
Sorafenib/2800Z Co-Loaded into Cholesterol and PEG Grafted Polylysine NPs for Liver Cancer Treatment

Abstract
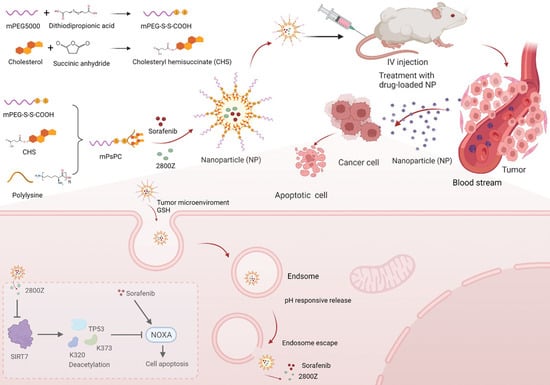
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Characterization of Nanomaterials
2.2. Characterization of Nanoparticles
2.3. Drug-Loading Efficiency and Release Rates of S2@PsPCs NPs
2.4. In Vitro Cell Uptake and the Distribution in Tumor Tissue
2.5. Anticancer Effects of S2@PsPCs NPS In Vitro
2.6. Anticancer Effects of S2@PsPCs NPs In Vivo
2.7. Evaluation of In Vivo Toxicity of S2@PsPCs NPS In Vivo
2.8. Immuno-Histopathology Analysis within the Tumor after S2@PsPCs NPS Treatment
3. Discussion
4. Materials and Methods
4.1. Cells and Animals
4.2. Antibodies and Chemicals
4.3. Synthesis of Succinate Acylated Cholesterol (CHS)
4.4. Synthesis of mPssCOOH
4.5. Synthesis of mPssPC
4.6. FTIR and NMR of Nanomaterials
4.7. Preparation of PsPC NPs and S2@PsPCs NPs
4.8. Size and Morphology Measurement of Nanoparticles
4.9. Evaluation of Drug-Loading Efficiency and Drug Release
4.10. Cellular Uptake Assessment In Vitro
4.11. Distribution of Nanoparticles In Vivo
4.12. Cell Counting Kit-8 (CCK8) Assay
4.13. Apoptosis Assay
4.14. Colony Formation Assay
4.15. Murine Xenograft Models
4.16. In Vivo Bioluminescence Imaging
4.17. Histological Analysis
4.18. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peacock, A.; Leung, J.; Larney, S.; Colledge, S.; Hickman, M.; Rehm, J.; Giovino, G.A.; West, R.; Hall, W.; Griffiths, P.; et al. Global statistics on alcohol, tobacco and illicit drug use: 2017 status report. Addiction 2018, 113, 1905–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Chan, S.L.; Galle, P.R.; Rimassa, L.; Sangro, B. Systemic treatment of hepatocellular carcinoma. An EASL position paper. J. Hepatol. 2021, 75, 960–974. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: Theoretical basis and therapeutic aspects. Signal Transduct. Target. Ther. 2020, 5, 87. [Google Scholar] [CrossRef]
- Ford, E.; Voit, R.; Liszt, G.; Magin, C.; Grummt, I.; Guarente, L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006, 20, 1075–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, M.F.; Michishita-Kioi, E.; Xi, Y.; Tasselli, L.; Kioi, M.; Moqtaderi, Z.; Tennen, R.I.; Paredes, S.; Young, N.L.; Chen, K.; et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 2012, 487, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.K.; Noh, J.H.; Jung, K.H.; Eun, J.W.; Bae, H.J.; Kim, M.G.; Chang, Y.G.; Shen, Q.; Park, W.S.; Lee, J.Y.; et al. Sirtuin7 oncogenic potential in human hepatocellular carcinoma and its regulation by the tumor suppressors MiR-125a-5p and MiR-125b. Hepatology 2013, 57, 1055–1067. [Google Scholar] [CrossRef]
- Malik, S.; Villanova, L.; Tanaka, S.; Aonuma, M.; Roy, N.; Berber, E.; Pollack, J.R.; Michishita-Kioi, E.; Chua, K.F. SIRT7 inactivation reverses metastatic phenotypes in epithelial and mesenchymal tumors. Sci. Rep. 2015, 5, 9841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.Y.; Ye, W.; Wu, J.X.; Meng, X.Q.; Liu, R.Y.; Ying, X.F.; Zhou, Y.; Wang, H.; Pan, C.C.; Huang, W.L. Overexpression of Sirt7 Exhibits Oncogenic Property and Serves as a Prognostic Factor in Colorectal Cancer. Clin. Cancer Res. 2014, 20, 3434–3445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.L.; Zhu, D.D.; Qin, S.H. SIRT7 suppresses the epithelial-to-mesenchymal transition in oral squamous cell carcinoma metastasis by promoting SMAD4 deacetylation. J. Exp. Clin. Cancer Res. 2018, 37, 148. [Google Scholar] [CrossRef]
- Li, H.; Tian, Z.; Qu, Y.; Yang, Q.; Guan, H.; Shi, B.; Ji, M.; Hou, P. SIRT7 promotes thyroid tumorigenesis through phosphorylation and activation of Akt and p70S6K1 via DBC1/SIRT1 axis. Oncogene 2019, 38, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wozniak, A.; Adams, A.; Cox, J.; Vittal, A.; Voss, J.; Bridges, B.; Weinman, S.A.; Li, Z. SIRT7 regulates hepatocellular carcinoma response to therapy by altering the p53-dependent cell death pathway. J. Exp. Clin. Cancer Res. 2019, 38, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Li, Y.Q.; Liu, B.H.; Ning, C.; Li, Y.M.; Wang, Y.; Li, Z. Discovery of SIRT7 Inhibitor as New Therapeutic Options Against Liver Cancer. Front. Cell Dev. Biol. 2022, 9, 813233. [Google Scholar] [CrossRef]
- Qi, S.S.; Sun, J.H.; Yu, H.H.; Yu, S.Q. Co-delivery nanoparticles of anti-cancer drugs for improving chemotherapy efficacy. Drug Deliv. 2017, 24, 1909–1926. [Google Scholar] [CrossRef] [Green Version]
- Cavalieri, F.; Beretta, G.L.; Cui, J.; Braunger, J.A.; Yan, Y.; Richardson, J.J.; Tinelli, S.; Folini, M.; Zaffaroni, N.; Caruso, F. Redox-Sensitive PEG-Polypeptide Nanoporous Particles for Survivin Silencing in Prostate Cancer Cells. Biomacromolecules 2015, 16, 2168–2178. [Google Scholar] [CrossRef] [Green Version]
- Bilensoy, E. Cationic nanoparticles for cancer therapy. Expert Opin. Drug Deliv. 2010, 7, 795–809. [Google Scholar] [CrossRef]
- Gallud, A.; Kloditz, K.; Ytterberg, J.; Ostberg, N.; Katayama, S.; Skoog, T.; Gogvadze, V.; Chen, Y.Z.; Xue, D.; Moya, S.; et al. Cationic gold nanoparticles elicit mitochondrial dysfunction: A multi-omics study. Sci. Rep. 2019, 9, 4366. [Google Scholar] [CrossRef]
- Bus, T.; Traeger, A.; Schubert, U.S. The great escape: How cationic polyplexes overcome the endosomal barrier. J. Mater. Chem. B 2018, 6, 6904–6918. [Google Scholar] [CrossRef] [PubMed]
- Malamas, A.S.; Gujrati, M.; Kummitha, C.M.; Xu, R.; Lu, Z.R. Design and evaluation of new pH-sensitive amphiphilic cationic lipids for siRNA delivery. J. Control. Release 2013, 171, 296–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amreddy, N.; Babu, A.; Muralidharan, R.; Panneerselvam, J.; Srivastava, A.; Ahmed, R.; Mehta, M.; Munshi, A.; Ramesh, R. Recent Advances in Nanoparticle-Based Cancer Drug and Gene Delivery. Adv. Cancer Res. 2018, 137, 115–170. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Tabei, Y. Role of oxidative stress in nanoparticles toxicity. Free Radic. Res. 2021, 55, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, H.; Huang, T.; Xiao, J.; Liao, Z.; Ouyang, J.; Dong, J.; Xian, C.J.; Hu, J.; Wang, L.; Ke, Y.; et al. The immuno-reactivity of polypseudorotaxane functionalized magnetic CDMNP-PEG-CD nanoparticles. J. Cell Mol. Med. 2021, 25, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; van der Meel, R.; Chen, X.; Lammers, T. The EPR effect and beyond: Strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics 2020, 10, 7921–7924. [Google Scholar] [CrossRef]
- He, Q.Y.; Chen, J.; Yan, J.H.; Cai, S.D.; Xiong, H.J.; Liu, Y.F.; Peng, D.M.; Mo, M.; Liu, Z.B. Tumor microenvironment responsive drug delivery systems. Asian J. Pharm. Sci. 2020, 15, 416–448. [Google Scholar] [CrossRef]
- Xiao, R.Z.; Zeng, Z.W.; Zhou, G.L.; Wang, J.J.; Li, F.Z.; Wang, A.M. Recent advances in PEG-PLA block copolymer nanoparticles. Int. J. Nanomed. 2010, 5, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Almoustafa, H.A.; Alshawsh, M.A.; Chik, Z. Technical aspects of preparing PEG-PLGA nanoparticles as carrier for chemotherapeutic agents by nanoprecipitation method. Int. J. Pharm. 2017, 533, 275–284. [Google Scholar] [CrossRef]
- Zhang, W.B.; Yu, L.L.; Ji, T.J.; Wang, C.X. Tumor Microenvironment-Responsive Peptide-Based Supramolecular Drug Delivery System. Front. Chem. 2020, 8, 549. [Google Scholar] [CrossRef]
- Guo, X.S.; Cheng, Y.; Zhao, X.T.; Luo, Y.L.; Chen, J.J.; Yuan, W.E. Advances in redox-responsive drug delivery systems of tumor microenvironment. J. Nanobiotechnol. 2018, 16, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Wan, L.; Yuan, Y.; Kuang, Y.; Xu, X.; Liao, T.; Liu, J.; Xu, Z.Q.; Jiang, B.; Li, C. pH/GSH-Dual-Sensitive Hollow Mesoporous Silica Nanoparticle-Based Drug Delivery System for Targeted Cancer Therapy. ACS Biomater. Sci. Eng. 2020, 6, 3375–3387. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Liu, H.; Chen, H.; Wang, G.; Teng, H.; Chang, Y. Polyphotosensitizer nanogels for GSH-responsive histone deacetylase inhibitors delivery and enhanced cancer photodynamic therapy. Colloids Surf. B Biointerfaces 2020, 188, 110753. [Google Scholar] [CrossRef]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Finkel, T.; Deng, C.X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar] [CrossRef] [Green Version]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435, 1262–1266. [Google Scholar] [CrossRef]
- Manuyakorn, A.; Paulus, R.; Farrell, J.; Dawson, N.A.; Tze, S.; Cheung-Lau, G.; Hines, O.J.; Reber, H.; Seligson, D.B.; Horvath, S.; et al. Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: Results from RTOG 9704. J. Clin. Oncol. 2010, 28, 1358–1365. [Google Scholar] [CrossRef] [Green Version]
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar] [CrossRef]
- Tang, X.; Shi, L.; Xie, N.; Liu, Z.; Qian, M.; Meng, F.; Xu, Q.; Zhou, M.; Cao, X.; Zhu, W.G.; et al. SIRT7 antagonizes TGF-beta signaling and inhibits breast cancer metastasis. Nat. Commun. 2017, 8, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Li, J.; Gu, P.; Fan, X. The application of nanoparticles in cancer immunotherapy: Targeting tumor microenvironment. Bioact. Mater. 2021, 6, 1973–1987. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Y.; Wang, K.; Chen, Y.; Shi, M.; Zhang, X.; Pan, W.; Li, N.; Tang, B. GSH-Responsive Nanoprodrug to Inhibit Glycolysis and Alleviate Immunosuppression for Cancer Therapy. Nano Lett. 2021, 21, 7862–7869. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, Y.; Wen, X.; Pan, Y.; Cheng, X.; An, R.; Gao, G.; Chen, H.-Y.; Ye, D. Responsive Trimodal Probes for In Vivo Imaging of Liver Inflammation by Coassembly and GSH-Driven Disassembly. Research 2020, 2020, 4087069. [Google Scholar] [CrossRef]
- Dai, J.; Dong, X.; Wang, Q.; Lou, X.; Xia, F.; Wang, S. PEG-Polymer Encapsulated Aggregation-Induced Emission Nanoparticles for Tumor Theranostics. Adv. Healthc. Mater. 2021, 10, e2101036. [Google Scholar] [CrossRef]
- Jin, K.; Luo, Z.; Zhang, B.; Pang, Z. Biomimetic nanoparticles for inflammation targeting. Acta Pharm. Sin. B 2018, 8, 23–33. [Google Scholar] [CrossRef]
- Shim, M.S.; Kwon, Y.J. Acid-transforming polypeptide micelles for targeted nonviral gene delivery. Biomaterials 2010, 31, 3404–3413. [Google Scholar] [CrossRef]
- Toshiyama, R.; Konno, M.; Eguchi, H.; Takemoto, H.; Noda, T.; Asai, A.; Koseki, J.; Haraguchi, N.; Ueda, Y.; Matsushita, K.; et al. Poly(ethylene glycol)-poly(lysine) block copolymer-ubenimex conjugate targets aminopeptidase N and exerts an antitumor effect in hepatocellular carcinoma stem cells. Oncogene 2019, 38, 244–260. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Duan, L.; Yang, L.; Jin, J.; Yang, F.; Liu, D.; Hu, K.; Wang, Q.; Yue, Y.; Gu, N. Micro/nano-bubble-assisted ultrasound to enhance the EPR effect and potential theranostic applications. Theranostics 2020, 10, 462–483. [Google Scholar] [CrossRef]
- Yuan, L.; Cao, Y.; Luo, Q.; Yang, W.; Wu, X.; Yang, X.; Wu, D.; Tan, S.; Qin, G.; Zhou, J.; et al. Pullulan-Based Nanoparticle-HSA Complex Formation and Drug Release Influenced by Surface Charge. Nanoscale Res. Lett. 2018, 13, 317. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Zhong, W.; Cao, Y.; Liu, B.; Tao, X.; Li, Z. Sorafenib/2800Z Co-Loaded into Cholesterol and PEG Grafted Polylysine NPs for Liver Cancer Treatment. Pharmaceuticals 2023, 16, 119. https://doi.org/10.3390/ph16010119
Zhang C, Zhong W, Cao Y, Liu B, Tao X, Li Z. Sorafenib/2800Z Co-Loaded into Cholesterol and PEG Grafted Polylysine NPs for Liver Cancer Treatment. Pharmaceuticals. 2023; 16(1):119. https://doi.org/10.3390/ph16010119
Chicago/Turabian StyleZhang, Chen, Wu Zhong, Ying Cao, Bohao Liu, Xiaojun Tao, and Zhuan Li. 2023. "Sorafenib/2800Z Co-Loaded into Cholesterol and PEG Grafted Polylysine NPs for Liver Cancer Treatment" Pharmaceuticals 16, no. 1: 119. https://doi.org/10.3390/ph16010119
APA StyleZhang, C., Zhong, W., Cao, Y., Liu, B., Tao, X., & Li, Z. (2023). Sorafenib/2800Z Co-Loaded into Cholesterol and PEG Grafted Polylysine NPs for Liver Cancer Treatment. Pharmaceuticals, 16(1), 119. https://doi.org/10.3390/ph16010119