
Preclinical Studies and Drug Combination of Low-Cost Molecules for Chagas Disease

 , ,
, ,  , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Toxicology
2.2.2. Analyzing the Potential Mechanisms of Actions
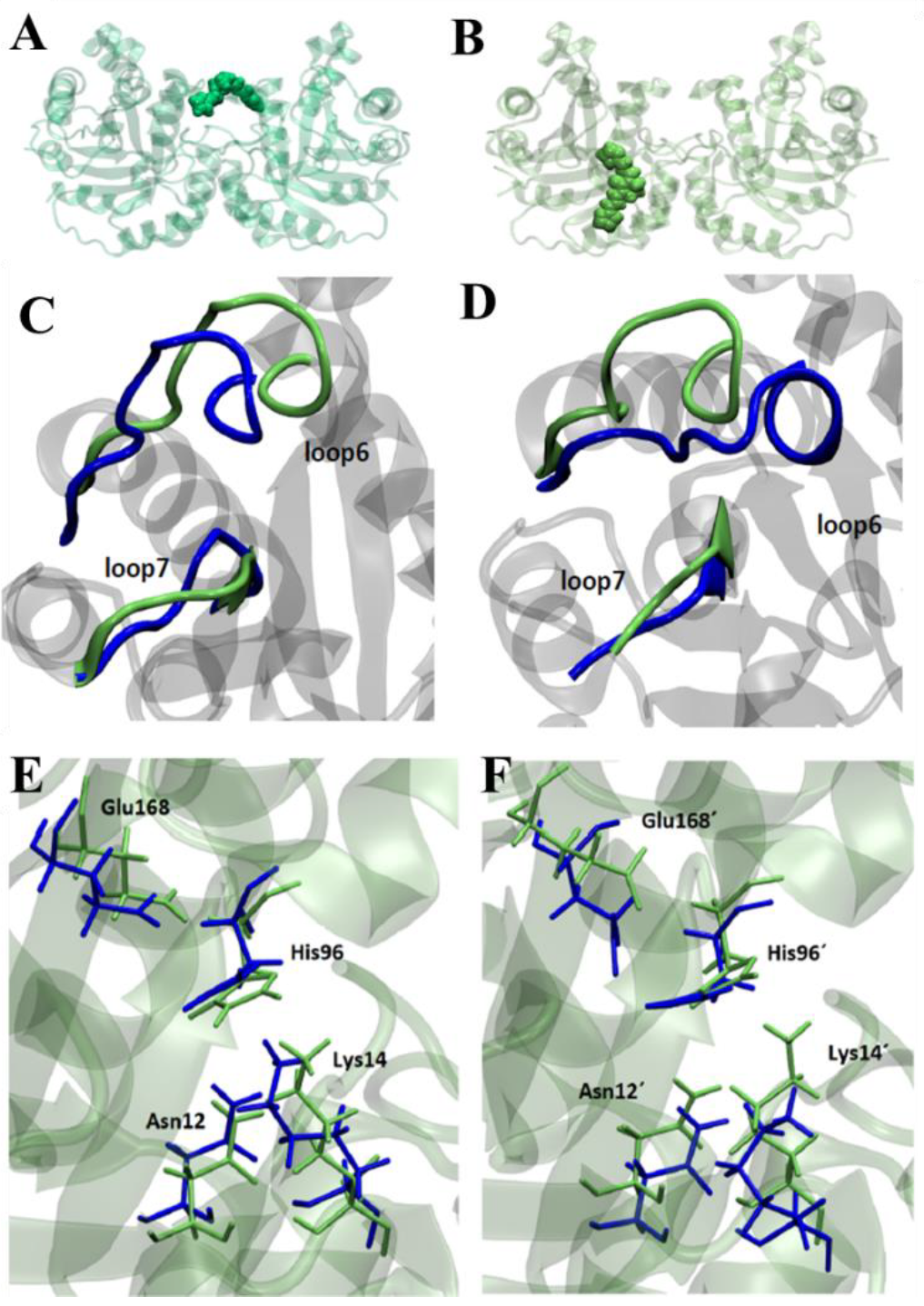
Triosephosphate Isomerase Mechanism of Inhibition
NMR Metabolomics Studies
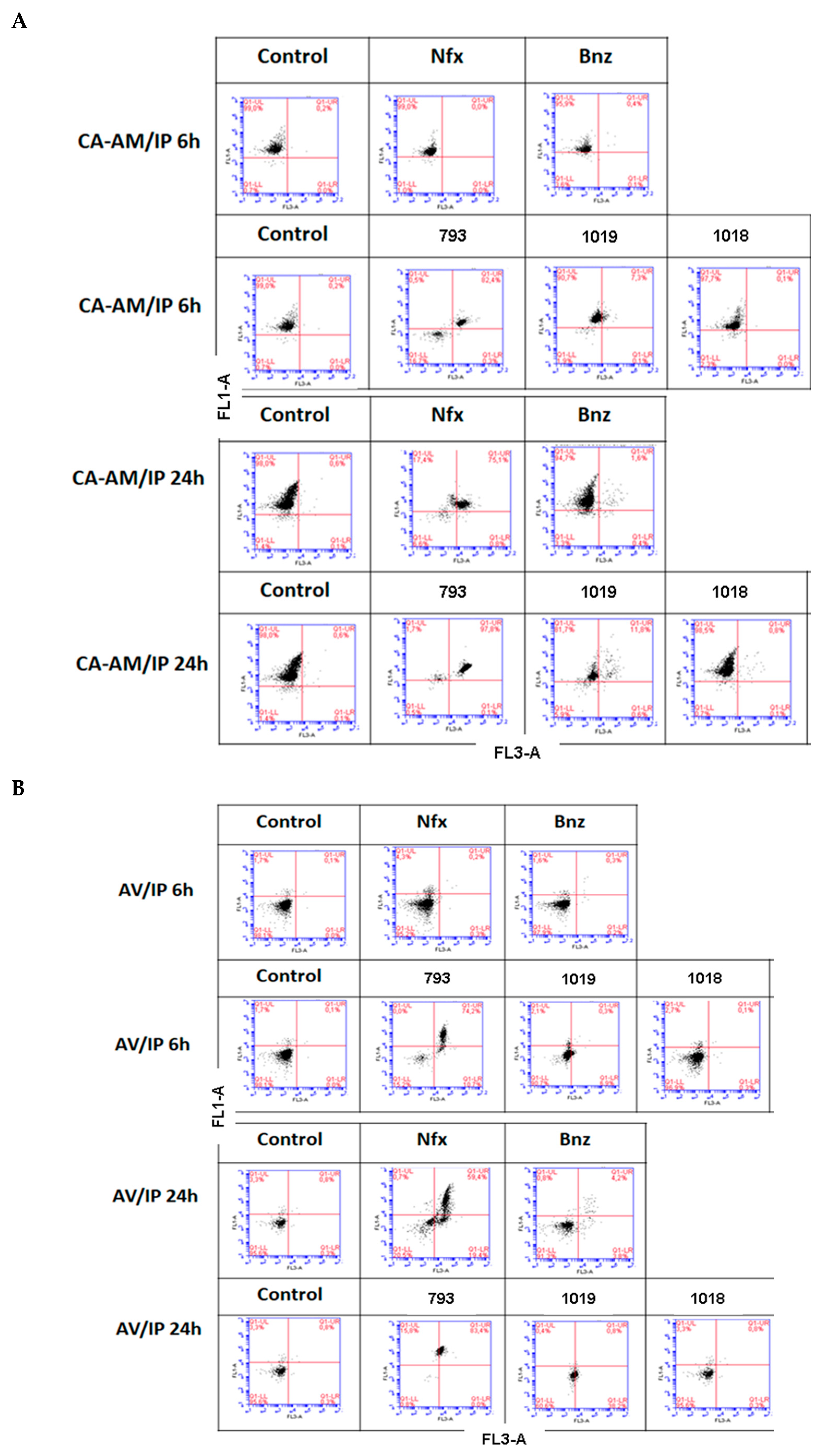
2.2.3. Mechanism of Death
2.2.4. In Vitro Metabolic Stability
2.2.5. Trypanosomicidal Activity in Other Forms of the Parasite
2.2.6. Compound Combinations and In Vitro Isobolograms
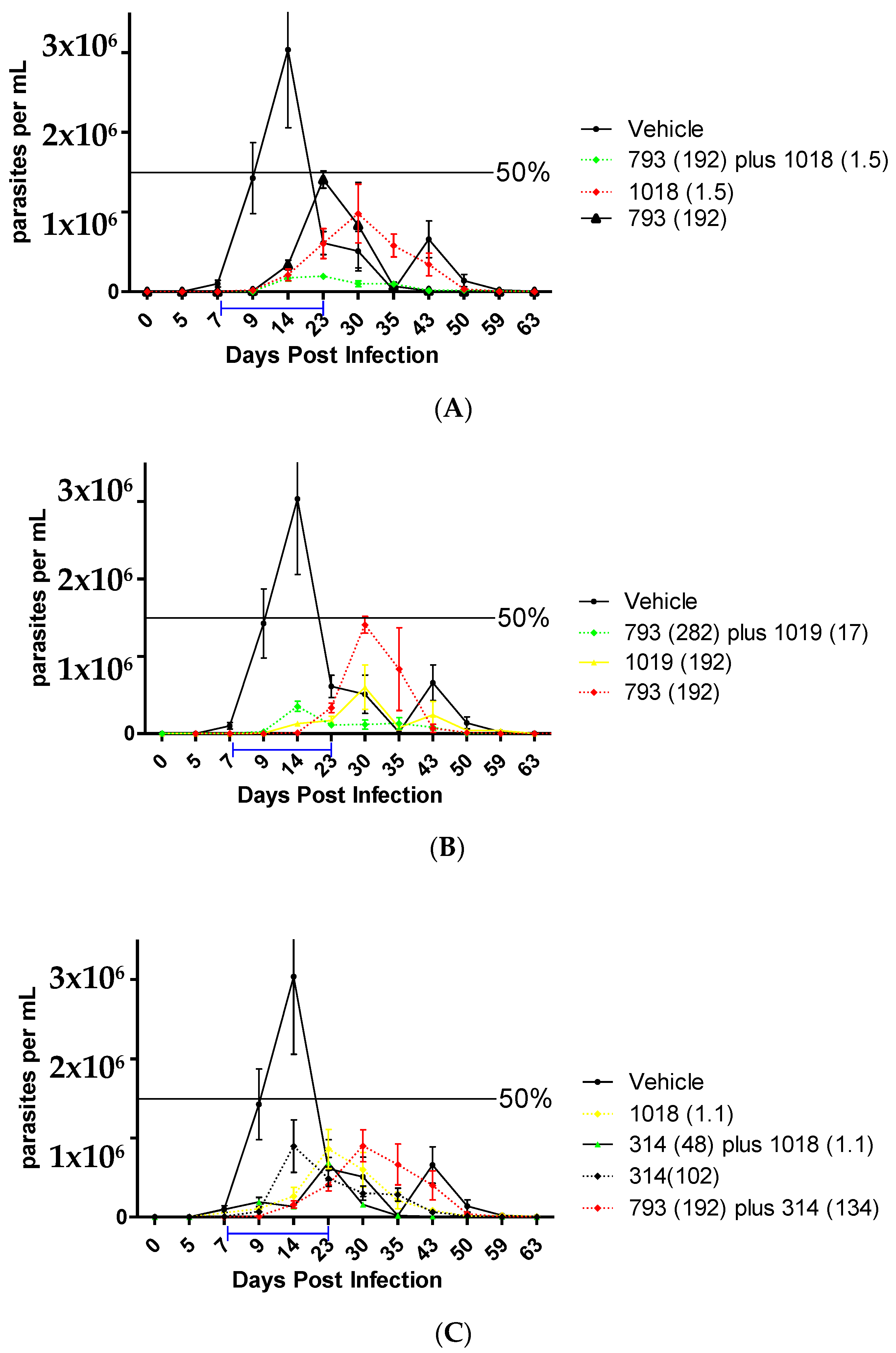
2.2.7. In Vivo Studies in the Acute Murine Model of Chagas
3. Material and Methods
3.1. Chemistry
3.2. Mutagenicity Tests (Ames Test)
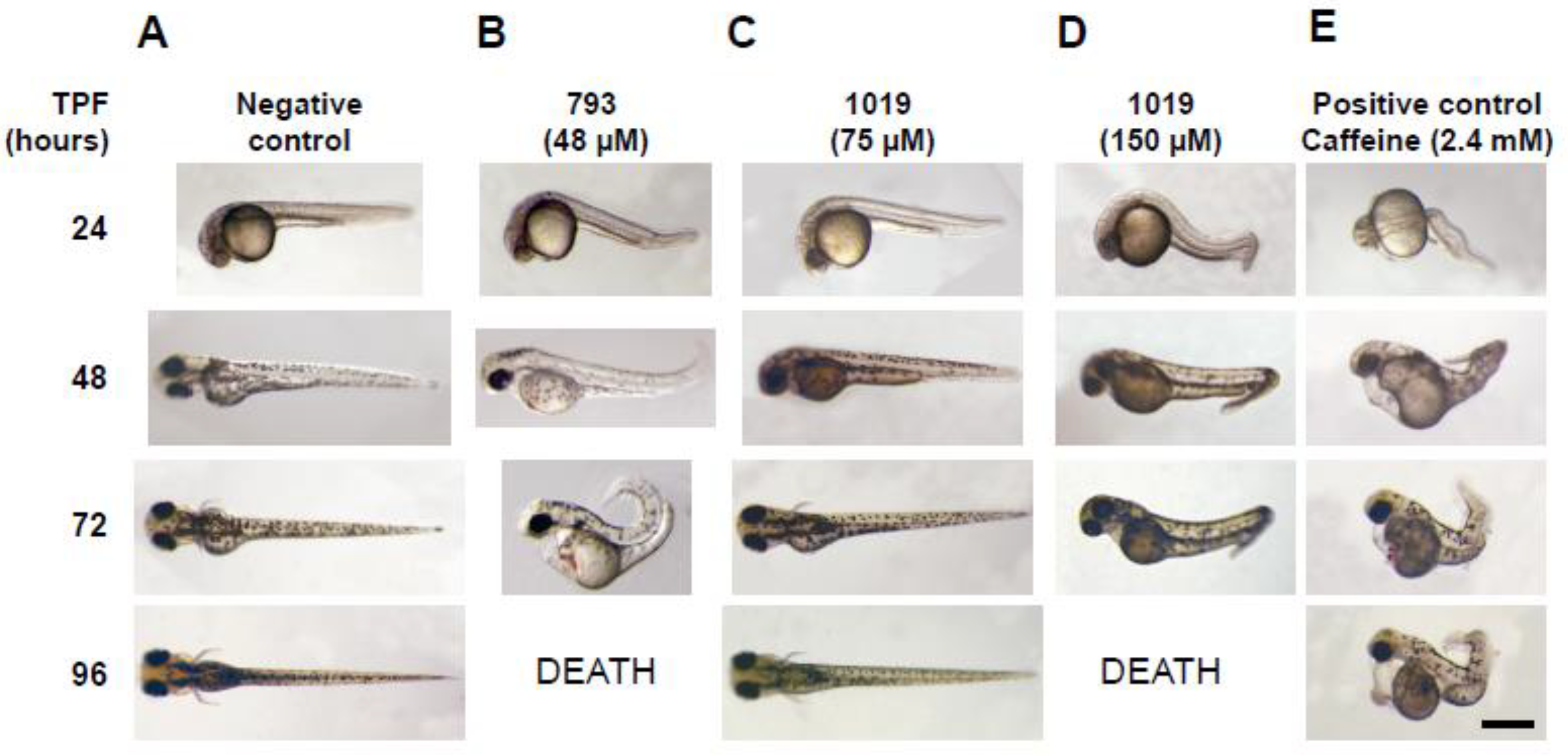
3.3. Teratogenicity in Zebrafish Embryos
3.4. Acute Oral Toxicity In Vivo in Mice (Up and Down Test)
3.5. Genotoxicity Evaluation by In Vivo Micronucleus Test in Mice
3.6. Molecular Dynamics Studies in TcTIM
3.7. Metabolomics Studies of T. cruzi Using 1H-NMR
3.8. Study of the Mechanism of Death by 1H-NMR in T. cruzi
3.9. Study of the Mechanism of Death by Flow Cytometry
3.10. Stability Studies with Microsomal and Cytosolic Fractions of Rat Hepatocytes
3.11. Trypanosomicidal Activity In Vitro on Epimastigotes of T. cruzi
3.12. Assay for the Combination of Compounds and Construction of the Isobolograms
3.13. Trypanosomicidal Activity in Trypomastigotes of T. cruzi
3.14. In Vivo Studies in the Acute Model of Chagas Disease in Mice
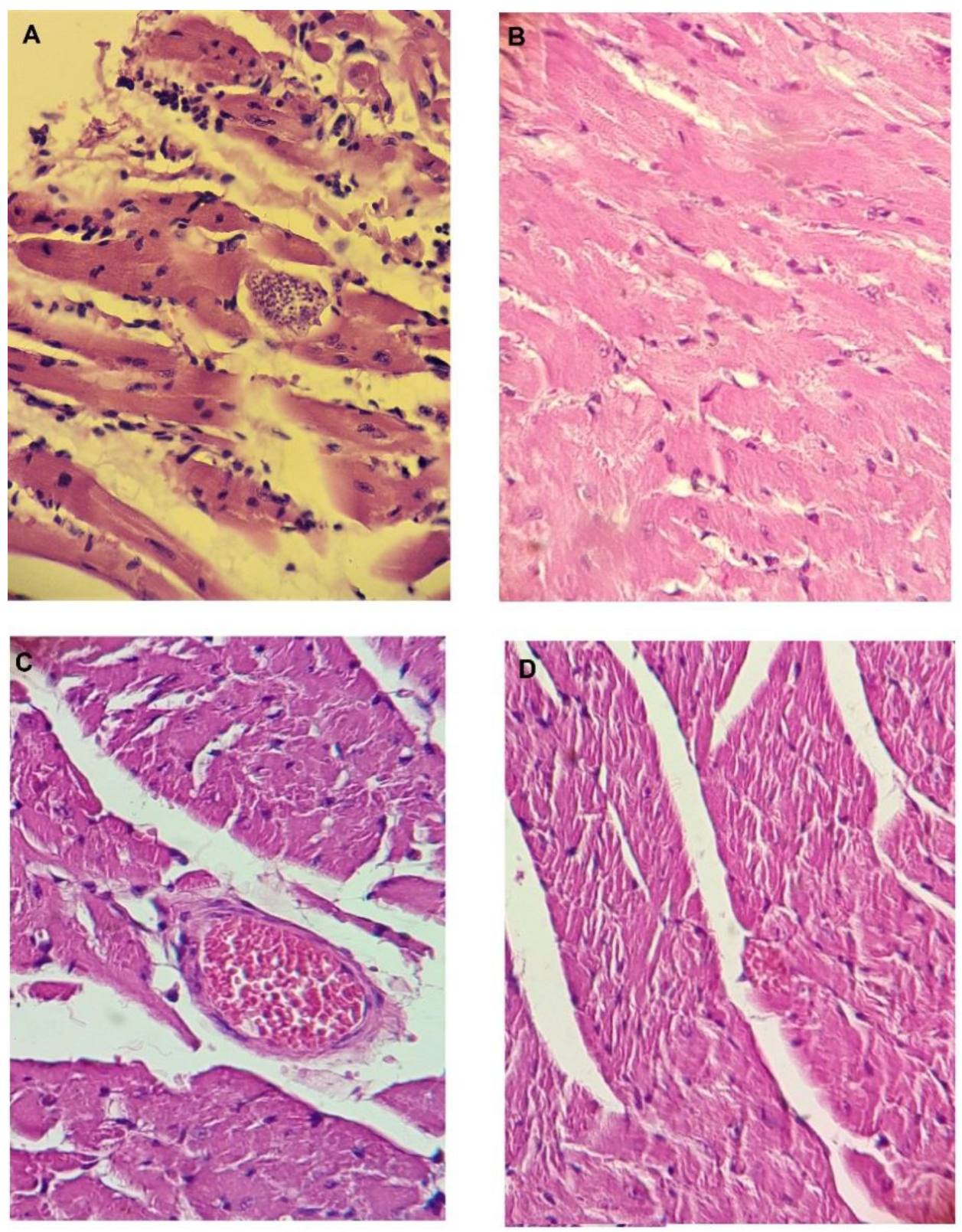
3.15. Histopathology of the Heart, Spleen, Intestine, Kidney, and Liver of the Mice at the End of the In Vivo Tests in the Acute Model of Chagas Disease
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Santos, S.S.; de Araújo, R.V.; Giarolla, J.; El Seoud, O.; Ferreira, E.I. Searching drugs for Chagas disease, leishmaniasis and schistosomiasis: A brief review. Int. J. Antimicrob. Agents 2020, 55, 105906. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.M. Chagas Disease: Epidemiology and Barriers to Treatment Chagas disease. Am. J. Med. 2020, 133, 1262–1265. [Google Scholar] [CrossRef] [PubMed]
- Ms, M.K.L.; Bossak, B.H.; Sandifer, P.A.; Watson, A.; Nolan, M.S. Contemporary autochthonous human Chagas disease in the USA. Acta Trop. 2020, 205, 105361. [Google Scholar]
- Pinazo, M.-J.; Gascon, J. Chagas disease: From Latin America to the world. Rep. Parasitol. 2015, 4, 7–14. [Google Scholar]
- Epting, C.L.; Coates, B.M.; Engman, D.M. Molecular mechanisms of host cell invasion by Trypanosoma cruzi. Exp. Parasitol. 2010, 126, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona, S.J.; Sartor, P.A.; Leguizamón, M.S.; Campetella, O.E.; Agüero, F. Diagnostic Peptide Discovery: Prioritization of Pathogen Diagnostic Markers Using Multiple Features. PLoS ONE 2012, 7, e50748. [Google Scholar] [CrossRef] [Green Version]
- Keenan, M.; Chaplin, J.H. A New Era for Chagas Disease Drug Discovery? 1st ed.; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Urbina, J.A. Specific chemotherapy of Chagas disease: Relevance, current limitations and new approaches. Acta Trop. 2009, 115, 55–68. [Google Scholar] [CrossRef]
- Martínez-Peinado, N.; Cortes-Serra, N.; Losada-Galvan, I.; Alonso-Vega, C.; Urbina, J.A.; Rodríguez, A.; VandeBerg, J.L.; Pinazo, M.J.; Gascon, J.; Alonso-Padilla, J. Emerging Agents for the Treatment of Chagas Disease: What Is in the Preclinical and Clinical Development Pipeline? Taylor & Francis: Abingdon, UK, 2020; Volume 29. [Google Scholar]
- Buckner, F.S.; Waters, N.C.; Avery, V.M. Recent highlights in anti-protozoan drug development and resistance research. Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 230–235. [Google Scholar] [CrossRef] [Green Version]
- Lescure, F.-X.; Le Loup, G.; Freilij, H.; Develoux, M.; Paris, L.; Brutus, L.; Pialoux, G. Chagas disease: Changes in knowledge and management. Lancet Infect. Dis. 2010, 10, 556–570. [Google Scholar] [CrossRef]
- Cerecetto, H.; González, M. Anti-T. cruzi agents: Our experience in the evaluation of more than five hundred compounds. Mini Rev. Med. Chem. 2008, 8, 1355–1383. [Google Scholar] [CrossRef]
- Soeiro, M.D.N.C.; de Castro, S.L. Screening of Potential anti-Trypanosoma cruzi Candidates: In Vitro and In Vivo Studies. Open Med. Chem. J. 2011, 5, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Moreno, M.; Sanz, A.M.; Gómez-Contreras, F.; Navarro, P.; Marín, C.; Ramírez-Macias, I.; Rosales, M.J.; Olmo, F.; Garcia-Aranda, I.; Campayo, L.; et al. In vivo trypanosomicidal activity of imidazole- or pyrazole-based benzo[g]phthalazine derivatives against acute and chronic phases of Chagas disease. J. Med. Chem. 2011, 54, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Fonseca, L.M.; Trujillo-Ferrara, J.G. Structural considerations for the rational design of selective anti-trypanosomal agents: The role of the aromatic clusters at the interface of triosephosphate isomerase dimer. Biochem. Biophys. Res. Commun. 2005, 328, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, M.L.; Gazzinelli, R.T.; Alves, R.O.; Urbina, J.A.; Romanha, A.J. The Anti-Trypanosoma cruzi activity of posaconazole in a murine model of acute Chagas’ disease is less dependent on gamma interferon than that of benznidazole. Antimicrob. Agents Chemother. 2007, 51, 1359–1364. [Google Scholar] [CrossRef] [Green Version]
- Fortes Francisco, A.; Lewis, M.D.; Jayawardhana, S.; Taylor, M.C.; Chatelain, E.; Kelly, J.M. The limited ability of posaconazole to cure both acute and chronic Trypanosoma cruzi infections revealed by highly sensitive in vivo imaging. Antimicrob. Agents Chemother. 2015, 59, 4653–4661. [Google Scholar] [CrossRef] [Green Version]
- Wierenga, R.K.; Kapetaniou, E.G.; Venkatesan, R. Triosephosphate isomerase: A highly evolved biocatalyst. Cell. Mol. Life Sci. 2010, 67, 3961–3982. [Google Scholar] [CrossRef]
- Gayosso-De-Lucio, J.; Torres-Valencia, M.; Rojo-Domínguez, A.; Nájera-Peña, H.; Aguirre-López, B.; Salas-Pacheco, J.; Avitia-Domínguez, C.; Téllez-Valencia, A. Selective inactivation of triosephosphate isomerase from Trypanosoma cruzi by brevifolin carboxylate derivatives isolated from Geranium bellum Rose. Bioorg. Med. Chem. Lett. 2009, 19, 5936–5939. [Google Scholar] [CrossRef]
- Maldonado, E.; Soriano-García, M.; Moreno, A.; Cabrera, N.; Garza-Ramos, G.; de Gómez-Puyou, M.; Gómez-Puyou, A.; Perez-Montfort, R. Differences in the intersubunit contacts in triosephosphate isomerase from two closely related pathogenic trypanosomes. J. Mol. Biol. 1998, 283, 193–203. [Google Scholar] [CrossRef]
- Vázquez-Raygoza, A.; Cano-González, L.; Velázquez-Martínez, I.; Trejo-Soto, P.J.; Castillo, R.; Hernández-Campos, A.; Hernández-Luis, F.; Oria-Hernández, J.; Castillo-Villanueva, A.; Avitia-Domínguez, C.; et al. Species-specific inactivation of triosephosphate isomerase from Trypanosoma brucei: Kinetic and molecular dynamics studies. Molecules 2017, 22, 2055. [Google Scholar] [CrossRef] [Green Version]
- Olivares-Illana, V.; Rodríguez-Romero, A.; Becker, I.; Berzunza, M.; García, J.; Pérez-Montfort, R.; Cabrera, N.; López-Calahorra, F.; de Gómez-Puyou, M.T.; Gómez-Puyou, A. Perturbation of the dimer interface of triosephosphate isomerase and its effect on Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2007, 1, e01–e08. [Google Scholar] [CrossRef] [Green Version]
- Michels, P.A.M.; Wierenga, R.K. Overexpression of trypanosomal triosephosphate isomerase in Escherichia coli and characterisation of a dimer-interface mutant. Eur. J. Biochem. 1993, 211, 703–710. [Google Scholar]
- Kumar, K.; Bhargava, P.; Roy, U. Cloning, overexpression and characterization of Leishmania donovani triosephosphate isomerase. Exp. Parasitol. 2012, 130, 430–436. [Google Scholar] [CrossRef]
- Aguilera, E.; Varela, J.; Birriel, E.; Serna, E.; Torres, S.; Yaluff, G.; de Bilbao, N.V.; Aguirre-López, B.; Cabrera, N.; Díaz Mazariegos, S.; et al. Potent and Selective Inhibitors of Trypanosoma cruzi Triosephosphate Isomerase with Concomitant Inhibition of Cruzipain: Inhibition of Parasite Growth through Multitarget Activity. ChemMedChem 2016, 11, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Battah, S.; Karachalias, N.; Babaei-Jadidi, R.; Horanyi, M.; Baroti, K.; Hollan, S.; Thornalley, P.J. Increased formation of methylglyoxal and protein glycation, oxidation and nitrosation in triosephosphate isomerase deficiency. Biochim. Biophys. Acta 2003, 1639, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnapp, A.R.; Eickhoff, C.S.; Sizemore, D.; Iii, R.C.; Hoft, D.F. Cruzipain Induces Both Mucosal and Systemic Protection against Trypanosoma cruzi in Mice. Infect. Inmunity 2002, 70, 5065–5074. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, N. Molecular basis of mammalian cell invasion by Trypanosoma cruzi. An. Acad. Bras. Cienc. 2006, 78, 87–111. [Google Scholar] [CrossRef] [Green Version]
- Cazorla, S.I.; Frank, F.M.; Becker, P.D.; Arnaiz, M.; Mirkin, G.A.; Corral, R.S.; Guzmán, C.A.; Malchiodi, E.L. Redirection of the immune response to the functional catalytic domain of the cystein proteinase cruzipain improves protective immunity against Trypanosoma cruzi infection. J. Infect. Dis. 2010, 202, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Ndao, M.; Beaulieu, C.; Black, W.C.; Isabel, E.; Vasquez-Camargo, F.; Nath-Chowdhury, M.; Massé, F.; Mellon, C.; Methot, N.; Nicoll-Griffith, D.A. Reversible cysteine protease inhibitors show promise for a chagas disease cure. Antimicrob. Agents Chemother. 2014, 58, 1167–1178. [Google Scholar] [CrossRef] [Green Version]
- Merlino, A.; Benitez, D.; Campillo, N.E.; Páez, J.A.; Tinoco, L.W.; González, M.; Cerecetto, H. Amidines bearing benzofuroxan or benzimidazole 1,3-dioxide core scaffolds as Trypanosoma cruzi-inhibitors: Structural basis for their interactions with cruzipain. Med. Chem. Commun. 2012, 3, 90–101. [Google Scholar] [CrossRef]
- Caputto, M.E.; Fabian, L.E.; Benítez, D.; Merlino, A.; Ríos, N.; Cerecetto, H.; Moltrasio, G.Y.; Moglioni, A.G.; González, M.; Finkielsztein, L.M. Thiosemicarbazones derived from 1-indanones as new anti-Trypanosoma cruzi agents. Bioorg. Med. Chem. 2011, 19, 6818–6826. [Google Scholar] [CrossRef]
- Diniz, L.D.F.; Urbina, J.A.; de Andrade, I.M.; Mazzeti, A.L.; Martins, T.A.F.; Caldas, I.S.; Talvani, A.; Ribeiro, I.; Bahia, M.T. Benznidazole and posaconazole in experimental Chagas disease: Positive interaction in concomitant and sequential treatments. PLoS Negl. Trop. Dis. 2013, 7, e2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, R.A.; Molina, J.; Payares, G.; Urbina, J.A. Experimental chemotherapy with combinations of ergosterol biosynthesis inhibitors in murine models of Chagas’ disease. Antimicrob. Agents Chemother. 1993, 37, 1353–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbina, J.A.; Lazardi, K.; Marchan, E.; Visbal, G.; Aguirre, T.; Piras, M.M.; Piras, R.; Maldonado, R.A.; Payares, G.; de Souza, W. Mevinolin (lovastatin) potentiates the antiproliferative effects of ketoconazole and terbinafine against Trypanosoma (Schizotrypanum) cruzi: In vitro and in vivo studies. Antimicrob. Agents Chemother. 1993, 37, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A.; Lazardi, K.; Aguirre, T.; Piras, M.M.; Piras, R. Antiproliferative synergism of the allylamine SF 86-327 and ketoconazole on epimastigotes and amastigotes of Trypanosoma (Schizotrypanum) cruzi. Antimicrob. Agents Chemother. 1988, 32, 1237–1242. [Google Scholar] [CrossRef] [Green Version]
- Capital, D. Susceptibilidad in vitro a Nifurtimox y Benznidazol de aislados de Trypanosoma cruzi obtenidos de pacientes venezolanos con enfermedad de Chagas infectados por mecanismos de transmisión oral y vectorial. Rev. Ibero-Latinoam. Parasitol. 2012, 71, 14–22. [Google Scholar]
- Echeverría, L.E.; González, C.I.; Hernandez, J.C.M.; Díaz, M.L.; Nieto, J.E.; López-Romero, L.A.; Rivera, J.D.; Suárez, E.U.; Ochoa, S.A.G.; Rojas, L.Z.; et al. Efficacy of the benznidazole+posaconazole combinatiotherapy in parasitemia reduction: An experimental murine model of acute chagas. Rev. Soc. Bras. Med. Trop. 2020, 53, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, G.; Perdomo, C.; Coronel, C.; Aguilera, E.; Varela, J.; Aparicio, G.; Zolessi, F.R.; Cabrera, N.; Vega, C.; Rolón, M.; et al. Multi-anti-parasitic activity of arylidene ketones and thiazolidene hydrazines against Trypanosoma cruzi and Leishmania spp. Molecules 2017, 22, 709. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, G.; Varela, J.; Cruces, E.; Fernández, M.; Gabay, M.; Leal, S.M.; Escobar, P.; Sanabria, L.; Serna, E.; Torres, S.; et al. Identification of a new amide-containing thiazole as a drug candidate for treatment of chagas’ disease. Antimicrob. Agents Chemother. 2015, 59, 1398–1404. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, E.; Perdomo, C.; Espindola, A.; Corvo, I.; Faral-Tello, P.; Robello, C.; Serna, E.; Benítez, F.; Riveros, R.; Torres, S.; et al. A nature-inspired design yields a new class of steroids against trypanosomatids. Molecules 2019, 24, 3800. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, G.; Varela, J.; Márquez, P.; Gabay, M.; Arias Rivas, C.E.; Cuchilla, K.; Echeverría, G.A.; Piro, O.E.; Chorilli, M.; Leal, S.M.; et al. Optimization of antitrypanosomatid agents: Identification of nonmutagenic drug candidates with in vivo activity. J. Med. Chem. 2014, 57, 3984–3999. [Google Scholar] [CrossRef]
- Klüver, N.; König, M.; Ortmann, J.; Massei, R.; Paschke, A.; Kuehne, R.; Scholz, S. The fish embryo toxicity test (FET)—Identification of compounds with weak toxicity and analysis of behavioral effects to improve prediction of acute toxicity for neurotoxic compounds. Environ. Sci. Technol. 2015, 49, 7002–7011. [Google Scholar] [CrossRef] [PubMed]
- OECD. Test No. 425: Acute Oral Toxicity—Up-and-Down Procedure; OECD Guidelines for the Testing of Chemicals, Section 4; OECD: Paris, France, 2001; pp. 1–29. [Google Scholar]
- Saramago, L.; Gomes, H.; Aguilera, E.; Cerecetto, H.; González, M.; Cabrera, M.; Alzugaray, M.; da Silva Vaz Junior, I.; Nunes da Fonseca, R.; Aguirre-López, B.; et al. Novel and Selective Rhipicephalus microplus Triosephosphate Isomerase Inhibitors with Acaricidal Activity. Vet. Sci. 2018, 5, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraro, F.; Corvo, I.; Bergalli, L.; Ilarraz, A.; Cabrera, M.; Gil, J.; Susuki, B.M.; Caffrey, C.R.; Timson, D.J.; Robert, X.; et al. Novel and selective inactivators of Triosephosphate isomerase with anti-trematode activity. Sci. Rep. 2020, 10, 2587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bringaud, F.; Ebikeme, C.; Boshart, M. Acetate and succinate production in amoebae, helminths, diplomonads, trichomonads and trypanosomatids: Common and diverse metabolic strategies used by parasitic lower eukaryotes. Parasitology 2010, 137, 1315–1331. [Google Scholar] [CrossRef]
- Rivière, L.; Van Weelden, S.W.H.; Glass, P.; Vegh, P.; Coustou, V.; Biran, M.; Van Hellemond, J.J.; Bringaud, F.; Tielens, A.G.M.; Boshart, M. Acetyl:succinate CoA-transferase in procyclic Trypanosoma brucei. Gene identification and role in carbohydrate metabolism. J. Biol. Chem. 2004, 279, 45337–45346. [Google Scholar] [CrossRef] [Green Version]
- Mikhailenko, V.M.; Philchenkov, A.A.; Zavelevich, M.P. Analysis of 1 H NMR-detectable mobile lipid domains for assessment of apoptosis induced by inhibitors of DNA synthesis and replication. Cell Biol. Int. 2005, 29, 33–39. [Google Scholar] [CrossRef]
- Benitez, D.; Pezaroglo, H.; Martínez, V.; Casanova, G.; Cabrera, G.; Galanti, N.; González, M.; Cerecetto, H. Study of Trypanosoma cruzi epimastigote cell death by NMR-visible mobile lipid analysis. Parasitology 2012, 139, 506–515. [Google Scholar] [CrossRef] [Green Version]
- Basmaciyan, L.; Azas, N.; Casanova, M. Calcein+/PI- as an early apoptotic feature in Leishmania. PLoS ONE 2017, 12, e0187756. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.A. Reactive Metabolites in the Biotransformation of Molecules Containing a Furan Ring. Chem. Res. Toxicol. 2013, 26, 6–25. [Google Scholar] [CrossRef] [Green Version]
- Osorio, Y.; Travi, B.L.; Renslo, A.R.; Peniche, A.G.; Melby, P.C. Identification of small molecule lead compounds for visceral leishmaniasis using a novel ex vivo splenic explant model system. PLoS Negl. Trop. Dis. 2011, 5, e962. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, C.; Moraca, F.; Medeiros, A.; Botta, M.; Hamilton, N.; Comini, M.A.; Schmidt, T.J. Binding mode and selectivity of steroids towards glucose-6-phosphate dehydrogenase from the pathogen Trypanosoma cruzi. Molecules 2016, 21, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014, 42, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 56531, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; Caldwell, J.; Wang, J.; Kollman, P. A Point-Charge Force Field for Molecular Mechanics Simulations of Proteins Based on Condensed-Phase. J. Comput. Chem. 2003, 24, 1999. [Google Scholar]
- Song, L.F.; Lee, T.; Zhu, C.; York, D.M.; Merz, K.M., Jr. Using AMBER18 for Relative Free Energy Calculations. J. Chem. Inf. Model. 2019, 176, 139–148. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Boiani, M.; Merlino, A.; Gerpe, A.; Porcal, W.; Croce, F.; Depaula, S.; Rodríguez, M.A. O-Nitroanilines as major metabolic products of in microsomal and cytosolic fractions of rat hepatocytes and in whole parasitic cells. Xenobiótica 2009, 39, 236–248. [Google Scholar] [CrossRef]
- Hallander, H.O.; Dornbusch, K.; Gezelius, L.; Jacobson, K.; Karlsson, I. Synergism between aminoglycosides and cephalosporins with antipseudomonal activity: Interaction index and killing curve method. Antimicrob. Agents Chemother. 1982, 22, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Machado, F.S.; Huang, H.; Oz, H.S.; Jelicks, L.A.; Prado, C.M.; Koba, W.; Fine, E.J.; Zhao, D.; Factor, S.M.; et al. Aspirin treatment of mice infected with Trypanosoma cruzi and implications for the pathogenesis of Chagas disease. PLoS ONE 2011, 6, e16959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldrich, C.; Bertozzi, C.; Georg, G.I.; Kiessling, L.; Lindsley, C.; Liotta, D.; Merz, K.M., Jr.; Schepartz, A.; Wang, S. The Ecstasy and Agony of Assay Interference Compounds. ACS Med. Chem. Lett. 2017, 8, 379–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risi, G.; Aguilera, E.; Ladós, G.; Suárez, G.; Carrera, I.; Álvarez, G.; Salinas, G. Caenorhabditis elegans Infrared-Based Motility Assay Identified New Hits for Nematicide Drug Development. Vet. Sci. 2019, 6, 29. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HITS | ||||
|---|---|---|---|---|
| Negative Control | Positive Control | 1018 | 1019 | 793 |
 |  |  | ||
| Toxicological profile | ||||
| Ames test (mutagenicity) | ||||
| NO | YES (one control per strain) | NO | NO | NO |
| Micronucleus test in mice (Genotoxicity) (mean of MnPE + ±SD +) | ||||
| 5 ± 1 | 36 ± 2 (cyclophosphamide) | 5 ± 1 | 6 ± 2 | 4 ± 1 |
| Teratogenicity in zebrafish (LD50 ± SD) * | ||||
| NA | 2.4 mM (caffeine) | >25 µM | 100 ± 12 µM | 22 ± 5 µM |
| Acute oral toxicity in mice (Up and Down test, LD50, mg/kg of body weight) | ||||
| NA | NA | >2000 | >2000 | 2000 |
| Glycine (Gly) | Succinate (Succ) | Pyruvate (Pyr) | Acetate (Ace) | Alanine (Ala) | Lactate (Lac) | Ethanol | |
|---|---|---|---|---|---|---|---|
| δ a (ppm) | 3.547 | 2.391 | 2.358 | 2.121 | 1.465 | 1.316 | 1.170 |
| Multiplicity | S b | S | S | S | D c | d | T d |
| Integration range | 3.55–3.54 | 2.40–2.38 | 2.37–2.35 | 2.12–2.12 | 1.49–1.44 | 1.34–1.30 | 1.20–1.14 |
| J(Hz) e | - c | - | - | - | 7.24 | 6.85 | 7.08 |
| Integration for 1019 | 1.53 ± 0.01 * | 7.08 ± 0.04 * | 6.3 ± 0.2 | 13.56 ± 0.03 | 10.3 ± 1.8 | 4.98 ± 0.03 | nq f |
| Integration of the baseline | 1.39 ± 0.03 | 5.24 ± 0.43 | 4.01 ± 0.94 | 11.5 ± 1.2 | 8.09 ± 0.40 | 3.99 ± 0.1 | nq |
| Condition | CH2/CH3 Ratio | Apparition of the Signal of Choline (3.10–3.30) ppm |
|---|---|---|
| Control | 0.27 | YES |
| Nfx | 0.25 | NO |
| Bnz | 0.50 | YES |
| 793 | 3.00 * | NO |
| 1019 | 0.18 | NO |
| 1018 | 0.06 | YES |
| Compound Combination | Effects | FICI | Optimal Molar Proportion |
|---|---|---|---|
| 793 plus 1018 | SYNERGISM | 0.5 | 125/1 |
| 793 plus 1019 | SYNERGISM | 0.75 | 8/1 |
| 793 plus 314 | SYNERGISM | 0.75 | 1/1.2 |
| 793 plus Bnz | SYNERGISM | 0.5 | 1/1 |
| 1018 plus 314 | SYNERGISM | 0.75 | 1/133 |
| 1018 plus 1019 | ANTAGONISM | 2 | - |
| 1018 plus Bnz | ANTAGONISM | 2 | - |
| 1260 plus 793 | ANTAGONISM | 2 | - |
| 1260 plus 1019 | ANTAGONISM | 2 | - |
| 1260 plus 1018 | ANTAGONISM | 1.5 | - |
| Bnz plus 314 | ADDITION | 1 | 1/2 |
| Treatment | Doses (µmoles/kg/day) | Reduction in the Parasitemia Peak (%) | Days Post-Infection of the First Parasitemia Peak | Survival (%) |
|---|---|---|---|---|
| Control | - | 0 | 21 | 50–100 |
| 793 [25] | 192 | 50 | 22 | 83 |
| 1019 [25] | 384 | 75 | 25 | 100 |
| 192 | 60 | 29 | 100 | |
| 1019 plus 793 | 17 + 282 (1/16) | 65 | 15 | 100 |
| 1018 [25] | 192 | 40 | 22 | 40 |
| 1.5 | 80 | 24 | 100 | |
| 1018 plus 793 | 1.5 + 192 (1/128) | 90 | 19 | 100 |
| 48 + 48 (1/1) | 50 | 30 | 88 | |
| Bnz | 48 | 95 | 43 | 100 |
| 38 | 90 | 15 | 100 | |
| 10 | 80 | 15 | 100 | |
| Bnz plus 793 | 38 + 192 | 83 | 30 | 100 |
| 314 | 102 | 77 | 20 | 100 |
| 314 plus 793 | 132 + 192 (1/1.4) | 68 | 26 | 88 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilera, E.; Sánchez, C.; Cruces, M.E.; Dávila, B.; Minini, L.; Mosquillo, F.; Pérez-Díaz, L.; Serna, E.; Torres, S.; Schini, A.; et al. Preclinical Studies and Drug Combination of Low-Cost Molecules for Chagas Disease. Pharmaceuticals 2023, 16, 20. https://doi.org/10.3390/ph16010020
Aguilera E, Sánchez C, Cruces ME, Dávila B, Minini L, Mosquillo F, Pérez-Díaz L, Serna E, Torres S, Schini A, et al. Preclinical Studies and Drug Combination of Low-Cost Molecules for Chagas Disease. Pharmaceuticals. 2023; 16(1):20. https://doi.org/10.3390/ph16010020
Chicago/Turabian StyleAguilera, Elena, Carina Sánchez, María Eugenia Cruces, Belén Dávila, Lucía Minini, Florencia Mosquillo, Leticia Pérez-Díaz, Elva Serna, Susana Torres, Alicia Schini, and et al. 2023. "Preclinical Studies and Drug Combination of Low-Cost Molecules for Chagas Disease" Pharmaceuticals 16, no. 1: 20. https://doi.org/10.3390/ph16010020
APA StyleAguilera, E., Sánchez, C., Cruces, M. E., Dávila, B., Minini, L., Mosquillo, F., Pérez-Díaz, L., Serna, E., Torres, S., Schini, A., Sanabria, L., Vera de Bilbao, N. I., Yaluff, G., Zolessi, F. R., Ceilas, L. F., Cerecetto, H., & Alvarez, G. (2023). Preclinical Studies and Drug Combination of Low-Cost Molecules for Chagas Disease. Pharmaceuticals, 16(1), 20. https://doi.org/10.3390/ph16010020