
Pyrrolyl and Indolyl α-γ-Diketo Acid Derivatives Acting as Selective Inhibitors of Human Carbonic Anhydrases IX and XII

 ,
, 
 ,
,  ,
,  ,
,  , , , ,
, , , ,  ,
, 
 , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. In Vitro Enzymatic Assay
2.3. Molecular Modeling
2.4. In Vitro Proliferation Assay
3. Materials and Methods
3.1. Chemistry
3.2. Molecular Modelling
3.3. Carbonic Anhydrase Inhibition
3.4. Biological Assays
3.4.1. Cell Cultures and Treatments
3.4.2. Cytotoxicity Assay
3.4.3. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Smith, K.S.; Jakubzick, C.; Whittam, T.S.; Ferry, J.G. Carbonic anhydrase is an ancient enzyme widespread in prokaryotes. Proc. Natl. Acad. Sci. USA 1999, 96, 15184–15189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, M.; Boone, C.D.; Kondeti, B.; McKenna, R. Structural annotation of human carbonic anhydrases. J. Enzyme Inhib. Med. Chem. 2013, 28, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.P.; Swenson, E.R. The distribution and physiological significance of carbonic anhydrase in vertebrate gas exchange organs. Respir. Physiol. 2000, 121, 1–12. [Google Scholar] [CrossRef]
- Esbaugh, A.J.; Tufts, B.L. The structure and function of carbonic anhydrase isozymes in the respiratory system of vertebrates. Respir. Physiol. Neurobiol. 2006, 154, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Occhipinti, R.; Boron, W.F. Role of Carbonic Anhydrases and Inhibitors in Acid–Base Physiology: Insights from Mathematical Modeling. Int. J. Mol. Sci. 2019, 20, 3841–3871. [Google Scholar] [CrossRef] [Green Version]
- Imtaiyaz Hassan, M.; Shajee, B.; Waheed, A.; Ahmad, F.; Sly, W.S. Structure, function and applications of carbonic anhydrase isozymes. Bioorganic Med. Chem. 2013, 21, 1570–1582. [Google Scholar] [CrossRef] [PubMed]
- Fisher, Z.; Hernandez Prada, J.A.; Tu, C.; Duda, D.; Yoshioka, C.; An, H.; Govindasamy, L.; Silverman, D.N.; McKenna, R. Structural and kinetic characterization of active-site histidine as a proton shuttle in catalysis by human carbonic anhydrase II. Biochemistry 2005, 44, 1097–1105. [Google Scholar] [CrossRef]
- Ismail, I.S. The Role of Carbonic Anhydrase in Hepatic Glucose Production. Curr. Diabetes Rev. 2018, 14, 108–112. [Google Scholar] [CrossRef]
- Singh, S.; Lomelino, C.L.; Mboge, M.Y.; Frost, S.C.; McKenna, R. Cancer drug development of carbonic anhydrase inhibitors beyond the active site. Molecules 2018, 23, 1045. [Google Scholar] [CrossRef] [Green Version]
- Krungkrai, S.R.; Krungkrai, J. Malaria parasite carbonic anhydrase: Inhibition of aromatic/heterocyclic sulfonamides and its therapeutic potential. Asian Pac. J. Trop. Biomed. 2011, 3, 233–242. [Google Scholar] [CrossRef]
- De Vita, D.; Angeli, A.; Pandolfi, F.; Bortolami, M.; Costi, R.; Di Santo, R.; Suffredini, E.; Ceruso, M.; Del Prete, S.; Capasso, C.; et al. Inhibition of the α-carbonic anhydrase from Vibrio cholerae with amides and sulfonamides incorporating imidazole moieties. J. Enzym. Inhib. Med. Chem. 2017, 32, 798–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez Covarrubias, A.; Larsson, A.M.; Högbom, M.; Lindberg, J.; Bergfors, T.; Björkelid, C.; Mowbray, S.L.; Unge, T.; Jones, T.A. Structure and function of carbonic anhydrases from Mycobacterium tuberculosis. J. Biol. Chem. 2005, 280, 18782–18789. [Google Scholar] [CrossRef] [Green Version]
- Klengel, T.; Liang, W.J.; Chaloupka, J.; Ruoff, C.; Schröppel, K.; Naglik, J.R.; Eckert, S.E.; Mogensen, E.G.; Haynes, K.; Tuite, M.F.; et al. Fungal adenylyl cyclase integrates CO2 sensing with cAMP signaling and virulence. Curr. Biol. 2005, 15, 2021–2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahn, Y.S.; Cox, G.M.; Perfect, J.R.; Heitman, J. Carbonic anhydrase and CO2 sensing during Cryptococcus neoformans growth, differentiation, and virulence. Curr. Biol. 2005, 15, 2013–2020. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, J.; Van Dijken, J.P.; De Winde, J.H.; Pronk, J.T. Carbonic anhydrase (Nce103p): An essential biosynthetic enzyme for growth of Saccharomyces cerevisiae at atmospheric carbon dioxide pressure. Biochem. J. 2005, 391, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia and cancer. Cancer Metastasis Rev. 2007, 26, 223–224. [Google Scholar] [CrossRef] [PubMed]
- Nasu, K.; Yamaguchi, K.; Takanashi, T.; Tamai, K.; Sato, I.; Ine, S.; Sasaki, O.; Satoh, K.; Tanaka, N.; Tanaka, Y.; et al. Crucial role of carbonic anhydrase IX in tumorigenicity of xenotransplanted adult T-cell leukemia-derived cells. Cancer Sci. 2017, 108, 435–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasu, K.; Yamaguchi, K.; Takanashi, Y.; Sugamura, K.; Harigae, H. Carbonic anhydrase IX promotes the tumorigenicity of adult T-cell leukemia/lymphoma-derived cells in NOG mice. Blood 2015, 126, 4822. [Google Scholar] [CrossRef]
- Lee, S.H.; Griffiths, J.R. How and why are cancers acidic? Carbonic anhydrase IX and the homeostatic control of tumour extracellular pH. Cancers 2020, 12, 1616. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Mojzych, M.; Marciniak, B.; Drozda, R.; Kontek, R. Targeting carbonic anhydrase IX and XII isoforms with small molecule inhibitors and monoclonal antibodies. J. Enzym. Inhib. Med. Chem. 2022, 37, 1278–1298. [Google Scholar] [CrossRef]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daunys, S.; Petrikaitė, V. The roles of carbonic anhydrases IX and XII in cancer cell adhesion, migration, invasion and metastasis. Biol. Cell 2020, 112, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Mboge, M.Y.; McKenna, R.; Frost, S.C. Advances in Anti-Cancer Drug Development Targeting Carbonic Anhydrase IX and XII. Top Anticancer. Res. 2015, 5, 3–42. [Google Scholar] [PubMed]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef]
- Ord, J.J.; Agrawal, S.; Thamboo, T.P.; Roberts, I.; Campo, L.; Turley, H.; Han, C.; Fawcett, D.W.; Kulkarni, R.P.; Cranston, D.; et al. An investigation into the prognostic significance of necrosis and hypoxia in high grade and invasive bladder cancer. J. Urol. 2007, 178, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev. 2007, 26, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, G.J.; Valentine, H.R.; Loncaster, J.A.; Davidson, S.E.; Hunter, R.D.; Roberts, S.A.; Harris, A.L.; Stratford, I.J.; Price, P.M.; West, C.M. Hypoxia-inducible factor 1alpha expression as an intrinsic marker of hypoxia: Correlation with tumor oxygen, pimonidazole measurements, and outcome in locally advanced carcinoma of the cervix. Clin. Cancer Res. 2004, 10, 8405–8412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Pastorek, J.; Karapantzos, I.; Gatter, K.C.; Harris, A.L. Tumour and angiogenesis research group. Hypoxia-activated tumor pathways of angiogenesis and pH regulation independent of anemia in head-and-neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.P.; Harris, A.L. Diagnostic, prognostic and therapeutic implications of carbonic anhydrases in cancer. Br. J. Cancer. 2003, 89, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.A.; Ganesan, R.; Reynolds, G.; Gross, L.; Stevens, A.; Pastorek, J.; Murray, P.G.; Perunovic, B.; Anwar, M.S.; Billingham, L.; et al. Hypoxia-regulated carbonic anhydrase IX expression is associated with poor survival in patients with invasive breast cancer. Br. J. Cancer 2007, 96, 104–109. [Google Scholar] [CrossRef]
- Swinson, D.E.; Jones, J.L.; Richardson, D.; Wykoff, C.; Turley, H.; Pastorek, J.; Taub, N.; Harris, A.L.; O’Byrne, K.J. Carbonic anhydrase IX expression, a novel surrogate marker of tumor hypoxia, is associated with a poor prognosis in non-small-cell lung cancer. J. Clin. Oncol. 2003, 21, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Dorai, T.; Sawczuk, I.; Pastorek, J.; Wiernik, P.H.; Dutcher, J.P. Role of carbonic anhydrases in the progression of renal cell carcinoma subtypes: Proposal of a unified hypothesis. Cancer Investig. 2006, 24, 754–779. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.H.C.; Lee, D.Y.; Lee, B.; Li, H.; Lim, J.C.T.; Lim, J.X.; Yeong, J.P.S.; Lau, H.Y.; Thike, A.A.; Tan, P.H.; et al. Hypoxia-regulated carbonic anhydrase IX (CAIX) protein is an independent prognostic indicator in triple negative breast cancer. Breast Cancer Res. 2022, 24, 38. [Google Scholar] [CrossRef]
- Trastour, C.; Benizri, E.; Ettore, F.; Ramaioli, A.; Chamorey, E.; Pouysségur, J.; Berra, E. HIF-1alpha and CA IX staining in invasive breast carcinomas: Prognosis and treatment outcome. Int. J. Cancer 2007, 120, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.S.; Lin, C.W.; Hsieh, Y.H.; Chien, M.H.; Chuang, C.Y.; Yang, S.F. Overexpression of carbonic anhydrase IX induces cell motility by activating matrix metalloproteinase-9 in human oral squamous cell carcinoma cells. Oncotarget 2017, 8, 83088–83099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viikilä, P.; Kivelä, A.J.; Mustonen, H.; Koskensalo, S.; Waheed, A.; Sly, W.S.; Pastorek, J.; Pastorekova, S.; Parkkila, S.; Haglund, C. Carbonic anhydrase enzymes II, VII, IX and XII in colorectal carcinomas. World J Gastroenterol. 2016, 22, 8168–8177. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase inhibitors. Bioorganic Med. Chem. Lett. 2010, 20, 3467–3474. [Google Scholar] [CrossRef]
- Ghiasi, M.; Gholami, S. Quantum mechanical study of human carbonic anhydrase II in complex with polyamines as novel inhibitors: Kinetic and thermodynamic investigation. Comput. Theor. Chem. 2020, 1186, 112911. [Google Scholar] [CrossRef]
- Moghoufei, L.; Mehrabi, M.; Adibi, H.; Khodarahmi, R. Synthesis of 4-hydroxy-L-proline derivatives as new non-classical inhibitors of human carbonic anhydrase II activity: An in vitro study. J. Biomol. Struct. Dyn. 2022, 40, 1–11. [Google Scholar] [CrossRef]
- Kirschberg, T.; Parrish, J. Metal chelators as antiviral agents. Curr. Opin. Drug Discov. Dev. 2007, 4, 460–472. [Google Scholar]
- Rouffet, M.; Cohen, S.M. Emerging trends in metalloprotein inhibition. Dalton Trans. 2011, 40, 3445–3454. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W. Rational design of artificial metalloproteins and metalloenzymes with metal clusters. Molecules 2019, 24, 2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.Y.; Adamek, R.N.; Dick, B.L.; Credille, C.V.; Morrison, C.N.; Cohen, S.M. Targeting metalloenzymes for therapeutic intervention. Chem. Rev. 2019, 119, 1323–1455. [Google Scholar] [CrossRef] [PubMed]
- Day, J.A.; Cohen, S.M. Investigating the selectivity of metalloenzyme inhibitors. J. Med. Chem. 2013, 56, 7997–8007. [Google Scholar] [CrossRef] [Green Version]
- Rogolino, D.; Carcelli, M.; Sechi, M.; Neamati, N. Viral enzymes containing magnesium: Metal binding as a successful strategy in drug design. Coord. Chem. Rev. 2012, 256, 3063–3086. [Google Scholar] [CrossRef]
- Prousis, K.C.; Athanasellis, G.; Stefanou, V.; Matiadis, D.; Kokalari, E.; Igglessi-Markopoulou, O.; McKee, V.; Markopoulos, J. Synthesis and crystal structure characterization of zinc (II) tetronic acid complexes. Bioinorg. Chem. Appl. 2010, 2010, 651652. [Google Scholar] [CrossRef] [Green Version]
- Corona, A.; Di Leva, F.S.; Rigogliuso, G.; Pescatori, L.; Madia, V.N.; Subra, F.; Delelis, O.; Esposito, F.; Cadeddu, M.; Costi, R.; et al. New insights into the interaction between pyrrolyl diketoacids and HIV-1 integrase active site and comparison with RNase, H. Antivir. Res. 2016, 134, 236–243. [Google Scholar] [CrossRef]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [CrossRef]
- Cuzzucoli Crucitti, G.; Metifiot, M.; Pescatori, L.; Messore, A.; Madia, V.N.; Pupo, G.; Saccoliti, F.; Scipione, L.; Tortorella, S.; Esposito, F.; et al. Structure-activity relationship of pyrrolyl diketo acid derivative as dual inhibitors of HIV-1 integrase and reverse transcriptase ribonuclease H domain. J. Med. Chem. 2015, 58, 1915–1928. [Google Scholar] [CrossRef]
- Messore, A.; Corona, A.; Madia, V.N.; Saccoliti, F.; Tudino, V.; De Leo, A.; Scipione, L.; De Vita, D.; Amendola, G.; Di Maro, S.; et al. Pyrrolyl pyrazoles as non-diketo acid inhibitors of the HIV-1 ribonuclease H function of reverse transcriptase. ACS Med. Chem. Lett. 2020, 11, 798–805. [Google Scholar] [CrossRef]
- Costi, R.; Cuzzucoli Crucitti, G.C.; Pescatori, L.; Messore, A.; Scipione, L.; Tortorella, S.; Amoroso, A.; Crespan, E.; Campiglia, P.; Maresca, B.; et al. New nucleotide-competitive non-nucleoside inhibitors of terminal deoxynucleotidyl transferase: Discovery, characterization, and crystal structure in complex with the target. J. Med. Chem. 2013, 56, 7431–7441. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Suite Release 2021-3. (a) Maestro v.12.9; (b) Epik, v.5.7; (c) Macromodel v.13.3; (d) Glide, v.9.2; (e) Desmond, v.6.7; (f) Prime, v.5.5; (g) Impact, v.9.2; (h) Jaguar, v.11.3; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Bonardi, A.; Nocentini, A.; Bua, S.; Combs, J.; Lomelino, C.; Andring, J.; Lucarini, L.; Sgambellone, S.; Masini, E.; McKenna, R.; et al. Sulfonamide Inhibitors of human carbonic anhydrases designed through a three-tails approach: Improving ligand/isoform matching and selectivity of action. J. Med. Chem. 2020, 63, 7422–7444. [Google Scholar] [CrossRef] [PubMed]
- Bonardi, A.; Micheli, L.; Di Cesare Mannelli, L.; Ghelardini, C.; Gratteri, P.; Nocentini, A.; Supuran, C.T. Development of hydrogen sulfide-releasing carbonic anhydrases IX- and XII-selective inhibitors with enhanced antihyperalgesic action in a rat model of arthritis. J. Med. Chem. 2022, 65, 13143–13157. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
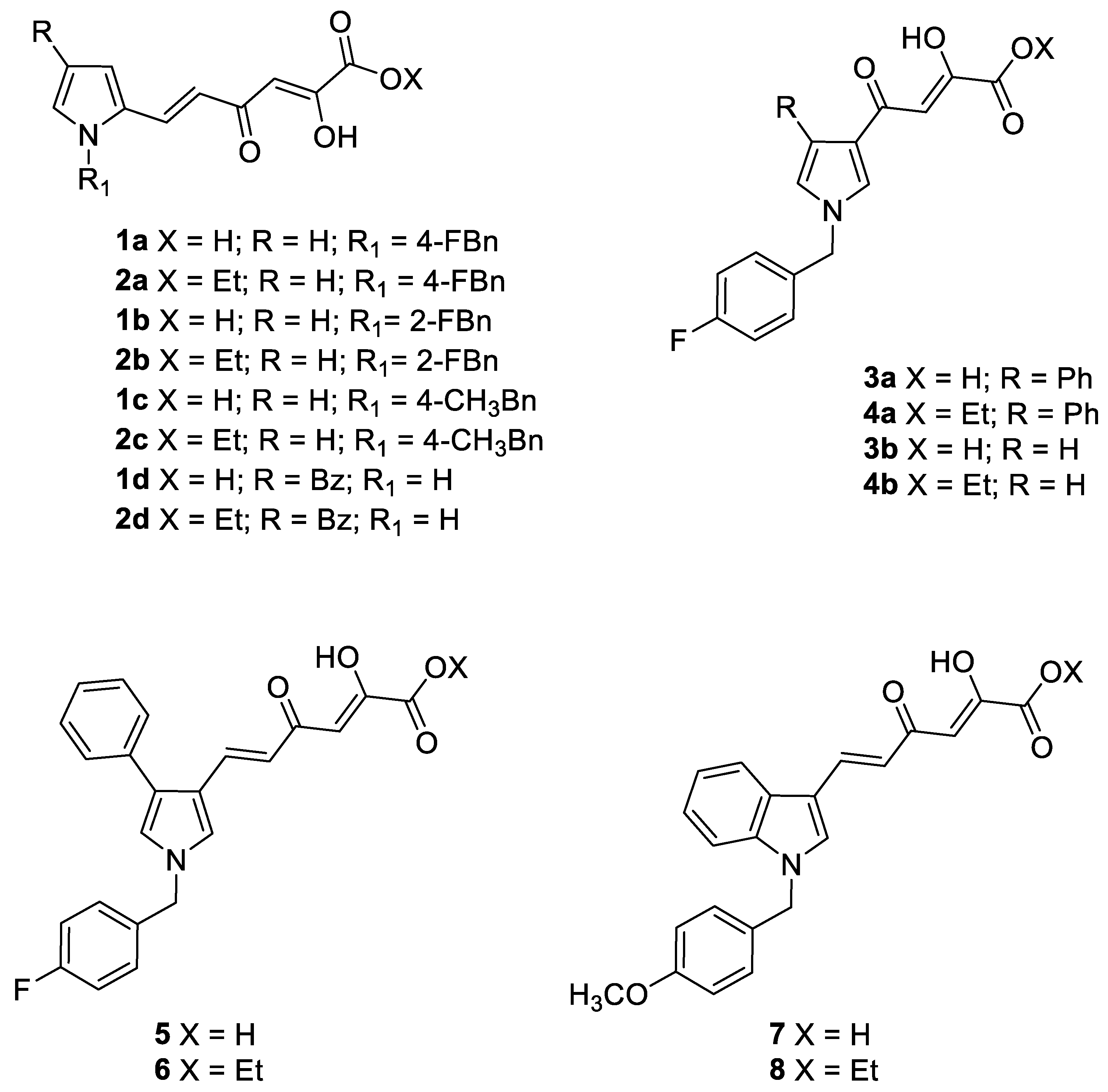
| Cpd | R | R1 | X | Ki (µM) 1 | |||
|---|---|---|---|---|---|---|---|
| CA I | CA II | CA IX | CA XII | ||||
| 1a | H | 4-FBn 2 | H | 81.9 ± 6.0 | 14.6 ± 0.9 | 2.0 ± 0.1 | 1.0 ± 0.1 |
| 2a | H | 4-FBn | Et | >100 | >100 | 90.1 ± 4.7 | 83.6 ± 5.4 |
| 1b | H | 2-FBn | H | 89.6 ± 4.8 | 14.0 ± 0.8 | 2.5 ± 0.1 | 2.2 ± 0.1 |
| 2b | H | 2-FBn | Et | >100 | >100 | 23.7 ± 1.6 | 80.5 ± 6.1 |
| 1c | H | 4-CH3Bn | H | 79.0 ± 3.9 | 44.4 ± 2.6 | 0.82 ± 0.05 | 0.90 ± 0.07 |
| 2c | H | 4-CH3Bn | Et | >100 | >100 | 90.8 ± 4.6 | 89.3 ± 5.3 |
| 1d | Bz 3 | - | H | 72.5 ± 5.1 | 38.7 ± 2.0 | 1.4 ± 0.1 | 1.1 ± 0.1 |
| 2d | Bz | - | Et | nd 5 | nd | nd | nd |
| 3a | Ph 4 | - | H | >100 | 77.6 ± 4.5 | 1.4 ± 0.1 | 0.33 ± 0.02 |
| 4a | Ph | - | Et | >100 | >100 | 57.3 ± 2.8 | 89.2 ± 5.2 |
| 3b | H | - | H | 50.9 ± 4.1 | 54.7 ± 2.9 | 18.4 ± 1.2 | 7.9 ± 0.6 |
| 4b | H | - | Et | >100 | >100 | >100 | 71.5 ± 4.3 |
| 5 | - | - | H | >100 | 62.5 ± 3.6 | 0.32 ± 0.02 | 0.24 ± 0.02 |
| 6 | - | - | Et | >100 | >100 | 43.1 ± 2.5 | 89.0 ± 6.1 |
| 7 | - | - | H | 91.7 ± 7.5 | 36.5 ± 1.9 | 1.1 ± 0.1 | 0.83 ± 0.06 |
| 8 | - | - | Et | >100 | >100 | 80.1 ± 5.3 | 92.0 ± 6.0 |
| AAZ | - | - | 0.250 | 0.012 | 0.025 | 0.0057 | |
| Cpd | EC50 (µM) 1 | |||
|---|---|---|---|---|
| MDA-MB231 | HT-29 | FaDu | SCC-15 | |
| 1b | nd 2 | 92 ± 9.9 (24 h) | 98 ± 8.6(24 h) | nd |
| nd | 75 ± 8.2 (48 h) | 90 ± 9.3(48 h) | nd | |
| 1c | 85 ± 9.1 (24 h) | 115 ± 10.8 (24 h) | 59 ± 4.5 (24 h) | 96 ± 8.5 (24 h) |
| 73 ± 5.5 (48 h) | 48 ± 4.1 (48 h) | 17 ± 0.9 (48 h) | 53 ± 5.9 (48 h) | |
| 5 | 120 ± 15.7 (24 h) | 98 ± 11.0 (24 h) | 103 ± 11.4 (24 h) | 102 ± 9.7 (24 h) |
| 105 ± 13.2 (48 h) | 21 ± 1.6 (48 h) | 23 ± 2.8 (48 h) | 58 ± 6.2 (48 h) | |
| 7 | 21 ± 1.8 (24 h) | 47 ± 5.0 (24 h) | 51 ± 4.7 (24 h) | 48 ± 3.9 (24 h) |
| 12 ± 1.1 (48 h) | 30 ± 2.8 (48 h) | 22 ± 1.2 (48 h) | 17 ± 0.8 (48 h) | |
| 8 | 23 ± 1.6 (24 h) | 75 ± 8.8(24 h) | 41 ± 3.3(24 h) | 44 ± 4.1 (24 h) |
| 17 ± 0.9 (48 h) | 44 ± 2.3 (48 h) | 18 ± 0.7(48 h) | 21 ± 1.9 (48 h) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ialongo, D.; Messore, A.; Madia, V.N.; Tudino, V.; Nocentini, A.; Gratteri, P.; Giovannuzzi, S.; Supuran, C.T.; Nicolai, A.; Scarpa, S.; et al. Pyrrolyl and Indolyl α-γ-Diketo Acid Derivatives Acting as Selective Inhibitors of Human Carbonic Anhydrases IX and XII. Pharmaceuticals 2023, 16, 188. https://doi.org/10.3390/ph16020188
Ialongo D, Messore A, Madia VN, Tudino V, Nocentini A, Gratteri P, Giovannuzzi S, Supuran CT, Nicolai A, Scarpa S, et al. Pyrrolyl and Indolyl α-γ-Diketo Acid Derivatives Acting as Selective Inhibitors of Human Carbonic Anhydrases IX and XII. Pharmaceuticals. 2023; 16(2):188. https://doi.org/10.3390/ph16020188
Chicago/Turabian StyleIalongo, Davide, Antonella Messore, Valentina Noemi Madia, Valeria Tudino, Alessio Nocentini, Paola Gratteri, Simone Giovannuzzi, Claudiu T. Supuran, Alice Nicolai, Susanna Scarpa, and et al. 2023. "Pyrrolyl and Indolyl α-γ-Diketo Acid Derivatives Acting as Selective Inhibitors of Human Carbonic Anhydrases IX and XII" Pharmaceuticals 16, no. 2: 188. https://doi.org/10.3390/ph16020188
APA StyleIalongo, D., Messore, A., Madia, V. N., Tudino, V., Nocentini, A., Gratteri, P., Giovannuzzi, S., Supuran, C. T., Nicolai, A., Scarpa, S., Taurone, S., Camarda, M., Artico, M., Papa, V., Saccoliti, F., Scipione, L., Di Santo, R., & Costi, R. (2023). Pyrrolyl and Indolyl α-γ-Diketo Acid Derivatives Acting as Selective Inhibitors of Human Carbonic Anhydrases IX and XII. Pharmaceuticals, 16(2), 188. https://doi.org/10.3390/ph16020188