
The Challenge of Diagnosing and Managing Pulmonary Arterial Hypertension in Systemic Sclerosis with Interstitial Lung Disease


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
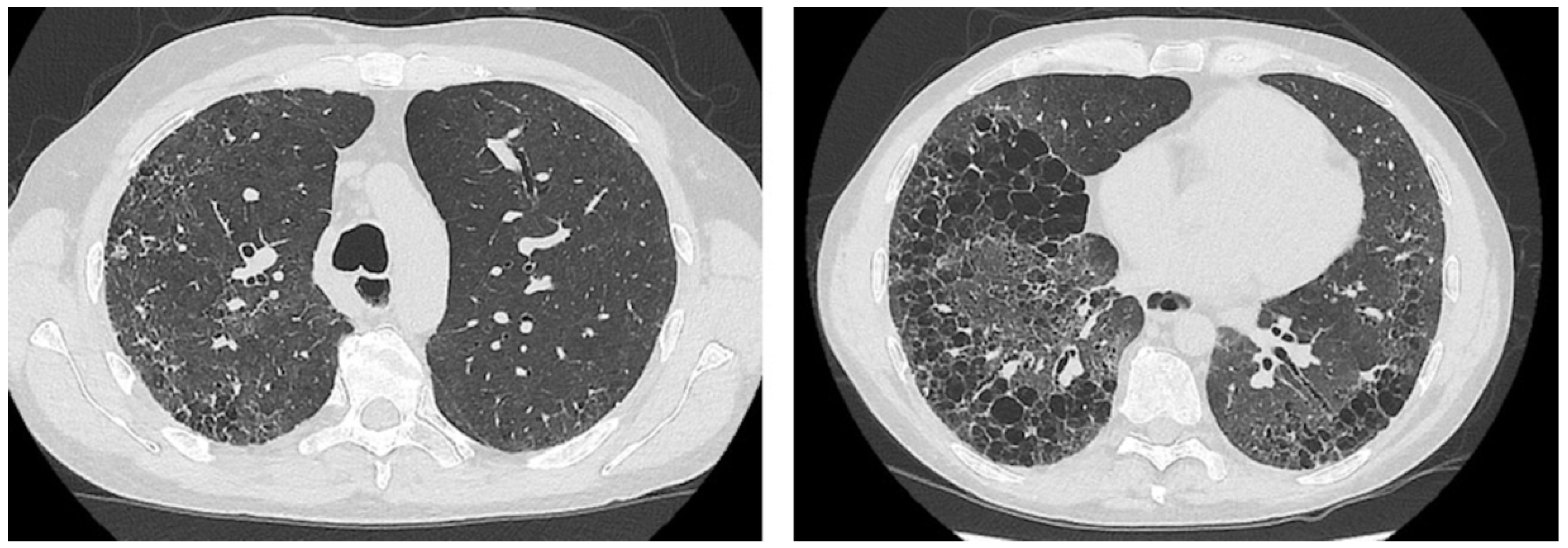
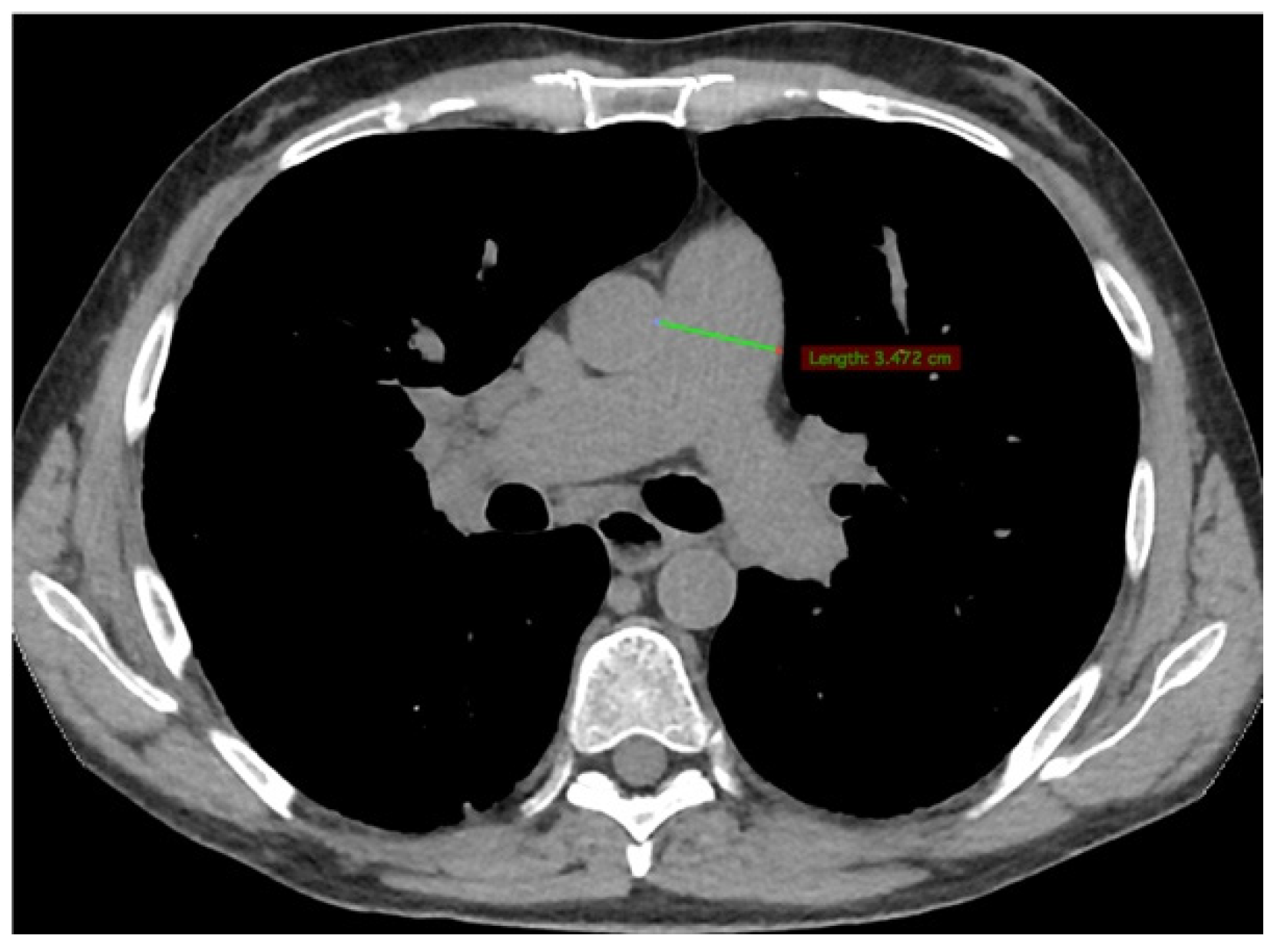
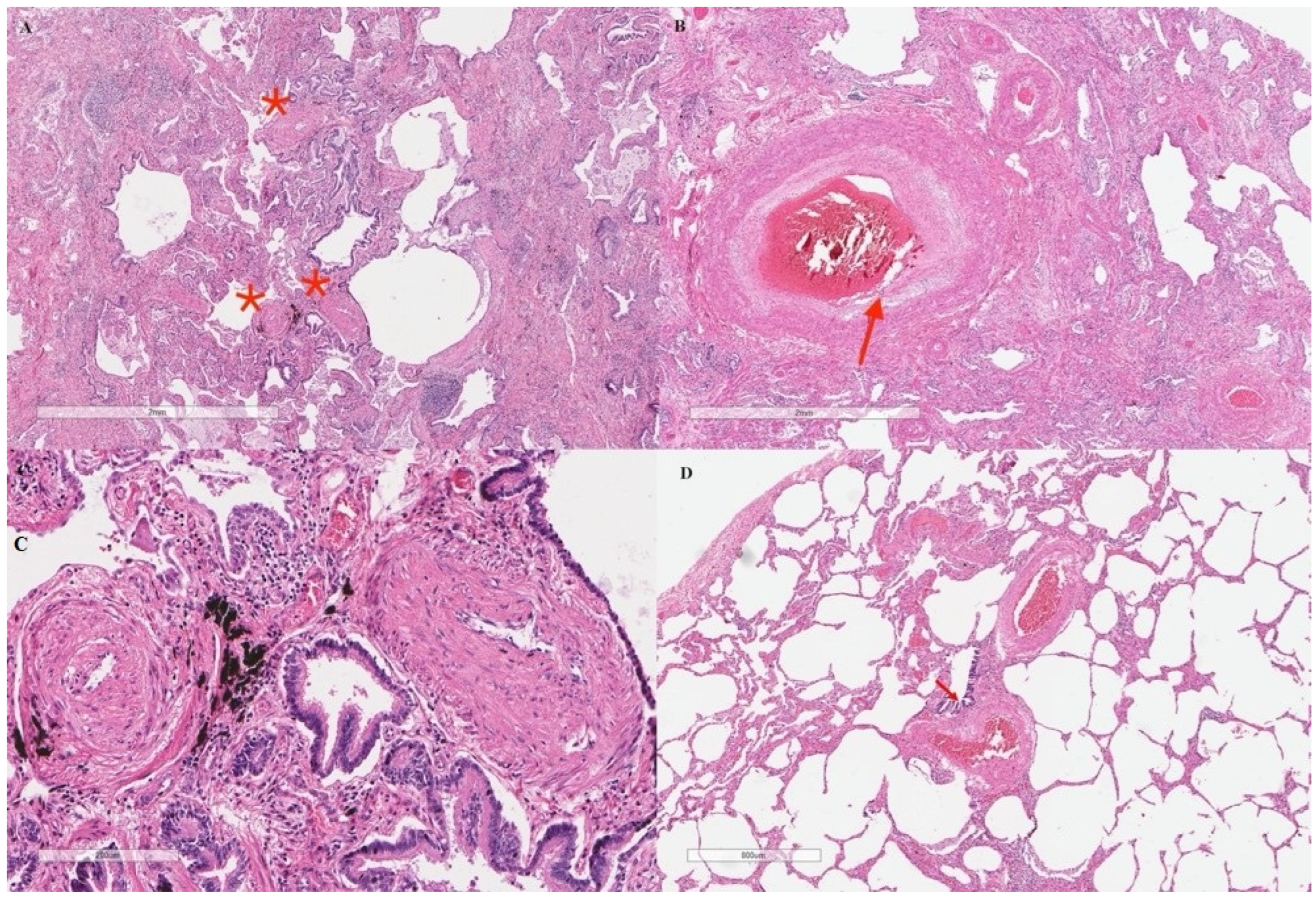
2. Case Presentation
3. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zanatta, E.; Polito, P.; Famoso, G.; Larosa, M.; De Zorzi, E.; Scarpieri, E.; Cozzi, F.; Doria, A. Pulmonary arterial hypertension in connective tissue disorders: Pathophysiology and treatment. Exp. Biol. Med. 2019, 244, 120–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruni, C.; De Luca, G.; Lazzaroni, M.G.; Zanatta, E.; Lepri, G.; Airò, P.; Dagna, L.; Doria, A.; Matucci-Cerinic, M. Screening for pulmonary arterial hypertension in systemic sclerosis: A systematic literature review. Eur. J. Intern. Med. 2020, 78, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Noordegraaf, A.V.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [PubMed]
- Launay, D.; Montani, D.; Hassoun, P.M.; Cottin, V.; Le Pavec, J.; Clerson, P.; Sitbon, O.; Jaïs, X.; Savale, L.; Weatherald, J.; et al. Clinical phenotypes and survival of pre-capillary pulmonary hypertension in systemic sclerosis. PLoS ONE 2018, 13, e0197112. [Google Scholar]
- Young, A.; Vummidi, D.; Visovatti, S.; Homer, K.; Wilhalme, H.; White, E.S.; Flaherty, K.; McLaughlin, V.; Khanna, D. Prevalence, Treatment, and Outcomes of Coexistent Pulmonary Hypertension and Interstitial Lung Disease in Systemic Sclerosis. Arthritis Rheumatol. 2019, 71, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Faccini, A.; Oppizzi, M.; Margonato, A.; Galderisi, M.; Sabbadini, M.G.; Franchini, S.; Camici, P.G. Coronary microvascular dysfunction in asymptomatic patients affected by systemic sclerosis—Limited vs. diffuse form. Circ. J. 2015, 79, 825–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elhai, M.; Meune, C.; Boubaya, M.; Avouac, J.; Hachulla, E.; Balbir-Gurman, A.; Riemekasten, G.; Airò, P.; Joven, B.; Vettori, S.; et al. Mapping and predicting mortality from systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S.; Preston, I.R. Right heart catheterization: Best practice and pitfalls in pulmonary hypertension. Eur. Respir. Rev. 2015, 24, 642–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Launay, D.; Savale, L.; Berezne, A.; Le Pavec, J.; Hachulla, E.; Mouthon, L.; Sitbon, O.; Lambert, B.; Gaudric, M.; Jais, X.; et al. Lung and heart-lung transplantation for systemic sclerosis patients. A monocentric experience of 13 patients, review of the literature and position paper of a multidisciplinary Working Group. Presse Med. 2014, 43, e345–e363. [Google Scholar] [CrossRef] [PubMed]
- Fayed, H.; Coghlan, J.G. Pulmonary Hypertension Associated with Connective Tissue Disease. Semin. Respir. Crit. Care Med. 2019, 40, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Ruaro, B.; Salton, F.; Baratella, E.; Confalonieri, P.; Geri, P.; Pozzan, R.; Torregiani, C.; Bulla, R.; Confalonieri, M.; Matucci-Cerinic, M.; et al. An Overview of Different Techniques for Improving the Treatment of Pulmonary Hypertension Secondary in Systemic Sclerosis Patients. Diagnostics 2022, 12, 616. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanatta, E.; Marra, M.P.; Famoso, G.; Balestro, E.; Giraudo, C.; Calabrese, F.; Rea, F.; Doria, A. The Challenge of Diagnosing and Managing Pulmonary Arterial Hypertension in Systemic Sclerosis with Interstitial Lung Disease. Pharmaceuticals 2022, 15, 1042. https://doi.org/10.3390/ph15091042
Zanatta E, Marra MP, Famoso G, Balestro E, Giraudo C, Calabrese F, Rea F, Doria A. The Challenge of Diagnosing and Managing Pulmonary Arterial Hypertension in Systemic Sclerosis with Interstitial Lung Disease. Pharmaceuticals. 2022; 15(9):1042. https://doi.org/10.3390/ph15091042
Chicago/Turabian StyleZanatta, Elisabetta, Martina Perazzolo Marra, Giulia Famoso, Elisabetta Balestro, Chiara Giraudo, Fiorella Calabrese, Federico Rea, and Andrea Doria. 2022. "The Challenge of Diagnosing and Managing Pulmonary Arterial Hypertension in Systemic Sclerosis with Interstitial Lung Disease" Pharmaceuticals 15, no. 9: 1042. https://doi.org/10.3390/ph15091042
APA StyleZanatta, E., Marra, M. P., Famoso, G., Balestro, E., Giraudo, C., Calabrese, F., Rea, F., & Doria, A. (2022). The Challenge of Diagnosing and Managing Pulmonary Arterial Hypertension in Systemic Sclerosis with Interstitial Lung Disease. Pharmaceuticals, 15(9), 1042. https://doi.org/10.3390/ph15091042