
Deconstructing Markush: Improving the R&D Efficiency Using Library Selection in Early Drug Discovery




Abstract
:1. Introduction
2. Results and Discussion
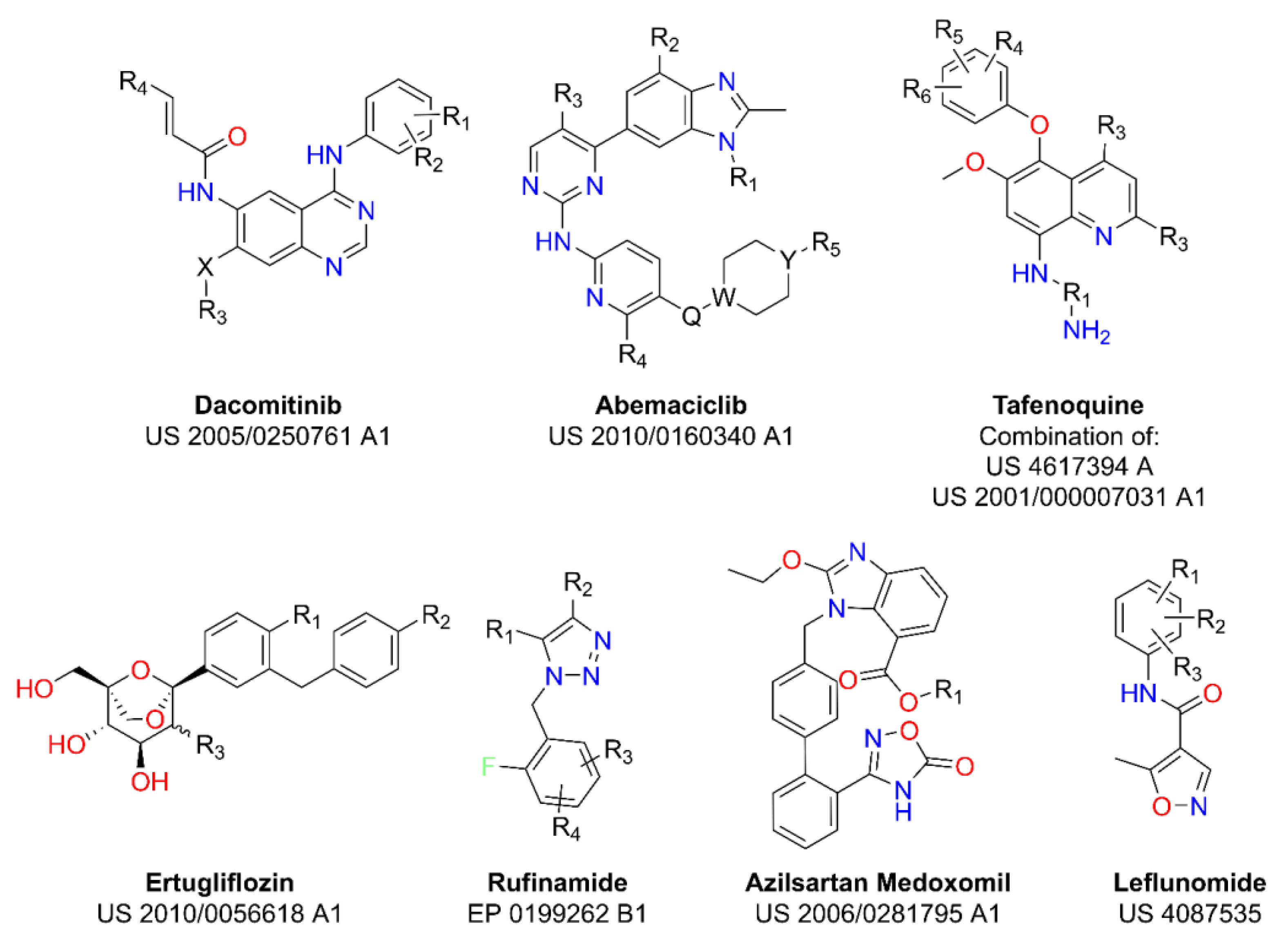
2.1. Markush Combinatorial Library
2.2. Bibliographical Database and Bibliographic Combinatorial Library
2.3. Clustering Methods for Chemical Space Exploration
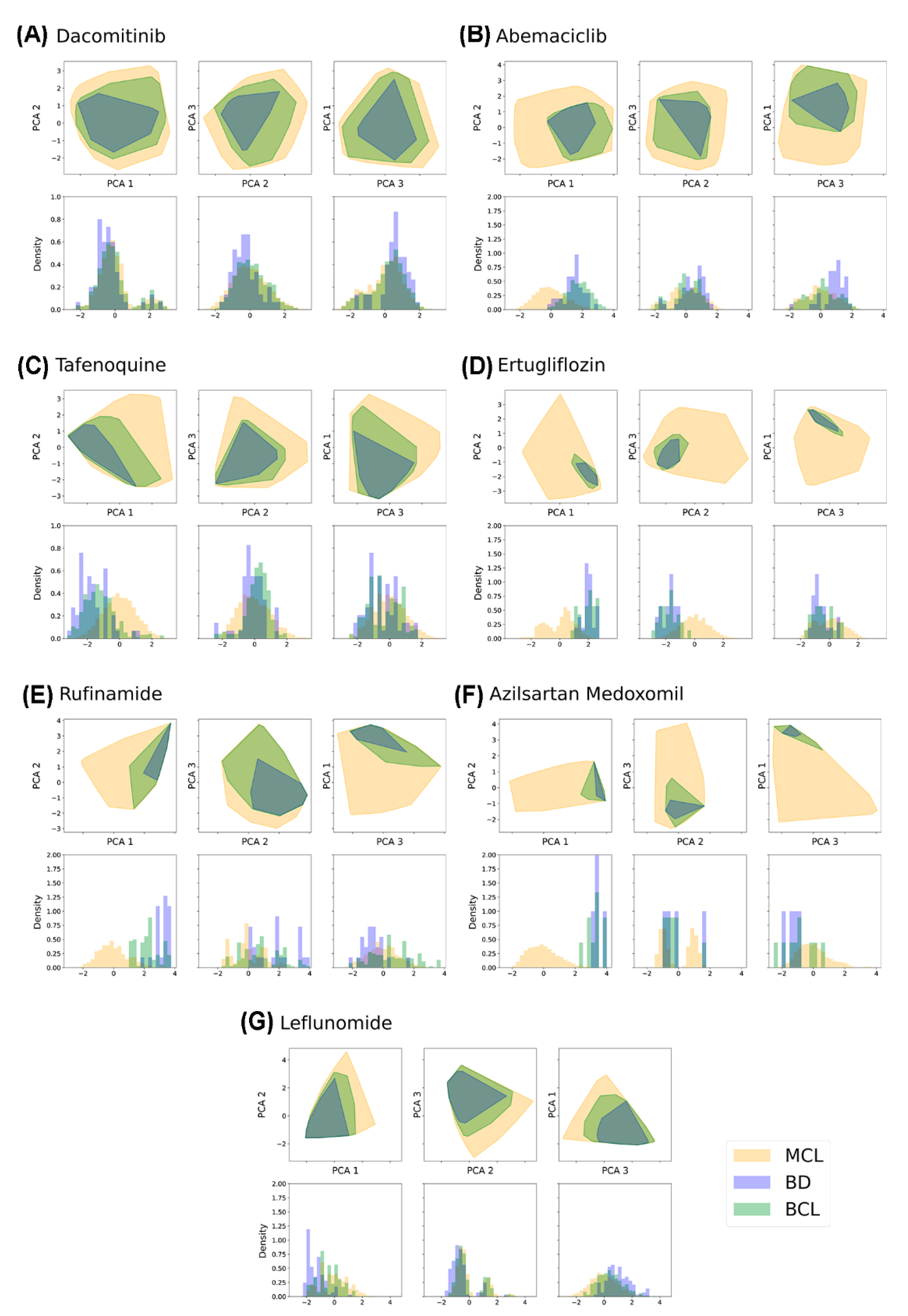
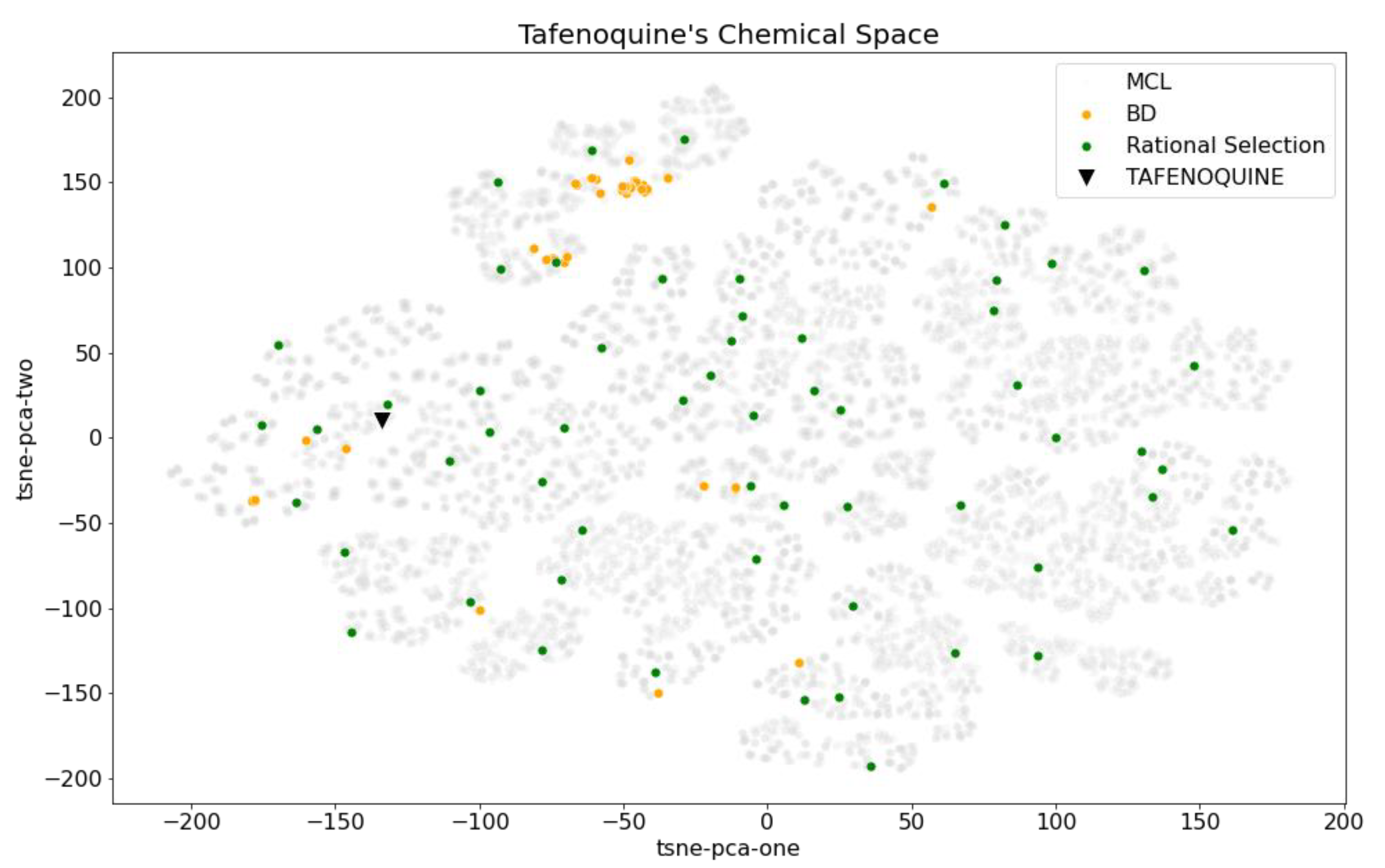
2.4. Bibliographical Representativeness in Its Chemical Space
2.5. Comparing the Chemical Space Described by MCL, BD, and BCL
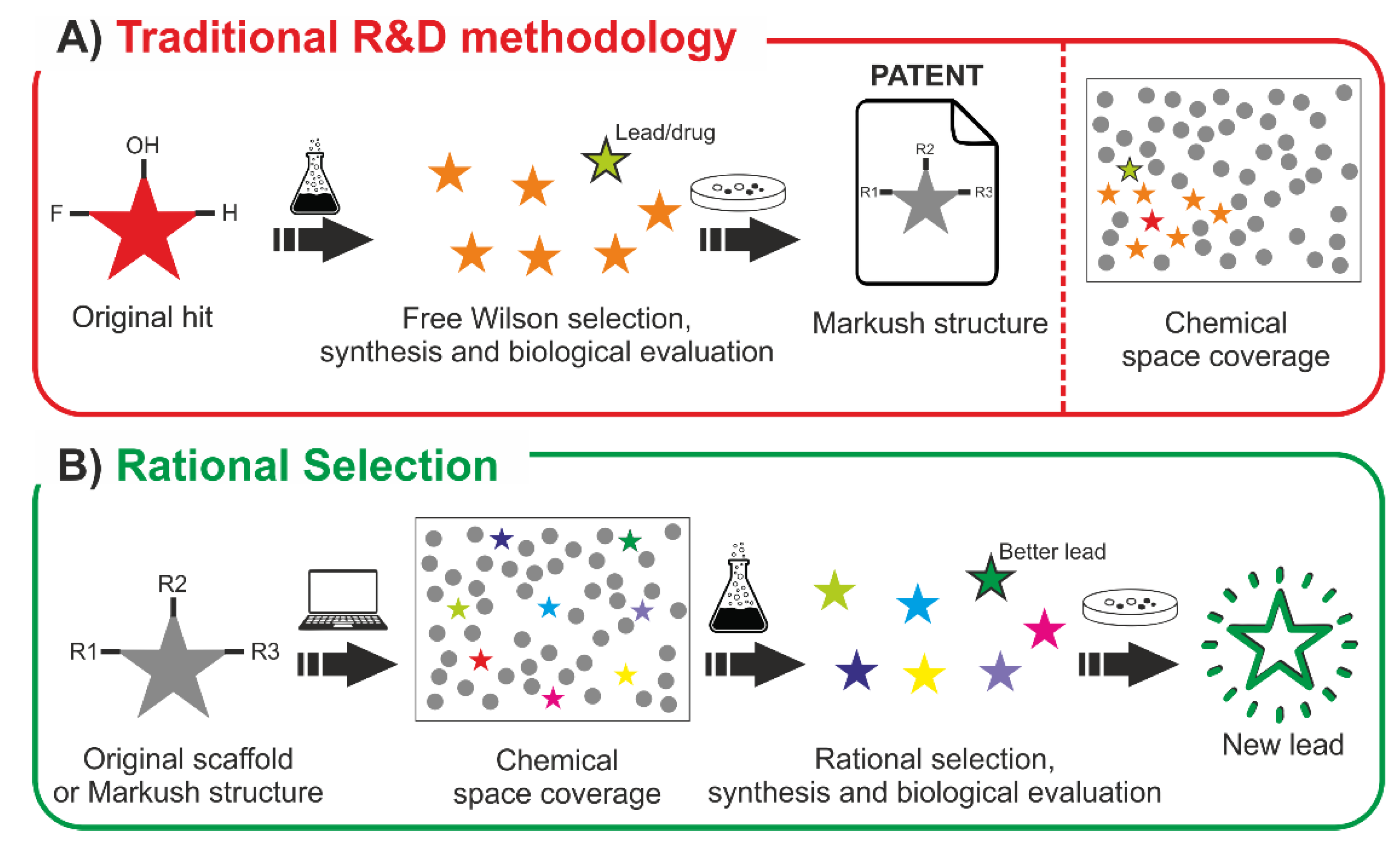
2.6. Towards a More Efficient Methodology
3. Materials and Methods
3.1. Enumeration of Combinatorial Libraries
3.2. Describing the Chemical Space
3.3. Clustering and Partitioning Methodologies
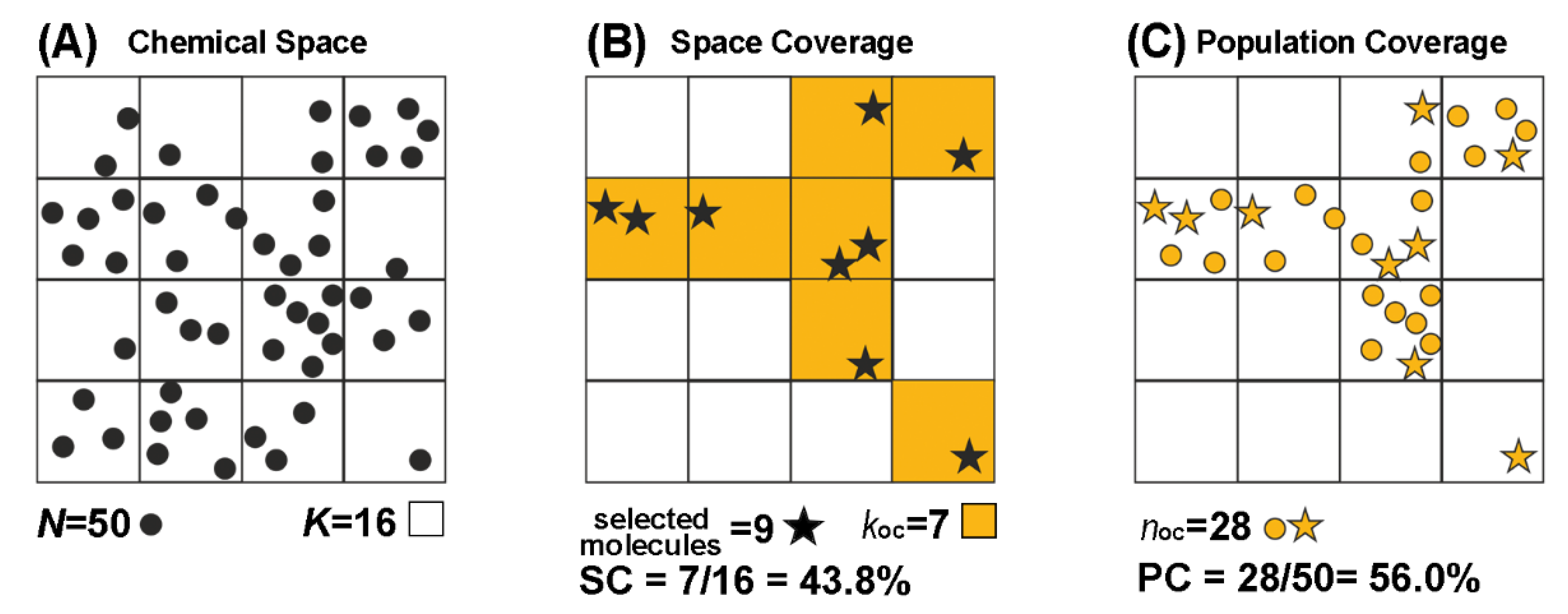
3.4. Space and Population Coverage
3.5. Number of Clusters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruffolo, R.R. Why Has R&D Productivity Declined in the Pharmaceutical Industry? Expert Opin. Drug Discov. 2006, 1, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the Decline in Pharmaceutical R&D Efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I.; Gottfries, J. Chemography: The Art of Navigating in Chemical Space. J. Comb. Chem. 2001, 3, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Bohacek, R.S.; Mcmartin, C.; Guida, W.C. The Art and Practice of Structure-Based Drug Design: A Molecular Modeling Perspective. Med. Res Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- Miller, M.A. Chemical Database Techniques in Drug Discovery. Nat. Rev. Drug Discov. 2002, 1, 220–227. [Google Scholar] [CrossRef]
- Willett, P.; Barnard, J.M.; Downs, G.M. Chemical Similarity Searching. J. Chem. Inf. Comput. Sci. 1998, 38, 983–996. [Google Scholar] [CrossRef]
- Vogt, M. How Do We Optimize Chemical Space Navigation? Expert Opin. Drug Discov. 2020, 15, 523–525. [Google Scholar] [CrossRef]
- Snarey, M.; Terrett, N.K.; Willett, P.; Wilton, D.J. Comparison of Algorithms for Dissimilarity-Based Compound Selection. J. Mol. Graph. Model. 1997, 15, 372–385. [Google Scholar] [CrossRef]
- Dunbar, J.B. Cluster-Based Selection. Perspect. Drug Discov. Des. 1997, 7, 51–63. [Google Scholar] [CrossRef]
- Rabal, O.; Pascual, R.; Borrell, J.I.; Teixidó, J. Cell-Integral-Diversity Criterion: A Proposal for Minimizing Cluster Artifact in Cell-Based Selections. J. Chem. Inf. Model. 2007, 47, 1886–1896. [Google Scholar] [CrossRef]
- Mason, J.S.; Picket, S.D. Partition-Based Selection. Perspect. Drug Discov. Des. 1997, 7, 85–114. [Google Scholar] [CrossRef]
- Gorse, A.-D. Diversity in Medicinal Chemistry Space. Curr. Top. Med. Chem. 2006, 6, 3–18. [Google Scholar] [CrossRef]
- Gilad, Y.; Nadassy, K.; Senderowitz, H. A Reliable Computational Workflow for the Selection of Optimal Screening Libraries. J. Cheminform. 2015, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.; Hopkins, A. Navigating Chemical Space for Biology and Medicine. Nature 2004, 432, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I. Chemical Space Navigation in Lead Discovery. Curr. Opin. Chem. Biol. 2002, 6, 384–389. [Google Scholar] [CrossRef]
- Reymond, J.; Van Deursen, R.; Blum, L.C.; Ruddigkeit, L. Chemical Space as a Source for New Drugs. MedChemComm 2010, 1, 30–38. [Google Scholar] [CrossRef]
- Zabolotna, Y.; Lin, A.; Horvath, D.; Marcou, G.; Volochnyuk, D.M.; Varnek, A. Chemography: Searching for Hidden Treasures. J. Chem. Inf. Model. 2021, 61, 179–188. [Google Scholar] [CrossRef]
- Lin, A.; Horvath, D.; Afonina, V.; Marcou, G.; Reymond, J.L.; Varnek, A. Mapping of the Available Chemical Space versus the Chemical Universe of Lead-Like Compounds. ChemMedChem 2018, 13, 540–554. [Google Scholar] [CrossRef]
- Fink, T.; Raymond, J.L. Virtual Exploration of the Chemical Universe up to 11 Atoms of C, N, O, F: Assembly of 26.4 Million Structures (110.9 Million Stereoisomers) and Analysis for New Ring Systems, Stereochemistry, Physicochemical Properties, Compound Classes, and Drug Discove. J. Chem. Inf. Model. 2007, 47, 342–353. [Google Scholar] [CrossRef]
- Ertl, P. Cheminformatics Analysis of Organic Substituents: Identification of the Most Common Substituents, Calculation of Substituent Properties, and Automatic Identification of Drug-like Bioisosteric Groups. J. Chem. Inf. Comput. Sci. 2003, 43, 374–380. [Google Scholar] [CrossRef]
- Warr, W.A.; Nicklaus, M.C.; Nicolaou, C.A.; Rarey, M. Exploration of Ultralarge Compound Collections for Drug Discovery. J. Chem. Inf. Model. 2022, 62(9), 2021–2034. [Google Scholar] [CrossRef] [PubMed]
- Pascual, R.; Mateu, M.; Gasteiger, J.; Borrell, J.I.; Teixidó, J. Design and Analysis of a Combinatorial Library of HEPT Analogues: Comparison of Selection Methodologies and Inspection of the Actually Covered Chemical Space. J. Chem. Inf. Comput. Sci. 2003, 43, 199–207. [Google Scholar] [CrossRef]
- Puig-De-La-Bellacasa, R.; Giménez, L.; Pettersson, S.; Pascual, R.; Gonzalo, E.; Esté, J.A.; Clotet, B.; Borrell, J.I.; Teixidó, J. Diverse Combinatorial Design, Synthesis and in Vitro Evaluation of New HEPT Analogues as Potential Non-Nucleoside HIV-1 Reverse Transcription Inhibitors. Eur. J. Med. Chem. 2012, 54, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Barnard, J.M. A Comparison of Different Approaches to Markush Structure Handling. J. Chem. Inf. Comput. Sci. 1991, 31, 64–68. [Google Scholar] [CrossRef]
- Simmons, E.S. The Grammar of Markush Structure Searching: Vocabulary vs.. Syntax. World Pat. Inf. 1991, 13, 173. [Google Scholar] [CrossRef]
- Free, S.M.; Wilson, J.W. A Mathematical Contribution to Structure-Activity Studies. J. Med. Chem. 1964, 7, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Kubinyi, H. Free Wilson Analysis. Theory, Applications and Its Relationship to Hansch Analysis. Quant. Struct. Relatsh. 1988, 7, 121–133. [Google Scholar] [CrossRef]
- Chen, H.; Carlsson, L.; Eriksson, M.; Varkonyi, P.; Norinder, U.; Nilsson, I. Beyond the Scope of Free-Wilson Analysis: Building Interpretable QSAR Models with Machine Learning Algorithms. J. Chem. Inf. Model. 2013, 53, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Craig, P.N. Comparison of the Hansch and Free-Wilson Approaches to Structure-Activity Correlation. Biol. Correl. Hansch Approach 1974, 114, 115–129. [Google Scholar]
- Pascual, R.; Borrell, J.I.; Teixidó, J. Analysis of Selection Methodologies for Combinatorial Library Design. Mol. Divers. 2003, 6, 121–133. [Google Scholar] [CrossRef]
- Willett, P. Dissimilarity-Based Algorithms for Selecting Structurally Diverse Sets of Compounds. J. Comput. Biol. 1999, 6, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zheng, S.; Luo, X.; Shen, J.; Zhu, W.; Liu, H.; Gui, C.; Zhang, J.; Zheng, M.; Chum, M.P.; et al. Focused Combinatorial Library Design Based on Structural Diversity, Druglikeness and Binding Affinity Score. J. Comb. Chem. 2005, 7, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Meinl, T.; Ostermann, C.; Berthold, M.R. Maximum-Score Diversity Selection for Early Drug Discovery. J. Chem. Inf. Model. 2011, 51, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Holliday, J.D.; Ranade, S.S.; Willett, P.A. Fast Algorithm for Selecting Sets of Dissimilar Molecules from Large Chemical Databases. Quant. Struct.−Act. Relat. 1995, 14, 501. [Google Scholar] [CrossRef]
- Willett, P. Similarity-Based Virtual Screening Using 2D Fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef]
- Groza, V.; Udrescu, M.; Bozdog, A.; Udrescu, L. Drug Repurposing Using Modularity Clustering in Drug-Drug Similarity Networks Based on Drug–Gene Interactions. Pharmaceutics 2021, 13, 2117. [Google Scholar] [CrossRef]
- Fakhoury, S.A.; Lee, H.T.; Reed, J.E.; Schlosser, K.M.; Sexton, K.E.; Tecle, H.; Winters, R.T. 4-phenylamino-quinazolin-6-yl-amides. U.S. Patent No. 7,772.243 B2, 2010. [Google Scholar]
- Mcchesney, J.; Nanayyakkra, D.N.; Bartlerr, M.; Ager, A.L. 8-aminoquinolines. U.S. Patent No. 2001/0007031 A1, 5 July 2001. [Google Scholar]
- Mascitti, V.; Collman, B.M. Dioxa-bicyclo[3.2.1]octane-2,3,4-triol dervatives. U.S. Patent No. 2010/0056618 A1, 4 March 2010. [Google Scholar]
- Coates, D.A.; Lawrence, M.; Knobeloch, J.M.; De Dios Magna, A.; De Prado Gonzalez, A.; Del Prado Catalina, M.F.; Garcia Paredes, M.C.; Martin de la Nava, M.; Martin Ortega Finger, M.D.; Martinez Perez, J.A.; et al. Protein Kinase Inhibitors. U.S. Patent No. 2010/0160340 A1, 24 June 2010. [Google Scholar]
- Heubach, G. 5-methyl-isoxazole-4-carboxylic Acid Anilides. U.S. Patent No. 4,087,535, 2 May 1978. [Google Scholar]
- Blumbergs, P.; LaMontagne, M.P. 4-methyl-5-(unsubstituted and substituted phenoxy)-2,6-dimethoxy-8- (aminoalkylamino) quinolines. U.S. Patent 4,617,394, 14 October 1986. [Google Scholar]
- Kuroita, T.; Sakamoto, H.; Ojima, M. Benzimidazole derivative and use as a receptor antagonist. U.S. Patent No. 7,572,920 B2, 11 August 2009. [Google Scholar]
- Meier, R. Fluorinated Benzyl Triazole Compounds. Patent No. EP 0199262, 15 April 1986. [Google Scholar]
- Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations. Available online: https://www.accessdata.fda.gov/scripts/cder/ob/index.cfm (accessed on 20 July 2021).
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2019, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Dutta, A.K.; Avery, B.A.; Wyandt, C.M. Development and Validation of a Stability-Indicating Reversed-Phase High Performance Liquid Chromatography Method for NPC 1161C, a Novel 8-Aminoquinoline Anti-Malarial Drug. J. Chromatogr. A 2006, 1110, 35–45. [Google Scholar] [CrossRef]
- Strube, R.E.; LaMontagne, M.P. 4-methyl-5-(Unsubstituted and Substituted Phenoxy)-6-methoxy-8-(aminoalkylamino)quinolines. U.S. Patent No. 4,431,807, 14 October 1984. [Google Scholar]
- Nodiff, E.A.; Tanabe, K.; Chen, E.H.; Saggiomo, A.J. Modifications of Primaquine as Antimalarials. 3. 5-Phenoxy Derivatives of Primaquine. J. Med. Chem. 1982, 4816, 1097–1101. [Google Scholar] [CrossRef]
- Azad, C.S.; Saxena, M.; Siddiqui, A.J.; Bhardwaj, J.; Puri, S.K.; Dutta, G.P.; Anand, N.; Saxena, A.K. Synthesis of Primaquine Glyco-Conjugates as Potential Tissue Schizontocidal Antimalarial Agents. Chem. Biol. Drug Des. 2017, 90, 254–261. [Google Scholar] [CrossRef]
- LaMontagne, M.P.; Blumbergs, P.; Strube, R.E. Antimalarials. 14. 5-(Aryloxy)-4-Methylprimaquine Analogues. A Highly Effective Series of Blood and Tissue Schizonticidal Agents. J. Med. Chem. 1982, 25, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.A.; Phyo, A.P. Drugs in Development for Malaria. Drugs 2018, 78, 861–879. [Google Scholar] [CrossRef] [PubMed]
- McChesney, J.; Nanayyakkra, D.N.; Bartlerr, M.; Ager, A.L. 8-aminoquinolines. Patent No. WO 97/36590, 28 March 1997. [Google Scholar]
- Chaurasiya, N.D.; Ganesan, S.; Nanayakkara, N.P.D.; Dias, L.R.S.; Walker, L.A.; Tekwani, B.L. Inhibition of Human Monoamine Oxidase A and B by 5-Phenoxy 8-Aminoquinoline Analogs. Bioorganic Med. Chem. Lett. 2012, 22, 1701–1704. [Google Scholar] [CrossRef] [PubMed]
- Brown, N. Chemoinformatics—An Introduction for Computer Scientists. ACM Comput. Surv. 2009, 41, 1–38. [Google Scholar] [CrossRef]
- Halkidi, M.; Vazirgiannis, M.; Balislakis, V. Quality Scheme Assessment in the Clustering Process. Lect. Notes Comput. Sci 2000, 1910, 265–276. [Google Scholar] [CrossRef]
- Rousseeuw, P.J. Silhouettes: A Graphical Aid to the Interpretation and Validation of Cluster Analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Mur, A.; Dormido, R.; Duro, N.; Dormido-Canto, S.; Vega, J. Determination of the Optimal Number of Clusters Using a Spectral Clustering Optimization. Expert Syst. Appl. 2016, 65, 304–314. [Google Scholar] [CrossRef]
- Hruschka, E.R.; Covões, T.F. Feature Selection for Cluster Analysis: An Approach Based on the Simplified Silhouette Criterion. In Proceedings of the International Conference on Computational Intelligence for Modelling, Control and Automation and CIMCA 2005 International Conference on Intelligent Agents, Web Technologies and Internet Commerce, Vienna, Austria, 28–30 November 2005; Volume 1, pp. 32–37. [Google Scholar] [CrossRef]
- ChemAxon—Software Solutions and Devices for Chemistry & Biology. Available online: https://chemaxon.com/ (accessed on 20 July 2021).
- Kim, S.; Thiessen, P.A.; Cheng, T.; Yu, B.; Bolton, E.E. An Update on PUG-REST: RESTful Interface for Programmatic Access to PubChem. Nucleic Acids Res. 2018, 46, W563–W570. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); Chemical Computing Group ULC: Montreal, QC, Canada, 2020.
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Jamois, E.A.; Hassan, M.; Waldman, M. Evaluation of Reagent-Based and Product-Based Strategies in the Design of Combinatorial Library Subsets. J. Chem. Inf. Comput. Sci. 2000, 40, 63–70. [Google Scholar] [CrossRef]
- Waldman, M.; Li, H.; Hassan, M. Novel Algorithms for the Optimization of Molecular Diversity of Combinatorial Libraries. J. Mol. Graph. Model. 2000, 18, 412–426. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Year | Applic | Disease | Status | ||||
|---|---|---|---|---|---|---|---|---|
| Dacomitinib | 2018 | Pfizer Inc. | Metastatic Non-Small-Cell Lung Cancer | ONP | 16,530 | 60 | 798 | 129 |
| Abemaciclib | 2017 | Eli Lilly | Breast Cancer | ONP | 45,696 | 41 | 736 | 214 |
| Tafenoquine | 2018 | GSK | Malaria | ONP | 25,472 | 58 | 600 | 160 |
| Ertugliflozin | 2017 | Merck | Diabetes | ONP | 14,194 | 21 | 56 | 120 |
| Rufinamide | 2008 | ESAI | Lennox-Gastaut Syndrome | OFP | 8959 | 22 | 144 | 95 |
| Azilsartan Medoxomil | 2011 | Takeda | Hypertension | ONP | 1110 | 4 | 9 | 34 |
| Leflunomide | 1998 | Sanofi | Rheumatoid Arthritis | OFP | 5641 | 114 | 2844 | 76 |
| HRC single | 318 ± 2582 | 28.7 | 159 ± 1816 | 26.2 | 16 ± 63 | 18.1 |
| HRC complete | 318 ± 184 | 0.0 | 159 ± 93 | 0.0 | 16 ± 11 | 0.0 |
| HRC median | 318 ± 847 | 0.0 | 159 ± 333 | 0.0 | 16 ± 23 | 5.7 |
| HRC average | 322 ± 348 | 0.0 | 159 ± 151 | 0.0 | 16 ± 14 | 0.7 |
| HRC centroid | 322 ± 2294 | 7.6 | 159 ± 1436 | 6.9 | 16 ± 25 | 7.4 |
| HRC Ward | 318 ± 113 | 0.0 | 159 ± 54 | 0.0 | 16 ± 6 | 0.0 |
| KMN | 318 ± 72 | 0.0 | 159 ± 38 | 0.0 | 16 ± 5 | 0.0 |
| KMED | 318 ± 125 | 0.0 | 159 ± 64 | 0.0 | 16 ± 7 | 0.2 |
| Binning | 411 ± 269 | 0.0 | 209 ± 165 | 0.0 | 20 ± 25 | 10.8 |
| OV binning | 509 ± 555 | 0.0 | 173 ± 201 | 0.7 | 20 ± 25 | 9.8 |
| Dacomitinib | Abemaciclib | Tafenoquine | |||||
|---|---|---|---|---|---|---|---|
| BD | Random | BD | Random | BD | Random | ||
| HRC average | SC | 26.7 | 41.1 | 22.0 | 35.1 | 17.2 | 46.8 |
| PC | 74.2 | 80.1 | 66.2 | 85.9 | 12.8 | 78.4 | |
| HRC Complete | SC | 50.0 | 54.0 | 34.1 | 52.0 | 25.9 | 59.3 |
| PC | 69.0 | 72.8 | 44.9 | 74.0 | 19.3 | 67.6 | |
| HRC Ward | SC | 41.7 | 53.5 | 34.1 | 58.6 | 17.2 | 61.8 |
| PC | 71.5 | 73.2 | 43.9 | 68.8 | 18.8 | 65.3 | |
| KMN | SC | 48.3 | 57.6 | 34.1 | 60.3 | 22.4 | 62.5 |
| PC | 67.3 | 69.4 | 39.1 | 67.4 | 21.0 | 64.7 | |
| KMED | SC | 55.0 | 57.6 | 34.1 | 59.1 | 24.1 | 61.4 |
| PC | 68.0 | 68.8 | 44.4 | 68.3 | 21.8 | 65.9 | |
| OV binning | SC | 25.0 | 42.9 | 28.1 | 51.8 | 24.0 | 49.2 |
| PC | 59.1 | 78.3 | 46.2 | 84.0 | 35.4 | 82.9 | |
| Ertugliflozin | Rufinamide | Azilsartan Medoxomil | Leflunomide | ||||||
|---|---|---|---|---|---|---|---|---|---|
| BD | Random | BD | Random | BD | Random | BD | Random | ||
| HRC average | SC | 9.5 | 19.8 | 36.4 | 30.6 | 50.0 | 26.4 | 27.2 | 50.1 |
| PC | 83.8 | 88.4 | 6.4 | 86.2 | 1.3 | 98.7 | 11.6 | 76.4 | |
| HRC Complete | SC | 23.8 | 38.7 | 22.7 | 43.6 | 50.0 | 26.3 | 26.3 | 55.3 |
| PC | 59.4 | 78.8 | 4.0 | 79.7 | 1.3 | 98.7 | 17.9 | 71.5 | |
| HRC Ward | SC | 19.0 | 57.7 | 13.6 | 70.8 | 50.0 | 41.0 | 15.8 | 60.6 |
| PC | 26.9 | 69.7 | 6.2 | 67.1 | 82.7 | 87.0 | 15.7 | 66.8 | |
| KMN | SC | 23.8 | 58.7 | 18.2 | 58.9 | 50.0 | 52.1 | 15.8 | 60.8 |
| PC | 23.5 | 69.3 | 7.9 | 69.4 | 59.9 | 84.8 | 13.1 | 66.7 | |
| KMED | SC | 19.0 | 57.7 | 27.3 | 61.6 | 50.0 | 68.3 | 18.4 | 59.8 |
| PC | 29.8 | 70.8 | 27.4 | 66.6 | 46.8 | 68.8 | 20.8 | 67.6 | |
| OV binning | SC | 13.3 | 65.6 | 28.6 | 50.3 | 25.0 | 56.6 | 26.4 | 53.0 |
| PC | 18.2 | 82.2 | 9.6 | 90.1 | 1.7 | 80.2 | 32.5 | 83.0 | |
| HRC Average | HRC Complete | OV Binning | |||||
|---|---|---|---|---|---|---|---|
| Tafenoquine | Selection Size | SC | PC | SC | PC | SC | PC |
| BD | 58 | 8.8 | 6.4 | 11.9 | 9.7 | 13.6 | 17.0 |
| Random BD | 58 | 27.0 | 45.4 | 29.1 | 37.1 | 27.1 | 53.6 |
| BCL | 600 | 38.8 | 33.2 | 33.8 | 28.3 | 36.0 | 49.3 |
| Random BCL | 600 | 80.4 | 95.6 | 90.6 | 96.1 | 74.7 | 96.1 |
| Random () | 160 | 50.9 | 74.8 | 57.7 | 69.1 | 48.2 | 79.8 |
| HRC Average | HRC Complete | OV Binning | |||||
|---|---|---|---|---|---|---|---|
| Dacomitinib | Selection size | SC | PC | SC | PC | SC | PC |
| BD | 60 | 20.9 | 50.3 | 27.1 | 42.1 | 16.8 | 52.0 |
| Random BD | 60 | 31.2 | 54.8 | 34.4 | 46.4 | 30.2 | 70.6 |
| BCL | 798 | 79.1 | 92.1 | 86.8 | 90.9 | 64.3 | 92.3 |
| Random BCL | 798 | 86.9 | 98.4 | 95.9 | 98.8 | 81.9 | 98.1 |
| Random () | 129 | 49.4 | 74.7 | 56.2 | 69.9 | 45.1 | 83.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manen-Freixa, L.; Borrell, J.I.; Teixidó, J.; Estrada-Tejedor, R. Deconstructing Markush: Improving the R&D Efficiency Using Library Selection in Early Drug Discovery. Pharmaceuticals 2022, 15, 1159. https://doi.org/10.3390/ph15091159
Manen-Freixa L, Borrell JI, Teixidó J, Estrada-Tejedor R. Deconstructing Markush: Improving the R&D Efficiency Using Library Selection in Early Drug Discovery. Pharmaceuticals. 2022; 15(9):1159. https://doi.org/10.3390/ph15091159
Chicago/Turabian StyleManen-Freixa, Leticia, José I. Borrell, Jordi Teixidó, and Roger Estrada-Tejedor. 2022. "Deconstructing Markush: Improving the R&D Efficiency Using Library Selection in Early Drug Discovery" Pharmaceuticals 15, no. 9: 1159. https://doi.org/10.3390/ph15091159
APA StyleManen-Freixa, L., Borrell, J. I., Teixidó, J., & Estrada-Tejedor, R. (2022). Deconstructing Markush: Improving the R&D Efficiency Using Library Selection in Early Drug Discovery. Pharmaceuticals, 15(9), 1159. https://doi.org/10.3390/ph15091159