


Advances of Artificial Intelligence in Anti-Cancer Drug Design: A Review of the Past Decade

Abstract
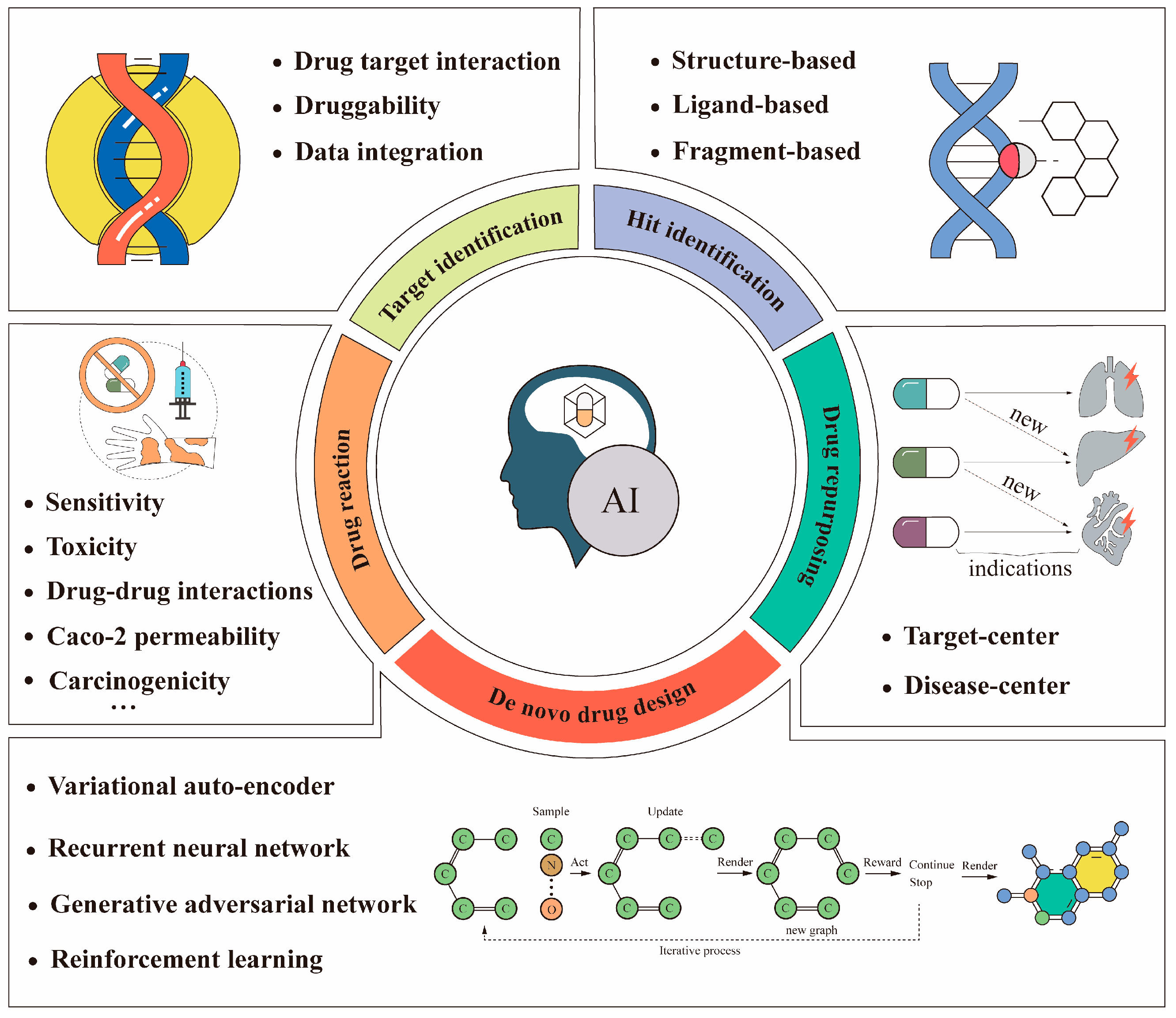
:1. Introduction
2. Method
3. Artificial Intelligence in Anti-Cancer Drug Target Identification
3.1. Artificial Intelligence Efficiently Elevates the Prediction Accuracy of DTI
3.2. Artificial Intelligence Could Integrate Data from Multiple Sources to Help with Anti-Drug Target Identification
3.3. Artificial Intelligence Could Help Predict the Druggability of Anti-Cancer Drug Targets
4. Artificial Intelligence in the Screening of Anti-Cancer Drug Hit Compounds
4.1. Structure-Based Screening of Hit Compounds Using Artificial Intelligence
4.2. Ligand-Based Screening of Hit Compounds Using Artificial Intelligence
4.3. Fragment-Based Screening of Hit Compounds Using Artificial Intelligence
5. Artificial Intelligence in De Novo Anti-Cancer Drug Design
5.1. Application of Variational Auto-Encoder to De Novo Design of Anticancer Drugs
5.2. Application of the Recurrent Neural Network to De Novo Design of Anti-Cancer Drugs
5.3. Application of Generative Adversarial Network to De Novo Design of Anti-Cancer Drugs
5.4. Application of Deep Reinforcement Learning to De Novo Design of Anti-Cancer Drugs
6. Artificial Intelligence in Anti-Cancer Drug Repurposing
6.1. Artificial Intelligence in Anti-Drug Repositioning Based on the Interaction between a Drug and a Target
6.2. Artificial Intelligence in Anti-Drug Repositioning Based on the Interaction between Drugs and Diseases
7. Artificial Intelligence-Assisted Accurate Prediction of Anti-Cancer Drug Reactions
7.1. Artificial Intelligence Aids in Predicting the ADMET Properties of Anti-Cancer Drugs
7.2. Artificial Intelligence Aids in Predicting Anti-Cancer Drug Sensitivity
7.3. Artificial Intelligence Aids in Predicting Toxicity of Anti-Cancer Drugs
7.4. Artificial Intelligence Can Predict Drug–Drug Interactions
8. Data Sources of Artificial Intelligence to Anti-Cancer Drug Designs
9. Successful Cases Applying AI in Anti-Cancer Drug Design
10. Conclusions and Prospects of Future Challenges
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moreau, P.; Garfall, A.L.; van de Donk, N.W.C.J.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Nooka, A.K.; Martin, T.; Rosinol, L.; Chari, A.; et al. Teclistamab in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2022, 387, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Avery, R.K.; Alain, S.; Alexander, B.D.; Blumberg, E.A.; Chemaly, R.F.; Cordonnier, C.; Duarte, R.F.; Florescu, D.F.; Kamar, N.; Kumar, D.; et al. Maribavir for Refractory Cytomegalovirus Infections With or Without Resistance Post-Transplant: Results From a Phase 3 Randomized Clinical Trial. Clin. Infect. Dis. 2022, 75, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Spetz, M.R.; Li, D.; Ho, M. Advances in immunotherapeutic targets for childhood cancers: A focus on glypican-2 and B7-H3. Pharmacol. Ther. 2021, 223, 107892. [Google Scholar] [CrossRef]
- Corti, C.; Cobanaj, M.; Marian, F.; Dee, E.C.; Lloyd, M.R.; Marcu, S.; Dombrovschi, A.; Biondetti, G.P.; Batalini, F.; Celi, L.A.; et al. Artificial intelligence for prediction of treatment outcomes in breast cancer: Systematic review of design, reporting standards, and bias. Cancer Treat. Rev. 2022, 108, 102410. [Google Scholar] [CrossRef]
- Chiu, H.-Y.; Chao, H.-S.; Chen, Y.-M. Application of Artificial Intelligence in Lung Cancer. Cancers 2022, 14, 1370. [Google Scholar] [CrossRef]
- Hamilton, W.; Walter, F.M.; Rubin, G.; Neal, R.D. Improving early diagnosis of symptomatic cancer. Nat. Rev. Clin. Oncol. 2016, 13, 740–749. [Google Scholar] [CrossRef]
- Fountzilas, E.; Tsimberidou, A.M.; Vo, H.H.; Kurzrock, R. Clinical trial design in the era of precision medicine. Genome. Med. 2022, 14, 101. [Google Scholar] [CrossRef]
- Bannigan, P.; Aldeghi, M.; Bao, Z.; Häse, F.; Aspuru-Guzik, A.; Allen, C. Machine learning directed drug formulation development. Adv. Drug Deliv. Rev. 2021, 175, 113806. [Google Scholar] [CrossRef]
- Olgen, S. Overview on Anticancer Drug Design and Development. Curr. Med. Chem. 2018, 25, 1704–1719. [Google Scholar] [CrossRef]
- Grandori, C.; Kemp, C.J. Personalized Cancer Models for Target Discovery and Precision Medicine. Trends Cancer 2018, 4, 634–642. [Google Scholar] [CrossRef]
- Hoelder, S.; Clarke, P.A.; Workman, P. Discovery of small molecule cancer drugs: Successes, challenges and opportunities. Mol. Oncol. 2012, 6, 155–176. [Google Scholar] [CrossRef]
- Schneider, P.; Walters, W.P.; Plowright, A.T.; Sieroka, N.; Listgarten, J.; Goodnow, R.A.; Fisher, J.; Jansen, J.M.; Duca, J.S.; Rush, T.S.; et al. Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 2020, 19, 353–364. [Google Scholar] [CrossRef]
- Pandiyan, S.; Wang, L. A comprehensive review on recent approaches for cancer drug discovery associated with artificial intelligence. Comput. Biol. Med. 2022, 150, 106140. [Google Scholar] [CrossRef]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Bleker de Oliveira, M.; Koshkin, V.; Liu, G.; Krylov, S.N. Analytical Challenges in Development of Chemoresistance Predictors for Precision Oncology. Anal. Chem. 2020, 92, 12101–12110. [Google Scholar] [CrossRef]
- Dentro, S.C.; Leshchiner, I.; Haase, K.; Tarabichi, M.; Wintersinger, J.; Deshwar, A.G.; Yu, K.; Rubanova, Y.; Macintyre, G.; Demeulemeester, J.; et al. Characterizing genetic intra-tumor heterogeneity across 2658 human cancer genomes. Cell 2021, 184. [Google Scholar] [CrossRef]
- Suhail, Y.; Cain, M.P.; Vanaja, K.; Kurywchak, P.A.; Levchenko, A.; Kalluri, R.; Kshitiz. Systems Biology of Cancer Metastasis. Cell Syst. 2019, 9, 109–127. [Google Scholar] [CrossRef]
- Fleming, N. How artificial intelligence is changing drug discovery. Nature 2018, 557, S55–S57. [Google Scholar] [CrossRef] [PubMed]
- Boniolo, F.; Dorigatti, E.; Ohnmacht, A.J.; Saur, D.; Schubert, B.; Menden, M.P. Artificial intelligence in early drug discovery enabling precision medicine. Expert. Opin. Drug Discov. 2021, 16. [Google Scholar] [CrossRef] [PubMed]
- Huwaimel, B.; Alobaida, A. Anti-Cancer Drug Solubility Development within a Green Solvent: Design of Novel and Robust Mathematical Models Based on Artificial Intelligence. Molecules 2022, 27, 5140. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem. Rev. 2019, 119, 10520–10594. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, I.M. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef]
- Hansen, S.G.; Wu, H.L.; Burwitz, B.J.; Hughes, C.M.; Hammond, K.B.; Ventura, A.B.; Reed, J.S.; Gilbride, R.M.; Ainslie, E.; Morrow, D.W.; et al. Broadly targeted CD8⁺ T cell responses restricted by major histocompatibility complex E. Science 2016, 351, 714–720. [Google Scholar] [CrossRef]
- Campos, K.R.; Coleman, P.J.; Alvarez, J.C.; Dreher, S.D.; Garbaccio, R.M.; Terrett, N.K.; Tillyer, R.D.; Truppo, M.D.; Parmee, E.R. The importance of synthetic chemistry in the pharmaceutical industry. Science 2019, 363. [Google Scholar] [CrossRef]
- Woo, M. An AI boost for clinical trials. Nature 2019, 573, S100–S102. [Google Scholar] [CrossRef]
- Zwicker, M. Understanding spatial environments from images. Science 2018, 360, 1188. [Google Scholar] [CrossRef]
- Cao, L.; Yang, J.; Rong, Z.; Li, L.; Xia, B.; You, C.; Lou, G.; Jiang, L.; Du, C.; Meng, H.; et al. A novel attention-guided convolutional network for the detection of abnormal cervical cells in cervical cancer screening. Med. Image Anal. 2021, 73, 102197. [Google Scholar] [CrossRef]
- Abriata, L.A.; Dal Peraro, M. State-of-the-art web services for de novo protein structure prediction. Brief. Bioinform. 2021, 22. [Google Scholar] [CrossRef]
- Xuan, P.; Zhang, Y.; Cui, H.; Zhang, T.; Guo, M.; Nakaguchi, T. Integrating multi-scale neighbouring topologies and cross-modal similarities for drug-protein interaction prediction. Brief. Bioinform. 2021, 22. [Google Scholar] [CrossRef]
- You, J.; Liu, B.; Ying, Z.; Pande, V.; Leskovec, J. Graph convolutional policy network for goal-directed molecular graph generation. Adv. Neural Inform. Process. Syst. 2018, 31. [Google Scholar] [CrossRef]
- Ghimire, A.; Tayara, H.; Xuan, Z.; Chong, K.T. CSatDTA: Prediction of Drug-Target Binding Affinity Using Convolution Model with Self-Attention. Int. J. Mol. Sci. 2022, 23, 8453. [Google Scholar] [CrossRef]
- Peng, J.; Wang, Y.; Guan, J.; Li, J.; Han, R.; Hao, J.; Wei, Z.; Shang, X. An end-to-end heterogeneous graph representation learning-based framework for drug-target interaction prediction. Brief. Bioinform. 2021, 22, bbaa430. [Google Scholar] [CrossRef]
- Shao, K.; Zhang, Y.; Wen, Y.; Zhang, Z.; He, S.; Bo, X. DTI-HETA: Prediction of drug-target interactions based on GCN and GAT on heterogeneous graph. Brief. Bioinform. 2022, 23. [Google Scholar] [CrossRef]
- Yang, Z.; Zhong, W.; Zhao, L.; Chen, C.Y.-C. ML-DTI: Mutual Learning Mechanism for Interpretable Drug-Target Interaction Prediction. J. Phys. Chem. Lett. 2021, 12, 4247–4261. [Google Scholar] [CrossRef]
- Dara, S.; Dhamercherla, S.; Jadav, S.S.; Babu, C.M.; Ahsan, M.J. Machine Learning in Drug Discovery: A Review. Artif. Intell. Rev. 2022, 55, 1947–1999. [Google Scholar] [CrossRef]
- Madhukar, N.S.; Khade, P.K.; Huang, L.; Gayvert, K.; Galletti, G.; Stogniew, M.; Allen, J.E.; Giannakakou, P.; Elemento, O. A Bayesian machine learning approach for drug target identification using diverse data types. Nat. Commun. 2019, 10, 5221. [Google Scholar] [CrossRef]
- Olayan, R.S.; Ashoor, H.; Bajic, V.B. DDR: Efficient computational method to predict drug-target interactions using graph mining and machine learning approaches. Bioinformatics 2018, 34, 1164–1173. [Google Scholar] [CrossRef]
- Raies, A.; Tulodziecka, E.; Stainer, J.; Middleton, L.; Dhindsa, R.S.; Hill, P.; Engkvist, O.; Harper, A.R.; Petrovski, S.; Vitsios, D. DrugnomeAI is an ensemble machine-learning framework for predicting druggability of candidate drug targets. Commun. Biol. 2022, 5, 1291. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, F.; Li, Y.; Wang, J.; Zhang, K.; Liu, Y.; Wu, M.; Zheng, J. KG4SL: Knowledge graph neural network for synthetic lethality prediction in human cancers. Bioinformatics 2021, 37, i418–i425. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhao, X.; Zhou, J.; Yang, J.; Zhang, Y.; Kuang, W.; Peng, J.; Chen, L.; Zeng, J. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 2017, 8, 573. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. [Google Scholar] [CrossRef]
- Tang, J.; Szwajda, A.; Shakyawar, S.; Xu, T.; Hintsanen, P.; Wennerberg, K.; Aittokallio, T. Making sense of large-scale kinase inhibitor bioactivity data sets: A comparative and integrative analysis. J. Chem. Inf. Model 2014, 54, 735–743. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Metz, J.T.; Johnson, E.F.; Soni, N.B.; Merta, P.J.; Kifle, L.; Hajduk, P.J. Navigating the kinome. Nat. Chem. Biol. 2011, 7, 200–202. [Google Scholar] [CrossRef]
- Chen, H.; Engkvist, O. Has Drug Design Augmented by Artificial Intelligence Become a Reality? Trends Pharmacol. Sci. 2019, 40, 806–809. [Google Scholar] [CrossRef]
- Lu, S.-H.; Wu, J.W.; Liu, H.-L.; Zhao, J.-H.; Liu, K.-T.; Chuang, C.-K.; Lin, H.-Y.; Tsai, W.-B.; Ho, Y. The discovery of potential acetylcholinesterase inhibitors: A combination of pharmacophore modeling, virtual screening, and molecular docking studies. J. Biomed. Sci. 2011, 18, 8. [Google Scholar] [CrossRef]
- Paricharak, S.; Méndez-Lucio, O.; Chavan Ravindranath, A.; Bender, A.; Ijzerman, A.P.; van Westen, G.J.P. Data-driven approaches used for compound library design, hit triage and bioactivity modeling in high-throughput screening. Brief. Bioinform. 2018, 19, 277–285. [Google Scholar] [CrossRef]
- Blay, V.; Tolani, B.; Ho, S.P.; Arkin, M.R. High-Throughput Screening: Today’s biochemical and cell-based approaches. Drug Discov. Today 2020, 25, 1807–1821. [Google Scholar] [CrossRef]
- Sheng, C.; Dong, G.; Miao, Z.; Zhang, W.; Wang, W. State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chem. Soc. Rev. 2015, 44, 8238–8259. [Google Scholar] [CrossRef]
- Lu, C.; Liu, S.; Shi, W.; Yu, J.; Zhou, Z.; Zhang, X.; Lu, X.; Cai, F.; Xia, N.; Wang, Y. Systemic evolutionary chemical space exploration for drug discovery. J. Cheminform. 2022, 14, 19. [Google Scholar] [CrossRef]
- Yasuo, N.; Sekijima, M. Improved Method of Structure-Based Virtual Screening via Interaction-Energy-Based Learning. J. Chem. Inf. Model. 2019, 59, 1050–1061. [Google Scholar] [CrossRef]
- Krasoulis, A.; Antonopoulos, N.; Pitsikalis, V.; Theodorakis, S. DENVIS: Scalable and High-Throughput Virtual Screening Using Graph Neural Networks with Atomic and Surface Protein Pocket Features. J. Chem. Inf. Model. 2022, 62, 4642–4659. [Google Scholar] [CrossRef]
- Li, B.-H.; Ge, J.-Q.; Wang, Y.-L.; Wang, L.-J.; Zhang, Q.; Bian, C. Ligand-Based and Docking-Based Virtual Screening of MDM2 Inhibitors as Potent Anticancer Agents. Comput. Math. Methods Med. 2021, 2021, 3195957. [Google Scholar] [CrossRef]
- Hutter, M.C. Differential Multimolecule Fingerprint for Similarity Search─Making Use of Active and Inactive Compound Sets in Virtual Screening. J. Chem. Inf. Model. 2022, 62, 2726–2736. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126. [Google Scholar] [CrossRef]
- Wang, Z.-Z.; Shi, X.-X.; Huang, G.-Y.; Hao, G.-F.; Yang, G.-F. Fragment-based drug design facilitates selective kinase inhibitor discovery. Trends Pharmacol. Sci. 2021, 42, 551–565. [Google Scholar] [CrossRef]
- Erlanson, D.A. Introduction to fragment-based drug discovery. Top. Curr. Chem. 2012, 317. [Google Scholar] [CrossRef]
- Bon, M.; Bilsland, A.; Bower, J.; McAulay, K. Fragment-based drug discovery-the importance of high-quality molecule libraries. Mol. Oncol. 2022, 16, 3761–3777. [Google Scholar] [CrossRef] [PubMed]
- Eguida, M.; Schmitt-Valencia, C.; Hibert, M.; Villa, P.; Rognan, D. Target-Focused Library Design by Pocket-Applied Computer Vision and Fragment Deep Generative Linking. J. Med. Chem. 2022, 65, 13771–13783. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zheng, S.; Su, S.; Zhao, C.; Xu, J.; Chen, H. SyntaLinker: Automatic fragment linking with deep conditional transformer neural networks. Chem. Sci. 2020, 11, 8312–8322. [Google Scholar] [CrossRef]
- Caburet, J.; Boucherle, B.; Bourdillon, S.; Simoncelli, G.; Verdirosa, F.; Docquier, J.-D.; Moreau, Y.; Krimm, I.; Crouzy, S.; Peuchmaur, M. A fragment-based drug discovery strategy applied to the identification of NDM-1 β-lactamase inhibitors. Eur. J. Med. Chem. 2022, 240, 114599. [Google Scholar] [CrossRef]
- Yang, T.; Li, Z.; Chen, Y.; Feng, D.; Wang, G.; Fu, Z.; Ding, X.; Tan, X.; Zhao, J.; Luo, X.; et al. DrugSpaceX: A large screenable and synthetically tractable database extending drug space. Nucleic. Acids Res. 2021, 49, D1170–D1178. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug. Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Paul, D.; Sanap, G.; Shenoy, S.; Kalyane, D.; Kalia, K.; Tekade, R.K. Artificial intelligence in drug discovery and development. Drug. Discov. Today 2021, 26, 80–93. [Google Scholar] [CrossRef]
- Henson, A.B.; Gromski, P.S.; Cronin, L. Designing Algorithms To Aid Discovery by Chemical Robots. ACS Cent. Sci. 2018, 4, 793–804. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in de Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef]
- Tong, X.; Liu, X.; Tan, X.; Li, X.; Jiang, J.; Xiong, Z.; Xu, T.; Jiang, H.; Qiao, N.; Zheng, M. Generative Models for De Novo Drug Design. J. Med. Chem. 2021, 64, 14011–14027. [Google Scholar] [CrossRef]
- Kingma, D.P.; Welling, M. Auto-encoding variational bayes. arXiv 2013. [Google Scholar] [CrossRef]
- Born, J.; Manica, M.; Oskooei, A.; Cadow, J.; Markert, G.; Rodríguez Martínez, M. PaccMann: De novo generation of hit-like anticancer molecules from transcriptomic data via reinforcement learning. iScience 2021, 24, 102269. [Google Scholar] [CrossRef]
- Hong, S.H.; Ryu, S.; Lim, J.; Kim, W.Y. Molecular Generative Model Based on an Adversarially Regularized Autoencoder. J. Chem. Inf. Model. 2020, 60, 29–36. [Google Scholar] [CrossRef]
- Samanta, B.; De, A.; Jana, G.; Gómez, V.; Chattaraj, P.K.; Ganguly, N.; Gomez-Rodriguez, M. Nevae: A deep generative model for molecular graphs. J. Mach. Learn. Res. 2020, 21, 1–33. [Google Scholar] [CrossRef]
- Kriegeskorte, N.; Golan, T. Neural network models and deep learning. Curr. Biol. 2019, 29, R231–R236. [Google Scholar] [CrossRef]
- Grisoni, F.; Moret, M.; Lingwood, R.; Schneider, G. Bidirectional Molecule Generation with Recurrent Neural Networks. J. Chem. Inf. Model. 2020, 60, 1175–1183. [Google Scholar] [CrossRef]
- Mou, L.; Yan, R.; Li, G.; Zhang, L.; Jin, Z. Backward and forward language modeling for constrained sentence generation. arXiv 2015. [Google Scholar] [CrossRef]
- Berglund, M.; Raiko, T.; Honkala, M.; Kärkkäinen, L.; Vetek, A.; Karhunen, J.T. Bidirectional recurrent neural networks as generative models. Adv. Neural Inform. Process. Syst. 2015, 28. [Google Scholar] [CrossRef]
- Krishnan, S.R.; Bung, N.; Bulusu, G.; Roy, A. Accelerating Drug Design against Novel Proteins Using Deep Learning. J. Chem. Inf. Model. 2021, 61, 621–630. [Google Scholar] [CrossRef]
- Moret, M.; Friedrich, L.; Grisoni, F.; Merk, D.; Schneider, G. Generative molecular design in low data regimes. Nature Mach. Intell. 2020, 2, 171–180. [Google Scholar] [CrossRef]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial networks. Commun. ACM 2020, 63, 139–144. [Google Scholar] [CrossRef]
- Maziarka, Ł.; Pocha, A.; Kaczmarczyk, J.; Rataj, K.; Danel, T.; Warchoł, M. Mol-CycleGAN: A generative model for molecular optimization. J. Cheminform. 2020, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.; Santos, B.P.; Pereira, T.C.; Sofia, R.; Monteiro, N.R.C.; Simões, C.J.V.; Brito, R.M.M.; Ribeiro, B.; Oliveira, J.L.; Arrais, J.P. Designing optimized drug candidates with Generative Adversarial Network. J. Cheminform. 2022, 14, 40. [Google Scholar] [CrossRef] [PubMed]
- Arulkumaran, K.; Deisenroth, M.P.; Brundage, M.; Bharath, A.A. Deep reinforcement learning: A brief survey. IEEE Signal Process. Magazine 2017, 34, 26–38. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef]
- Goel, M.; Raghunathan, S.; Laghuvarapu, S.; Priyakumar, U.D. MoleGuLAR: Molecule Generation Using Reinforcement Learning with Alternating Rewards. J. Chem. Inf. Model. 2021, 61, 5815–5826. [Google Scholar] [CrossRef]
- Cheng, F.; Lu, W.; Liu, C.; Fang, J.; Hou, Y.; Handy, D.E.; Wang, R.; Zhao, Y.; Yang, Y.; Huang, J.; et al. A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat. Commun. 2019, 10, 3476. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Chen, S.; Wang, J. DeepDRK: A deep learning framework for drug repurposing through kernel-based multi-omics integration. Brief. Bioinform. 2021, 22, bbab048. [Google Scholar] [CrossRef]
- Zhang, H.; Cui, H.; Zhang, T.; Cao, Y.; Xuan, P. Learning multi-scale heterogenous network topologies and various pairwise attributes for drug-disease association prediction. Brief. Bioinform. 2022, 23, bbac009. [Google Scholar] [CrossRef]
- Jarada, T.N.; Rokne, J.G.; Alhajj, R. SNF-NN: Computational method to predict drug-disease interactions using similarity network fusion and neural networks. BMC Bioinform. 2021, 22, 28. [Google Scholar] [CrossRef]
- Luo, H.; Wang, J.; Li, M.; Luo, J.; Peng, X.; Wu, F.-X.; Pan, Y. Drug repositioning based on comprehensive similarity measures and Bi-Random walk algorithm. Bioinformatics 2016, 32, 2664–2671. [Google Scholar] [CrossRef]
- Sadeghi, S.; Lu, J.; Ngom, A. An Integrative Heterogeneous Graph Neural Network-Based Method for Multi-Labeled Drug Repurposing. Front. Pharmacol. 2022, 13, 908549. [Google Scholar] [CrossRef]
- Doshi, S.; Chepuri, S.P. A computational approach to drug repurposing using graph neural networks. Comput. Biol. Med. 2022, 150, 105992. [Google Scholar] [CrossRef]
- Hodgson, J. ADMET--turning chemicals into drugs. Nat. Biotechnol. 2001, 19, 722–726. [Google Scholar] [CrossRef]
- Niu, J.; Straubinger, R.M.; Mager, D.E. Pharmacodynamic Drug-Drug Interactions. Clin. Pharmacol. Ther. 2019, 105, 1395–1406. [Google Scholar] [CrossRef]
- Selvaraj, C.; Chandra, I.; Singh, S.K. Artificial intelligence and machine learning approaches for drug design: Challenges and opportunities for the pharmaceutical industries. Mol. Divers 2022, 26, 1893–1913. [Google Scholar] [CrossRef]
- Li, T.; Tong, W.; Roberts, R.; Liu, Z.; Thakkar, S. DeepCarc: Deep Learning-Powered Carcinogenicity Prediction Using Model-Level Representation. Front. Artif. Intell. 2021, 4, 757780. [Google Scholar] [CrossRef]
- Vatansever, S.; Schlessinger, A.; Wacker, D.; Kaniskan, H.Ü.; Jin, J.; Zhou, M.-M.; Zhang, B. Artificial intelligence and machine learning-aided drug discovery in central nervous system diseases: State-of-the-arts and future directions. Med. Res. Rev. 2021, 41, 1427–1473. [Google Scholar] [CrossRef]
- Mulpuru, V.; Mishra, N. In Silico Prediction of Fraction Unbound in Human Plasma from Chemical Fingerprint Using Automated Machine Learning. ACS Omega 2021, 6, 6791–6797. [Google Scholar] [CrossRef]
- Chawla, S.; Rockstroh, A.; Lehman, M.; Ratther, E.; Jain, A.; Anand, A.; Gupta, A.; Bhattacharya, N.; Poonia, S.; Rai, P.; et al. Gene expression based inference of cancer drug sensitivity. Nat. Commun. 2022, 13, 5680. [Google Scholar] [CrossRef] [PubMed]
- Gogleva, A.; Polychronopoulos, D.; Pfeifer, M.; Poroshin, V.; Ughetto, M.; Martin, M.J.; Thorpe, H.; Bornot, A.; Smith, P.D.; Sidders, B.; et al. Knowledge graph-based recommendation framework identifies drivers of resistance in EGFR mutant non-small cell lung cancer. Nat. Commun. 2022, 13, 1667. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, H.; Casado, P.; Dokal, A.; Hijazi, M.; Akhtar, N.; Osuntola, R.; Rajeeve, V.; Fitzgibbon, J.; Travers, J.; Britton, D.; et al. Drug ranking using machine learning systematically predicts the efficacy of anti-cancer drugs. Nat. Commun. 2021, 12, 1850. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Clark, N.R.; Ma’ayan, A. Drug-induced adverse events prediction with the LINCS L1000 data. Bioinformatics 2016, 32, 2338–2345. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zheng, K.; Li, Y.; Wang, J. A novel graph attention model for predicting frequencies of drug-side effects from multi-view data. Brief. Bioinform. 2021, 22, bbab239. [Google Scholar] [CrossRef]
- Zhu, J.; Liu, Y.; Zhang, Y.; Chen, Z.; Wu, X. Multi-Attribute Discriminative Representation Learning for Prediction of Adverse Drug-Drug Interaction. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 44, 10129–10144. [Google Scholar] [CrossRef]
- Deng, Y.; Xu, X.; Qiu, Y.; Xia, J.; Zhang, W.; Liu, S. A multimodal deep learning framework for predicting drug-drug interaction events. Bioinformatics 2020, 36, 4316–4322. [Google Scholar] [CrossRef]
- He, H.; Chen, G.; Yu-Chian Chen, C. 3DGT-DDI: 3D graph and text based neural network for drug-drug interaction prediction. Brief Bioinform. 2022, 23, bbac134. [Google Scholar] [CrossRef]
- Wunnava, S.; Qin, X.; Kakar, T.; Sen, C.; Rundensteiner, E.A.; Kong, X. Adverse Drug Event Detection from Electronic Health Records Using Hierarchical Recurrent Neural Networks with Dual-Level Embedding. Drug Saf. 2019, 42, 113–122. [Google Scholar] [CrossRef]
- Zhang, T.; Leng, J.; Liu, Y. Deep learning for drug-drug interaction extraction from the literature: A review. Brief. Bioinform. 2020, 21, 1609–1627. [Google Scholar] [CrossRef]
- Basile, A.O.; Yahi, A.; Tatonetti, N.P. Artificial Intelligence for Drug Toxicity and Safety. Trends Pharmacol. Sci. 2019, 40, 624–635. [Google Scholar] [CrossRef]
- Jing, Y.; Bian, Y.; Hu, Z.; Wang, L.; Xie, X.-Q. Deep Learning for Drug Design: An Artificial Intelligence Paradigm for Drug Discovery in the Big Data Era. AAPS J. 2018, 20, 58. [Google Scholar] [CrossRef]
- Harari, Y.N. Reboot for the AI revolution. Nature 2017, 550, 324–327. [Google Scholar] [CrossRef]
- Welling, D.B.; Collier, K.A.; Burns, S.S.; Oblinger, J.L.; Shu, E.; Miles-Markley, B.A.; Hofmeister, C.C.; Makary, M.S.; Slone, H.W.; Blakeley, J.O.; et al. Early phase clinical studies of AR-42, a histone deacetylase inhibitor, for neurofibromatosis type 2-associated vestibular schwannomas and meningiomas. Laryngoscope Investig. Otolaryngol. 2021, 6, 1008–1019. [Google Scholar] [CrossRef]
- Casaletto, J.; Maglic, D.; Toure, B.B.; Taylor, A.; Schoenherr, H.; Hudson, B.; Bruderek, K.; Zhao, S.; O’Hearn, P.; Gerami-Moayed, N. RLY-4008, a novel precision therapy for FGFR2-driven cancers designed to potently and selectively inhibit FGFR2 and FGFR2 resistance mutations. Cancer Res. 2021, 81, 1455. [Google Scholar] [CrossRef]
- Sun, J.; Patel, C.B.; Jang, T.; Merchant, M.; Chen, C.; Kazerounian, S.; Diers, A.R.; Kiebish, M.A.; Vishnudas, V.K.; Gesta, S.; et al. High levels of ubidecarenone (oxidized CoQ10) delivered using a drug-lipid conjugate nanodispersion (BPM31510) differentially affect redox status and growth in malignant glioma versus non-tumor cells. Sci. Rep. 2020, 10, 13899. [Google Scholar] [CrossRef]
- Vladimer, G.; Alt, I.; Sehlke, R.; Lobley, A.; Baumgärtler, C.; Stulic, M.; Hackner, K.; Dzurillova, L.; Petru, E.; Hadjari, L. 23P Enriching for response: Patient selection criteria for A2AR inhibition by EXS-21546 through ex vivo modelling in primary patient material. Immuno-Oncol. Technol. 2022, 16, 100128. [Google Scholar] [CrossRef]
- Park, S.J.; Chang, S.-J.; Suh, D.H.; Kong, T.W.; Song, H.; Kim, T.H.; Kim, J.-W.; Kim, H.S.; Lee, S.-J. A phase IA dose-escalation study of PHI-101, a new checkpoint kinase 2 inhibitor, for platinum-resistant recurrent ovarian cancer. BMC Cancer 2022, 22, 28. [Google Scholar] [CrossRef]


{kind=link}
| Strategy | Descriptors Used |
|---|---|
| #1 | (“cancer” [Title/Abstract] AND “artificial intelligence” [Title/Abstract] AND “drug” [Title/Abstract]) AND (y_10[Filter]) |
| #2 | (“cancer” [Title/Abstract] AND “drug discovery” [Title/Abstract] AND “AI” [Title/Abstract]) AND (y_10[Filter]) |
| #3 | (“cancer” [Title/Abstract] AND “drug design” [Title/Abstract] AND “machine learning” [Title/Abstract]) AND (y_10[Filter]) |
| #4 | (“database” [Title/Abstract] AND “drug” [Title/Abstract] AND “artificial intelligence” [Title/Abstract]) AND (y_10[Filter]) |
| Model | Data Source | Code | References |
|---|---|---|---|
| EEG-DTI | Luo dataset [44], Yamanishi dataset [45] | https://github.com/MedicineBiology-AI/EEG-DTI (5 July 2022) | [35] |
| DTI-HETA | Yamanishi dataset | https://github.com/ZhangyuXM/DTI-HETA (13 October 2022) | [36] |
| ML-DTI | Metz dataset, KIBA dataset, Davis dataset, [46,47,48], Drugbank | https://github.com/guaguabujianle/ML-DTI.git (19 June 2021) | [37] |
| DDR | Yamanishi dataset, KEGG BRITE, BRENDA, SuperTarget, DrugBank | https://bitbucket.org/RSO24/ddr/(22 November 2017) | [40] |
| DrugnomeAI | TCRD, StringDB, CTDbase, InterPro, OMIM | https://github.com/astrazeneca-cgr-publications/DrugnomeAI-release (4 November 2022) | [41] |
| KG4SL | SynLethDB | https://github.com/JieZheng-ShanghaiTech/KG4SL (12 September 2021) | [43] |
| Model | Data Source | Code | References |
|---|---|---|---|
| SECSE | PDB | http://github.com/KeenThera/SECSE (15 July 2022) | [54] |
| SIEVE-Score | ChEMBL, ZINC | https://github.com/sekijima-lab/SIEVE-Score (15 November 2019) | [55] |
| DENVIS | PDB, DUD-E, LIT-PCBA | https://github.com/deeplab-ai/denvis (3 October 2022) | [56] |
| DMMFP | ChEMBL, DUD-E, ZINC | https://github.com/michahutter/multimolecule_fingerprints (28 April 2022) | [58] |
| POEM | ChEMBL, ZINC, PDB | https://github.com/kimeguida/POEM (30 December 2022) | [63] |
| SyntaLinker | ChEMBL | https://github.com/YuYaoYang2333/SyntaLinker (18 June 2021) | [64] |
| Model | Data Source | Code | References |
|---|---|---|---|
| PaccMannRL | TCGA, ChEMBL, GDSC, CCLE | https://github.com/PaccMann/ (10 February 2022) | [73] |
| ACGT | QM9, ZINC | https://github.com/gicsaw/ARAE_SMILES (14 October 2022) | [74] |
| NEVAE | QM9, ZINC | https://github.com/Networks-Learning/nevae (22 November 2019) | [75] |
| BIMODAL | ChEMBL | https://github.com/ETHmodlab/BIMODAL (3 June 2020) | [77] |
| Mol-CycleGAN | ChEMBL, ZINC | https://github.com/ardigen/mol-cycle-gan (6 February 2019) | [83] |
| GAN-Drug-Generator | ChEMBL, ZINC | https://github.com/larngroup/GAN-Drug-Generator (13 April 2022) | [84] |
| ReLeaSE | ChEMBL, ZINC | https://github.com/isayev/ReLeaSE (9 December 2021) | [86] |
| MoleGuLAR | ChEMBL, ZINC | https://github.com/devalab/MoleGuLAR (21 October 2021) | [87] |
| Model | Data Source | Code | References |
|---|---|---|---|
| GPSnet | DrugBank, TTD, PharmGKB, ChEMBL, BindingDB, UniProt, TCGA | https://github.com/ChengF-Lab/GPSnet (16 December 2018) | [88] |
| DeepDRK | CTRP, GDSC, TCGA, DrugBank, KEGG | https://github.com/wangyc82/DeepDRK (16 January 2021) | [89] |
| MBiRW | Drugbank, OMIM | http://github.com//bioinfomaticsCSU/MBiRW (19 December 2016) | [92] |
| DR-HGNN | Drugbank, CTD, SIDER | https://github.com/sshaghayeghs/DR_HGNN (26 April 2022) | [93] |
| GDRnet | Drugbank, Hetionet, GNBR, STRING, IntAct, DGIdb | https://github.com/siddhant-doshi/GDRnet (27 December 2021) | [94] |
| Model | Data Source | Code | References |
|---|---|---|---|
| DeepCarc | CPDB, Pubchem, Drugbank | https://github.com/TingLi2016/DeepCarc (6 July 2022) | [98] |
| Precily | CCLE, MSigDB, GDSC, Pubchem | https://github.com/SmritiChawla/Precily (26 August 2022) | [101] |
| DRUML | PharmacoDB, DepMap portal, PRIDE dataset, DrugBank, ChEMBL | https://github.com/CutillasLab/DRUMLR (24 March 2022) | [103] |
| MGPred | SIDER, STITCH, DrugBank, PubChem | https://github.com/zhc940702/MGPred (6 May 2018) | [105] |
| DLADE | cTAKES, EHR and PubMed article | https://github.com/qinxiao (7 October 2022) | [109] |
| MADRL | KEEG, SIDER, CTD, DrugBank | https://github.com/AdverseDDI/MADRL (18 January 2022) | [106] |
| DDIMDL | DrugBank, KEGG | https://github.com/YifanDengWHU/DDIMDL (1 May 2021) | [107] |
| 3DGT-DDI | DrugBank, DDI extraction 2013 | https://github.com/hehh77/3DGT-DDI (21 February 2022) | [108] |
| Name | Chemical Structure | Company | Therapeutic Area | Target/Function | Phase |
|---|---|---|---|---|---|
| REC-2282 |  | Recursion | Neurofibromatosis Type 2 | HDAC | Phase 2/3 |
| RLY-4008 | not disclosed | Relay Therapeutics | Solid tumor | FGFR | Phase 2 |
| BPM31510 |  | Berg | Solid tumor | Protein cbcl2 modulators | Phase 2 |
| EXS-21546 | not disclosed | Exscientia | Solid tumor | A2aR | Phase 1 |
| PHI-101 |  | Pharos iBio | Ovarian cancer Breast cancer | Flt3 tyrosine kinase inhibitor | Phase 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Song, Y.; Wang, H.; Zhang, X.; Wang, M.; He, J.; Li, S.; Zhang, L.; Li, K.; Cao, L. Advances of Artificial Intelligence in Anti-Cancer Drug Design: A Review of the Past Decade. Pharmaceuticals 2023, 16, 253. https://doi.org/10.3390/ph16020253
Wang L, Song Y, Wang H, Zhang X, Wang M, He J, Li S, Zhang L, Li K, Cao L. Advances of Artificial Intelligence in Anti-Cancer Drug Design: A Review of the Past Decade. Pharmaceuticals. 2023; 16(2):253. https://doi.org/10.3390/ph16020253
Chicago/Turabian StyleWang, Liuying, Yongzhen Song, Hesong Wang, Xuan Zhang, Meng Wang, Jia He, Shuang Li, Liuchao Zhang, Kang Li, and Lei Cao. 2023. "Advances of Artificial Intelligence in Anti-Cancer Drug Design: A Review of the Past Decade" Pharmaceuticals 16, no. 2: 253. https://doi.org/10.3390/ph16020253
APA StyleWang, L., Song, Y., Wang, H., Zhang, X., Wang, M., He, J., Li, S., Zhang, L., Li, K., & Cao, L. (2023). Advances of Artificial Intelligence in Anti-Cancer Drug Design: A Review of the Past Decade. Pharmaceuticals, 16(2), 253. https://doi.org/10.3390/ph16020253