
A Peptide Inhibitor of the Human Cytomegalovirus Core Nuclear Egress Complex


 ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
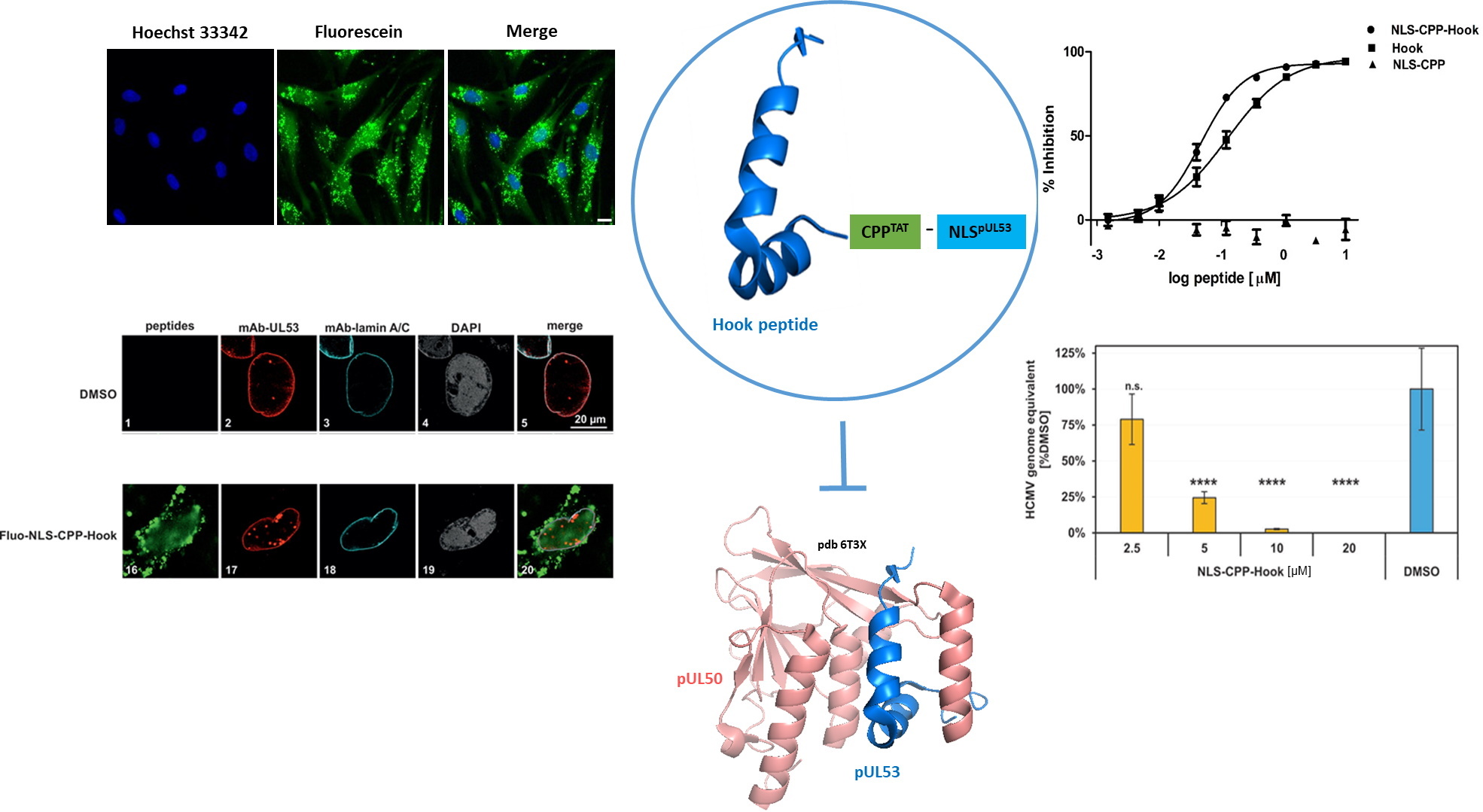
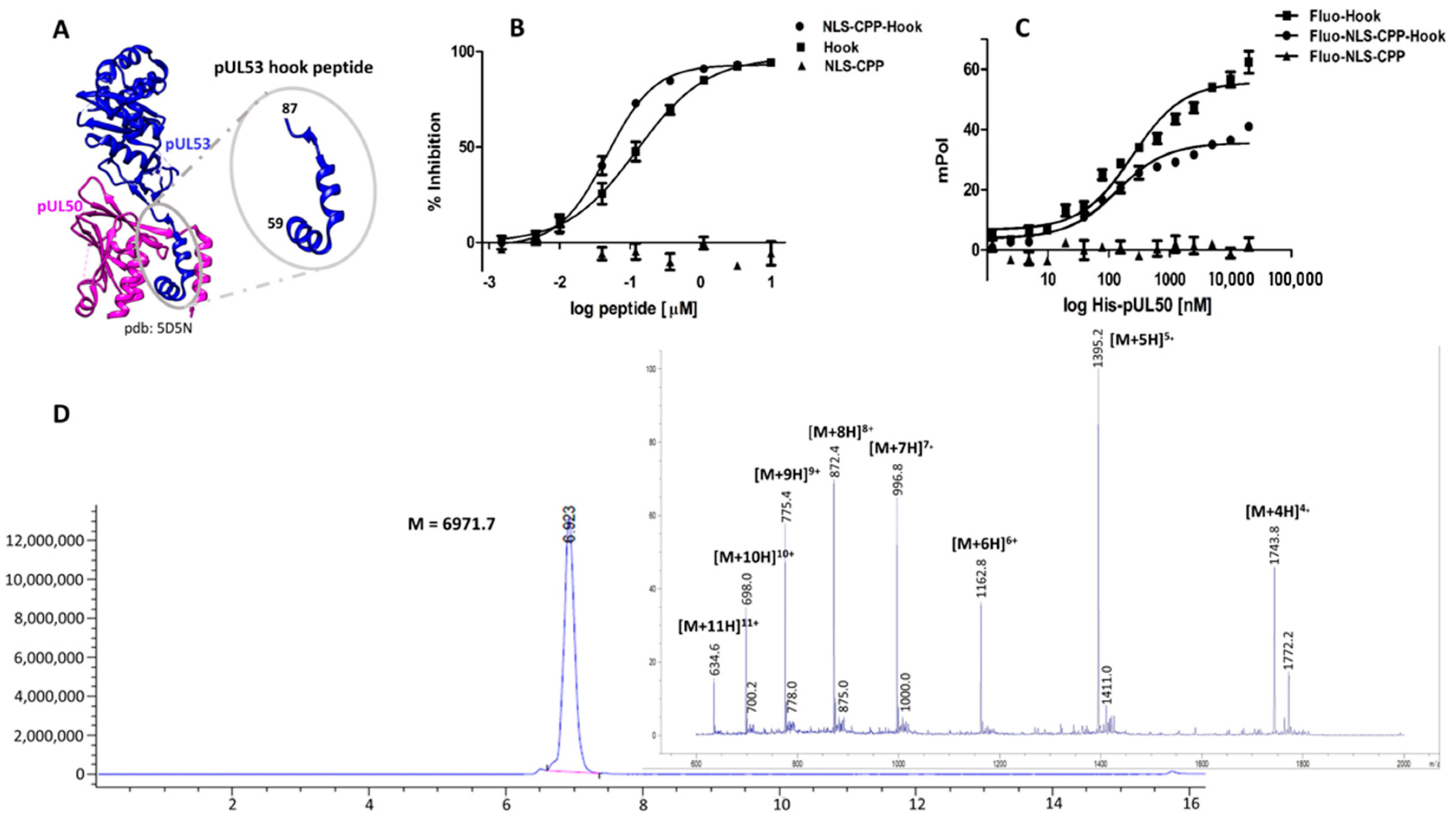
2.1. Design and Evaluation of the pUL53 Hook Fusion Peptide

2.2. Cellular and Nuclear Peptide Uptake
2.3. Delocalization of pUL53 from the Nuclear Rim
2.4. Antiviral Activity and Cytotoxicity
3. Materials and Methods
3.1. Peptide Synthesis
3.2. pUL50 Binding and pUL50-pUL53 Inhibition Assay
3.3. Cellular and Nuclear Uptake of Peptides
3.4. Cell Culture and Virus Infection
3.5. Indirect Immunofluorescence Assay and Confocal Laser-Scanning Microscopy
3.6. Quantitative Polymerase Chain Reaction (qPCR)
3.7. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schlick, K.; Grundbichler, M.; Auberger, J.; Kern, J.M.; Hell, M.; Hohla, F.; Hopfinger, G.; Greil, R. Cytomegalovirus reactivation and its clinical impact in patients with solid tumors. Infect. Agents Cancer 2015, 10, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schottstedt, V.; Blümel, J.; Burger, R.; Drosten, C.; Gröner, A.; Gürtler, L.; Heiden, M.; Hildebrandt, M.; Jansen, B.; Montag-Lessing, T.; et al. Human Cytomegalovirus (HCMV)—Revised. Transfus. Med. Hemother. 2010, 37, 365–375. [Google Scholar] [PubMed] [Green Version]
- Andrei, G.; De Clercq, E.; Snoeck, R. Drug Targets in Cytomegalovirus Infection. Infect. Disord. Drug Targets 2009, 9, 201–222. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Kosugi, I.; Meguro, S.; Iwashita, T. Pathogenesis of developmental anomalies of the central nervous system induced by congenital cytomegalovirus infection. Pathol. Int. 2017, 67, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Brizić, I.; Hiršl, L.; Britt, W.J.; Krmpotić, A.; Jonjić, S. Immune responses to congenital cytomegalovirus infection. Microbes Infect. 2018, 20, 543–551. [Google Scholar] [CrossRef]
- Gilden, D.H.; Mahalingam, R.; Cohrs, R.J.; Tyler, K.L. Herpesvirus infections of the nervous system. Nat. Clin. Pract. Neurol. 2007, 3, 82–94. [Google Scholar] [CrossRef]
- Perera, M.R.; Wills, M.R.; Sinclair, J.H. HCMV Antivirals and Strategies to Target the Latent Reservoir. Viruses 2021, 13, 817. [Google Scholar] [CrossRef]
- Britt, W.J.; Prichard, M.N. New therapies for human cytomegalovirus infections. Antivir. Res. 2018, 159, 153–174. [Google Scholar] [CrossRef]
- Shigle, T.L.; Handy, V.W.; Chemaly, R.F. Letermovir and its role in the prevention of cytomegalovirus infection in seropositive patients receiving an allogeneic hematopoietic cell transplant. Ther. Adv. Hematol. 2020, 11, 2040620720937150. [Google Scholar] [CrossRef]
- Kang, C. Maribavir: First Approval. Drugs 2022, 82, 335–340. [Google Scholar] [CrossRef]
- Gandhi, R.G.; Kotton, C.N. Evaluating the Safety of Maribavir for the Treatment of Cytomegalovirus. Ther. Clin. Risk Manag. 2022, 18, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Lurain, N.S.; Chou, S. Antiviral Drug Resistance of Human Cytomegalovirus. Clin. Microbiol. Rev. 2010, 23, 689–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coen, D.M.; Whitley, R.J. Antiviral drugs and antiviral drug resistance. Curr. Opin. Virol. 2011, 1, 545–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-P.; Chen, M.-R. Escape of herpesviruses from the nucleus. Rev. Med. Virol. 2010, 20, 214–230. [Google Scholar] [CrossRef]
- Roller, R.J.; Baines, J.D. Herpesvirus Nuclear Egress. Adv. Anat. Embryol. Cell Biol. 2017, 223, 143–169. [Google Scholar]
- Molenberghs, F.; Bogers, J.J.; De Vos, W.H. Confined no more: Viral mechanisms of nuclear entry and egress. Int. J. Biochem. Cell Biol. 2020, 129, 105875. [Google Scholar] [CrossRef]
- Marschall, M.; Muller, Y.A.; Diewald, B.; Sticht, H.; Milbradt, J. The human cytomegalovirus nuclear egress complex unites multiple functions: Recruitment of effectors, nuclear envelope rearrangement, and docking to nuclear capsids. Rev. Med. Viol. 2017, 27, e1934. [Google Scholar] [CrossRef]
- Lye, M.F.; Wilkie, A.R.; Filman, D.J.; Hogle, J.M.; Coen, D.M. Getting to and through the inner nuclear membrane during herpesvirus nuclear egress. Curr. Opin. Cell Biol. 2017, 46, 9–16. [Google Scholar] [CrossRef]
- Bigalke, J.M.; Heldwein, E.E. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J. 2015, 34, 2921–2936. [Google Scholar] [CrossRef]
- Draganova, E.B.; Thorsen, M.K.; Heldwein, E.E. Nuclear Egress. Curr. Issues Mol. Biol. 2021, 41, 125–170. [Google Scholar] [CrossRef]
- Bigalke, J.M.; Heldwein, E.E. The Great (Nuclear) Escape: New Insights into the Role of the Nuclear Egress Complex of Herpesviruses. J. Virol. 2015, 89, 9150–9153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camozzi, D.; Pignatelli, S.; Valvo, C.; Lattanzi, G.; Capanni, C.; Dal Monte, P.; Landini, M.P. Remodelling of the nuclear lamina during human cytomegalovirus infection: Role of the viral proteins pUL50 and pUL53. J. Gen. Virol. 2008, 89, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Marschall, M.; Hage, S.; Conrad, M.; Alkhashrom, S.; Kicuntod, J.; Schweininger, J.; Kriegel, M.; Losing, J.; Tillmanns, J.; Neipel, F.; et al. Nuclear Egress Complexes of HCMV and Other Herpesviruses: Solving the Puzzle of Sequence Coevolution, Conserved Structures and Subfamily-Spanning Binding Properties. Viruses 2020, 12, 683. [Google Scholar] [CrossRef] [PubMed]
- Alkhashrom, S.; Kicuntod, J.; Häge, S.; Schweininger, J.; Muller, Y.A.; Lischka, P.; Marschall, M.; Eichler, J. Exploring the Human Cytomegalovirus Core Nuclear Egress Complex as a Novel Antiviral Target: A New Type of Small Molecule Inhibitors. Viruses 2021, 13, 471. [Google Scholar] [CrossRef] [PubMed]
- Walzer, S.A.; Egerer-Sieber, C.; Sticht, H.; Sevvana, M.; Hohl, K.; Milbradt, J.; Muller, Y.A.; Marschall, M. Crystal Structure of the Human Cytomegalovirus pUL50-pUL53 Core Nuclear Egress Complex Provides Insight into a Unique Assembly Scaffold for Virus-Host Protein Interactions. J. Biol. Chem. 2015, 290, 27452–27458. [Google Scholar] [CrossRef] [Green Version]
- Muller, Y.A.; Hage, S.; Alkhashrom, S.; Hollriegl, T.; Weigert, S.; Dolles, S.; Hof, K.; Walzer, S.A.; Egerer-Sieber, C.; Conrad, M.; et al. High-resolution crystal structures of two prototypical beta- and gamma-herpesviral nuclear egress complexes unravel the determinants of subfamily specificity. J. Biol. Chem. 2020, 295, 3189–3201. [Google Scholar] [CrossRef]
- Yang, N.J.; Hinner, M.J. Getting Across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. Methods Mol. Biol. 2015, 1266, 29–53. [Google Scholar]
- Feni, L.; Neundorf, I. The Current Role of Cell-Penetrating Peptides in Cancer Therapy. Adv. Exp. Med. Biol. 2017, 1030, 279–295. [Google Scholar]
- Reissmann, S. Cell penetration: Scope and limitations by the application of cell-penetrating peptides. J. Pept. Sci. 2014, 20, 760–784. [Google Scholar] [CrossRef]
- Xu, J.; Khan, A.R.; Fu, M.; Wang, R.; Ji, J.; Zhai, G. Cell-penetrating peptide: A means of breaking through the physiological barriers of different tissues and organs. J. Control. Release 2019, 309, 106–124. [Google Scholar] [CrossRef]
- Madani, F.; Lindberg, S.; Langel, Ü.; Futaki, S.; Gräslund, A. Mechanisms of Cellular Uptake of Cell-Penetrating Peptides. Biophys 2011, 2011, 414729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vale, N.; Duarte, D.; Silva, S.; Correia, A.S.; Costa, B.; Gouveia, M.J.; Ferreira, A. Cell-penetrating peptides in oncologic pharmacotherapy: A review. Pharmacol. Res. 2020, 162, 105231. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, J.D.; Flynn, N.H. Cell-penetrating peptides transport therapeutics into cells. Pharmacol. Ther. 2015, 154, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habault, J.; Poyet, J.L. Recent Advances in Cell Penetrating Peptide-Based Anticancer Therapies. Molecules 2019, 24, 927. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Wu, T.; Zhang, B.; Liu, S.; Song, W.; Qiao, J.; Ruan, H. Types of nuclear localization signals and mechanisms of protein import into the nucleus. Cell Commun. Signal. 2021, 19, 60. [Google Scholar] [CrossRef]
- Lange, A.; Mills, R.E.; Lange, C.J.; Stewart, M.; Devine, S.E.; Corbett, A.H. Classical Nuclear Localization Signals: Definition, Function, and Interaction with Importin alpha. J. Biol. Chem. 2007, 282, 5101–5105. [Google Scholar] [CrossRef] [Green Version]
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Peng, Q.; Wang, P.; Zhou, B. Progress in Research and Application of HIV-1 TAT-Derived Cell-Penetrating Peptide. J. Membr. Biol. 2017, 250, 115–122. [Google Scholar] [CrossRef]
- Ciobanasu, C.; Siebrasse, J.P.; Kubitscheck, U. Cell-penetrating HIV1 TAT peptides can generate pores in model membranes. Biophys. J. 2010, 99, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Ciobanasu, C.; Harms, E.; Tunnemann, G.; Cardoso, M.C.; Kubitscheck, U. Cell-penetrating HIV1 TAT peptides float on model lipid bilayers. Biochemistry 2009, 48, 4728–4737. [Google Scholar] [CrossRef] [Green Version]
- Schmeiser, C.; Borst, E.; Sticht, H.; Marschall, M.; Milbradt, J. The cytomegalovirus egress proteins pUL50 and pUL53 are translocated to the nuclear envelope through two distinct modes of nuclear import. J. Gen. Virol. 2013, 94, 2056–2069. [Google Scholar] [CrossRef] [PubMed]
- Usui, K.; Kikuchi, T.; Mie, M.; Kobatake, E.; Mihara, H. Systematic screening of the cellular uptake of designed alpha-helix peptides. Bioorg. Med. Chem. 2013, 21, 2560–2567. [Google Scholar] [CrossRef] [PubMed]
- Scheller, A.; Oehlke, J.; Wiesner, B.; Dathe, M.; Krause, E.; Beyermann, M.; Melzig, M.; Bienert, M. Structural requirements for cellular uptake of α-helical amphipathic peptides. J. Pept. Sci. 1999, 5, 185–194. [Google Scholar] [CrossRef]
- Gronewold, A.; Horn, M.; Neundorf, I. Design and biological characterization of novel cell-penetrating peptides preferentially targeting cell nuclei and subnuclear regions. Beilstein J. Org. Chem. 2018, 14, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Weißenborn, L.; Richel, E.; Hüseman, H.; Welzer, J.; Beck, S.; Schäfer, S.; Sticht, H.; Überla, K.; Eichler, J. Smaller, Stronger, More Stable: Peptide Variants of a SARS-CoV-2 Neutralizing Miniprotein. Int. J. Mol. Sci. 2022, 23, 6309. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | IC50 [nM] | Kd [nM] |
|---|---|---|---|
| (Fluo a-)Hook | Ac b(Fluo-Aoa c)-59 dLTLHDLHDIFREHPELELKYLNMMKMAIT87-NH2 | 125 ± 16.9 e | 228.4 ± 32.22 |
| (Fluo-)NLS-CPP | Ac(Fluo-Aoa)-18RSLLRKRRQR27-Aoa-47YGRKKRRQRRRPP59-NH2 | >10,000 | >10,000 |
| (Fluo-)NLS-CPP-Hook | Ac(Fluo-Aoa)-RSLLRKRRQR-Aoa-YGRKKRRQRRRPP-Aoa-LTLHDLHDIFREHPELELKYLNMMKMAIT-NH2 | 46.9 ± 4.7 | 122.4 ± 20.6 |
| Peptide | EC50 [µM] | CC50 [µM] | SI b |
|---|---|---|---|
| Hook peptide | >10 | >100 | - |
| NLS-CPP | >10 | >100 | - |
| NLS-CPP-Hook | 5.6 ± 0.78 a | 44.2 ± 6.9 | 7.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkhashrom, S.; Kicuntod, J.; Stillger, K.; Lützenburg, T.; Anzenhofer, C.; Neundorf, I.; Marschall, M.; Eichler, J. A Peptide Inhibitor of the Human Cytomegalovirus Core Nuclear Egress Complex. Pharmaceuticals 2022, 15, 1040. https://doi.org/10.3390/ph15091040
Alkhashrom S, Kicuntod J, Stillger K, Lützenburg T, Anzenhofer C, Neundorf I, Marschall M, Eichler J. A Peptide Inhibitor of the Human Cytomegalovirus Core Nuclear Egress Complex. Pharmaceuticals. 2022; 15(9):1040. https://doi.org/10.3390/ph15091040
Chicago/Turabian StyleAlkhashrom, Sewar, Jintawee Kicuntod, Katharina Stillger, Tamara Lützenburg, Christian Anzenhofer, Ines Neundorf, Manfred Marschall, and Jutta Eichler. 2022. "A Peptide Inhibitor of the Human Cytomegalovirus Core Nuclear Egress Complex" Pharmaceuticals 15, no. 9: 1040. https://doi.org/10.3390/ph15091040
APA StyleAlkhashrom, S., Kicuntod, J., Stillger, K., Lützenburg, T., Anzenhofer, C., Neundorf, I., Marschall, M., & Eichler, J. (2022). A Peptide Inhibitor of the Human Cytomegalovirus Core Nuclear Egress Complex. Pharmaceuticals, 15(9), 1040. https://doi.org/10.3390/ph15091040