomized phase III study of matrix metalloproteinase inhibitor BMS-275291 in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: National Cancer Institute of Canada-Clinical Trials Group Study BR.18. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 2831–2839. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Saphner, T.J.; Waterhouse, D.M.; Chen, T.T.; Rush-Taylor, A.; Sparano, J.A.; Wolff, A.C.; Cobleigh, M.A.; Galbraith, S.; Sledge, G.W. A randomized phase II feasibility trial of BMS-275291 in patients with early stage breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 1971–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirte, H.; Vergote, I.B.; Jeffrey, J.R.; Grimshaw, R.N.; Coppieters, S.; Schwartz, B.; Tu, D.; Sadura, A.; Brundage, M.; Seymour, L. A phase III randomized trial of BAY 12-9566 (tanomastat) as maintenance therapy in patients with advanced ovarian cancer responsive to primary surgery and paclitaxel/platinum containing chemotherapy: A National Cancer Institute of Canada Clinical Trials Group Study. Gynecol. Oncol. 2006, 102, 300–308. [Google Scholar] [CrossRef]
- Wong, M.S.; Sidik, S.M.; Mahmud, R.; Stanslas, J. Molecular targets in the discovery and development of novel antimetastatic agents: Current progress and future prospects. Clin. Exp. Pharmacol. Physiol. 2013, 40, 307–319. [Google Scholar] [CrossRef]
- Philips, N.; Conte, J.; Chen, Y.J.; Natrajan, P.; Taw, M.; Keller, T.; Givant, J.; Tuason, M.; Dulaj, L.; Leonardi, D.; et al. Beneficial regulation of matrixmetalloproteinases and their inhibitors, fibrillar collagens and transforming growth factor-beta by Polypodium leucotomos, directly or in dermal fibroblasts, ultraviolet radiated fibroblasts, and melanoma cells. Arch. Dermatol. Res. 2009, 301, 487–495. [Google Scholar] [CrossRef]
- Philips, N.; Smith, J.; Keller, T.; Gonzalez, S. Predominant effects of Polypodium leucotomos on membrane integrity, lipid peroxidation, and expression of elastin and matrixmetalloproteinase-1 in ultraviolet radiation exposed fibroblasts, and keratinocytes. J. Dermatol. Sci. 2003, 32, 1–9. [Google Scholar] [CrossRef]
- Philips, N.; Samuel, M.; Arena, R.; Chen, Y.J.; Conte, J.; Natarajan, P.; Haas, G.; Gonzalez, S. Direct inhibition of elastase and matrixmetalloproteinases and stimulation of biosynthesis of fibrillar collagens, elastin, and fibrillins by xanthohumol. J. Cosmet. Sci. 2010, 61, 125–132. [Google Scholar] [CrossRef]
- Astner, S.; Wu, A.; Chen, J.; Philips, N.; Rius-Diaz, F.; Parrado, C.; Mihm, M.C.; Goukassian, D.A.; Pathak, M.A.; Gonzalez, S. Dietary lutein/zeaxanthin partially reduces photoaging and photocarcinogenesis in chronically UVB-irradiated Skh-1 hairless mice. Ski. Pharm. Physiol. 2007, 20, 283–291. [Google Scholar] [CrossRef]
- Philips, N.; Keller, T.; Hendrix, C.; Hamilton, S.; Arena, R.; Tuason, M.; Gonzalez, S. Regulation of the extracellular matrix remodeling by lutein in dermal fibroblasts, melanoma cells, and ultraviolet radiation exposed fibroblasts. Arch. Dermatol. Res. 2007, 299, 373–379. [Google Scholar] [CrossRef]
- Chan, B.Y.H.; Roczkowsky, A.; Cho, W.J.; Poirier, M.; Sergi, C.; Keschrumrus, V.; Churko, J.M.; Granzier, H.; Schulz, R. MMP inhibitors attenuate doxorubicin cardiotoxicity by preventing intracellular and extracellular matrix remodelling. Cardiovasc. Res. 2021, 117, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Parente, J.M.; Blascke de Mello, M.M.; Silva, P.; Omoto, A.C.M.; Pernomian, L.; Oliveira, I.S.; Mahmud, Z.; Fazan, R., Jr.; Arantes, E.C.; Schulz, R.; et al. MMP inhibition attenuates hypertensive eccentric cardiac hypertrophy and dysfunction by preserving troponin I and dystrophin. Biochem. Pharmacol. 2021, 193, 114744. [Google Scholar] [CrossRef] [PubMed]
- Tessone, A.; Feinberg, M.S.; Barbash, I.M.; Reich, R.; Holbova, R.; Richmann, M.; Mardor, Y.; Leor, J. Effect of matrix metalloproteinase inhibition by doxycycline on myocardial healing and remodeling after myocardial infarction. Cardiovasc. Drugs Ther. 2005, 19, 383–390. [Google Scholar] [CrossRef]
- Novak, M.J.; Johns, L.P.; Miller, R.C.; Bradshaw, M.H. Adjunctive benefits of subantimicrobial dose doxycycline in the management of severe, generalized, chronic periodontitis. J. Periodontol. 2002, 73, 762–769. [Google Scholar] [CrossRef]
- Mata, K.M.; Tefe-Silva, C.; Floriano, E.M.; Fernandes, C.R.; Rizzi, E.; Gerlach, R.F.; Mazzuca, M.Q.; Ramos, S.G. Interference of doxycycline pretreatment in a model of abdominal aortic aneurysms. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2015, 24, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Antonio, R.C.; Ceron, C.S.; Rizzi, E.; Coelho, E.B.; Tanus-Santos, J.E.; Gerlach, R.F. Antioxidant effect of doxycycline decreases MMP activity and blood pressure in SHR. Mol. Cell. Biochem. 2014, 386, 99–105. [Google Scholar] [CrossRef]
- Castro, M.M.; Rizzi, E.; Ceron, C.S.; Guimaraes, D.A.; Rodrigues, G.J.; Bendhack, L.M.; Gerlach, R.F.; Tanus-Santos, J.E. Doxycycline ameliorates 2K-1C hypertension-induced vascular dysfunction in rats by attenuating oxidative stress and improving nitric oxide bioavailability. Nitric Oxide Biol. Chem. Off. J. Nitric Oxide Soc. 2012, 26, 162–168. [Google Scholar] [CrossRef]
- Castro, M.M.; Rizzi, E.; Figueiredo-Lopes, L.; Fernandes, K.; Bendhack, L.M.; Pitol, D.L.; Gerlach, R.F.; Tanus-Santos, J.E. Metalloproteinase inhibition ameliorates hypertension and prevents vascular dysfunction and remodeling in renovascular hypertensive rats. Atherosclerosis 2008, 198, 320–331. [Google Scholar] [CrossRef]
- Guimaraes, D.A.; Rizzi, E.; Ceron, C.S.; Oliveira, A.M.; Oliveira, D.M.; Castro, M.M.; Tirapelli, C.R.; Gerlach, R.F.; Tanus-Santos, J.E. Doxycycline dose-dependently inhibits MMP-2-mediated vascular changes in 2K1C hypertension. Basic Clin. Pharmacol. Toxicol. 2011, 108, 318–325. [Google Scholar] [CrossRef]
- Rizzi, E.; Castro, M.M.; Prado, C.M.; Silva, C.A.; Fazan, R., Jr.; Rossi, M.A.; Tanus-Santos, J.E.; Gerlach, R.F. Matrix metalloproteinase inhibition improves cardiac dysfunction and remodeling in 2-kidney, 1-clip hypertension. J. Card. Fail. 2010, 16, 599–608. [Google Scholar] [CrossRef]
- Villarreal, F.J.; Griffin, M.; Omens, J.; Dillmann, W.; Nguyen, J.; Covell, J. Early short-term treatment with doxycycline modulates postinfarction left ventricular remodeling. Circulation 2003, 108, 1487–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerisano, G.; Buonamici, P.; Valenti, R.; Sciagra, R.; Raspanti, S.; Santini, A.; Carrabba, N.; Dovellini, E.V.; Romito, R.; Pupi, A.; et al. Early short-term doxycycline therapy in patients with acute myocardial infarction and left ventricular dysfunction to prevent the ominous progression to adverse remodelling: The TIPTOP trial. Eur. Heart J. 2014, 35, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.L.; Desai, K.K.; Vakili, B.A.; Nouneh, C.; Lee, H.M.; Golub, L.M. Clinical and biochemical results of the metalloproteinase inhibition with subantimicrobial doses of doxycycline to prevent acute coronary syndromes (MIDAS) pilot trial. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 733–738. [Google Scholar] [CrossRef] [Green Version]
- Schulze, C.J.; Castro, M.M.; Kandasamy, A.D.; Cena, J.; Bryden, C.; Wang, S.H.; Koshal, A.; Tsuyuki, R.T.; Finegan, B.A.; Schulz, R. Doxycycline reduces cardiac matrix metalloproteinase-2 activity but does not ameliorate myocardial dysfunction during reperfusion in coronary artery bypass patients undergoing cardiopulmonary bypass. Crit. Care Med. 2013, 41, 2512–2520. [Google Scholar] [CrossRef]
- Hudson, M.P.; Armstrong, P.W.; Ruzyllo, W.; Brum, J.; Cusmano, L.; Krzeski, P.; Lyon, R.; Quinones, M.; Theroux, P.; Sydlowski, D.; et al. Effects of selective matrix metalloproteinase inhibitor (PG-116800) to prevent ventricular remodeling after myocardial infarction: Results of the PREMIER (Prevention of Myocardial Infarction Early Remodeling) trial. J. Am. Coll. Cardiol. 2006, 48, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Bencsik, P.; Kupai, K.; Gorbe, A.; Kenyeres, E.; Varga, Z.V.; Paloczi, J.; Gaspar, R.; Kovacs, L.; Weber, L.; Takacs, F.; et al. Development of Matrix Metalloproteinase-2 Inhibitors for Cardioprotection. Front. Pharmacol. 2018, 9, 296. [Google Scholar] [CrossRef] [PubMed]
- Gomori, K.; Szabados, T.; Kenyeres, E.; Pipis, J.; Foldesi, I.; Siska, A.; Dorman, G.; Ferdinandy, P.; Gorbe, A.; Bencsik, P. Cardioprotective Effect of Novel Matrix Metalloproteinase Inhibitors. Int. J. Mol. Sci. 2020, 21, 6990. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.B.; Cadete, V.J.; Sra, B.; Sawicka, J.; Chen, Z.; Bekar, L.K.; Cayabyab, F.; Sawicki, G. Inhibition of MMP-2 expression with siRNA increases baseline cardiomyocyte contractility and protects against simulated ischemic reperfusion injury. BioMed Res. Int. 2014, 2014, 810371. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Chung, J.J.; Li, E.C.; Uman, S.; Atluri, P.; Burdick, J.A. Injectable and protease-degradable hydrogel for siRNA sequestration and triggered delivery to the heart. J. Control. Release Off. J. Control. Release Soc. 2018, 285, 152–161. [Google Scholar] [CrossRef]
- Nakaya, R.; Uzui, H.; Shimizu, H.; Nakano, A.; Mitsuke, Y.; Yamazaki, T.; Ueda, T.; Lee, J.D. Pravastatin suppresses the increase in matrix metalloproteinase-2 levels after acute myocardial infarction. Int. J. Cardiol. 2005, 105, 67–73. [Google Scholar] [CrossRef]
- Shirakabe, A.; Asai, K.; Hata, N.; Yokoyama, S.; Shinada, T.; Kobayashi, N.; Tomita, K.; Tsurumi, M.; Matsushita, M.; Mizuno, K. Immediate administration of atorvastatin decreased the serum MMP-2 level and improved the prognosis for acute heart failure. J. Cardiol. 2012, 59, 374–382. [Google Scholar] [CrossRef] [Green Version]
- Tousoulis, D.; Andreou, I.; Tentolouris, C.; Antoniades, C.; Papageorgiou, N.; Gounari, P.; Kotrogiannis, I.; Miliou, A.; Charakida, M.; Trikas, A.; et al. Comparative effects of rosuvastatin and allopurinol on circulating levels of matrix metalloproteinases and tissue inhibitors of metalloproteinases in patients with chronic heart failure. Int. J. Cardiol. 2010, 145, 438–443. [Google Scholar] [CrossRef]
- Cortese, F.; Gesualdo, M.; Cortese, A.; Carbonara, S.; Devito, F.; Zito, A.; Ricci, G.; Scicchitano, P.; Ciccone, M.M. Rosuvastatin: Beyond the cholesterol-lowering effect. Pharmacol. Res. 2016, 107, 1–18. [Google Scholar] [CrossRef]
- Mason, R.P. Molecular basis of differences among statins and a comparison with antioxidant vitamins. Am. J. Cardiol. 2006, 98, 34P–41P. [Google Scholar] [CrossRef]
- Gong, W.; Ma, Y.; Li, A.; Shi, H.; Nie, S. Trimetazidine suppresses oxidative stress, inhibits MMP-2 and MMP-9 expression, and prevents cardiac rupture in mice with myocardial infarction. Cardiovasc. Ther. 2018, 36, e12460. [Google Scholar] [CrossRef] [Green Version]
- Mendes, A.S.; Blascke de Mello, M.M.; Parente, J.M.; Omoto, A.C.M.; Neto-Neves, E.M.; Fazan, R., Jr.; Tanus-Santos, J.E.; Castro, M.M. Verapamil decreases calpain-1 and matrix metalloproteinase-2 activities and improves hypertension-induced hypertrophic cardiac remodeling in rats. Life Sci. 2020, 244, 117153. [Google Scholar] [CrossRef]
- Skrzypiec-Spring, M.; Urbaniak, J.; Sapa-Wojciechowska, A.; Pietkiewicz, J.; Orda, A.; Karolko, B.; Danielewicz, R.; Bil-Lula, I.; Wozniak, M.; Schulz, R.; et al. Matrix Metalloproteinase-2 Inhibition in Acute Ischemia-Reperfusion Heart Injury-Cardioprotective Properties of Carvedilol. Pharmaceuticals 2021, 14, 1276. [Google Scholar] [CrossRef]
- Dang, Y.; Gao, N.; Niu, H.; Guan, Y.; Fan, Z.; Guan, J. Targeted Delivery of a Matrix Metalloproteinases-2 Specific Inhibitor Using Multifunctional Nanogels to Attenuate Ischemic Skeletal Muscle Degeneration and Promote Revascularization. ACS Appl. Mater. Interfaces 2021, 13, 5907–5918. [Google Scholar] [CrossRef]
- Harwood, S.L.; Nielsen, N.S.; Diep, K.; Jensen, K.T.; Nielsen, P.K.; Yamamoto, K.; Enghild, J.J. Development of selective protease inhibitors via engineering of the bait region of human alpha2-macroglobulin. J. Biol. Chem. 2021, 297, 100879. [Google Scholar] [CrossRef]
- Higashi, S.; Hirose, T.; Takeuchi, T.; Miyazaki, K. Molecular design of a highly selective and strong protein inhibitor against matrix metalloproteinase-2 (MMP-2). J. Biol. Chem. 2013, 288, 9066–9076. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.H.; Chu, P.Y.; Chen, J.L.; Huang, C.T.; Huang, C.C.; Tsai, Y.F.; Wang, Y.L.; Lien, P.J.; Tseng, L.M.; Liu, C.Y. Expression pattern and prognostic impact of glycoprotein non-metastatic B (GPNMB) in triple-negative breast cancer. Sci. Rep. 2021, 11, 12171. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, P.; Li, Z.; Ren, W.; Wang, S.; Shao, S.; Sun, J.; Ren, X.; Perkins, N.G.; Guo, Z.; Chang, C.A.; et al. Inhibiting Matrix Metalloproteinase-2 Activation by Perturbing Protein-Protein Interactions Using a Cyclic Peptide. J. Med. Chem. 2020, 63, 6979–6990. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.H.; Gao, B.T.; Goldsmith, Z.K.; Irvine, A.S.; Saleh, N.; Lee, R.P.; Lendermon, J.B.; Bheemreddy, R.; Zhang, Q.; Brennan, R.C.; et al. Inhibition of MMP-2 and MMP-9 decreases cellular migration, and angiogenesis in in vitro models of retinoblastoma. BMC Cancer 2017, 17, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Classification | Left Ventricle Ejection Fraction (LVEF) | Main Cardiac Alterations (Ecodoppler) |
|---|---|---|
| HFrEF | <40% | Structural change and systolic dysfunction |
| HFpEF | ≥50% | Structural change and diastolic dysfunction |
| HFiEF | 41% to 49% | Structural change and diastolic dysfunction |
| Class | General Description | Patient Symptoms |
|---|---|---|
| I | Asymptomatic | No limitation of physical activity; regular physical activity does not cause undue fatigue, palpitation and dyspnea. |
| II | Mild symptoms | Slight limitation of physical activity; comfortable at rest; activity results in fatigue, palpitation and dyspnea. |
| III | Moderate symptoms | Marked limitation of physical activity; comfortable at rest; regular exercise causes fatigue, palpitation and dyspnea. |
| IV | Severe symptoms | Unable to perform any physical activity without discomfort; HF symptoms at rest; if any physical activity is performed, the pain increases. |
| Class of MMP Inhibitors | Inhibitor (Alternative Names) | Characteristics |
|---|---|---|
| Endogenous inhibitors | α2-macroglobulin and TIMPs | It traps MMPs in the plasma, preventing them from degrading their substrates. It inhibits tissue MMPs and has four members: TIMP-1 to 4. TIMPs can inhibit all MMPs, but with different specificities. |
| Hydroxamate-based inhibitors | Batimastat and Marimastat | They are designed to mimic the natural peptide substrate (collagen) of MMPs. It targets the catalytic site of MMPs. |
| The new generation of hydroxamate-based inhibitors | Cipemastat and MMI-166 | They were developed with a sulfonamide and a zinc-binding hydroxamate group, in addition to the substitution of an aryl group, generating a compound with more specificity. It targets the catalytic site of MMPs. |
| Non-hydroxamate inhibitors | Rebimastat and Tanomastat | They were designed with various peptidomimetics and non-mimetics, not limited to mimicking the substrate of MMPs. It targets the catalytic site of MMPs. |
| Inhibitors targeting alternative binding sites | BMS-275291 and specific MMP-13 inhibitor (provided by Pfizer, Ann Arbor, MI, USA) | Highly selective, unlike previous MMP inhibitors, because it does not bind to catalytic zinc ion and is not competitive for substrate binding. They target alternative, less conserved binding sites. |
| Non-Selective Inhibitor | Species | Disease | Comments | References |
|---|---|---|---|---|
| Doxycycline | Rats | Renovascular hypertension with HF | Prevented the conversion of concentric hypertrophy to eccentric hypertrophy in the LV, associated with decreased MMP-2 activity and reduced troponin I and dystrophin proteolysis | [84] |
| Doxycycline | Mice | Model of acute myocardial infarction with HF | It has not reduced scar thinning and compensatory LV hypertrophy, despite having decreased MMP-2 and MMP-9 activity | [85] |
| Doxycycline (Adjuvant therapy) | Humans | Acute myocardial infarction (40% of patients with HFrEF) | Improved diastolic function and reduced infarct area | [94] |
| Doxycycline (Adjuvant therapy) | Humans | Coronary artery disease and atherosclerosis | There was no improvement in cardiac dysfunction parameters and sudden death outcomes | [95,96] |
| PG-116800 (Adjuvant therapy) | Humans | Acute myocardial infarction (HFpEF) with HF | No improvement in heart function and death rates Development of musculoskeletal toxicity | [97] |
| MMP-2 selective inhibitor | ||||
| ONO-4817 | Mice | Ischemia and reperfusion model with HF | Shown to improve contractile dysfunction associated with decreased MMP-2 activity and titin proteolysis | [50] |
| ONO-4817 | Mice | Model of doxorubicin-induced cardiotoxicity | Attenuated LV remodeling and myocardial fibrosis | [83] |
| TISAM | Mice | Model of acute myocardial infarction with HF | It improved survival rate by preventing cardiac rupture and delaying post-infarction remodeling | [56] |
| MMPI-1154, MMPI-1260 and MMPI-1248 (Chemical Modeling) | Mice | Model of acute myocardial infarction with HF | They showed inhibitory activity on MMP-2, associated with a reduction in the infarct area | [98,99] |
| siRNA for MMP-2 | Mice | Ischemia and reperfusion model with HF | It prevented contractile dysfunction associated with decreased degradation of MLC1/2 | [100] |
| Hydrogel encapsulated siRNA for MMP-2 | Mice | Model of acute myocardial infarction | Improved cardiac output and ejection fraction | [101] |
| Statins (Atorvastatin, Rosuvastatin and Pravastatin) | Humans | Acute myocardial infarction (HFrEF) | Decreased serum MMP-2 levels are associated with a reduced number of deaths and hospital readmission | [102,103,104] |
| Antihypertensive drugs (Verapamil, Carvedilol and Trimethazine) | Mice and rats | Ischemia/reperfusion model; Model of HF induced by hypertension and Myocardial Infarction Model | Positive effects on cardiac function and remodeling associated with decreased activity and expression of MMP-2 | [107,108,109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonçalves, P.R.; Nascimento, L.D.; Gerlach, R.F.; Rodrigues, K.E.; Prado, A.F. Matrix Metalloproteinase 2 as a Pharmacological Target in Heart Failure. Pharmaceuticals 2022, 15, 920. https://doi.org/10.3390/ph15080920
Gonçalves PR, Nascimento LD, Gerlach RF, Rodrigues KE, Prado AF. Matrix Metalloproteinase 2 as a Pharmacological Target in Heart Failure. Pharmaceuticals. 2022; 15(8):920. https://doi.org/10.3390/ph15080920
Chicago/Turabian StyleGonçalves, Pricila Rodrigues, Lisandra Duarte Nascimento, Raquel Fernanda Gerlach, Keuri Eleutério Rodrigues, and Alejandro Ferraz Prado. 2022. "Matrix Metalloproteinase 2 as a Pharmacological Target in Heart Failure" Pharmaceuticals 15, no. 8: 920. https://doi.org/10.3390/ph15080920
APA StyleGonçalves, P. R., Nascimento, L. D., Gerlach, R. F., Rodrigues, K. E., & Prado, A. F. (2022). Matrix Metalloproteinase 2 as a Pharmacological Target in Heart Failure. Pharmaceuticals, 15(8), 920. https://doi.org/10.3390/ph15080920