
Nintedanib in Idiopathic Pulmonary Fibrosis: Tolerability and Safety in a Real Life Experience in a Single Centre in Patients also Treated with Oral Anticoagulant Therapy

 ,
, 

 , ,
, ,  ,
,  , , and
, , and
Abstract
:1. Introduction
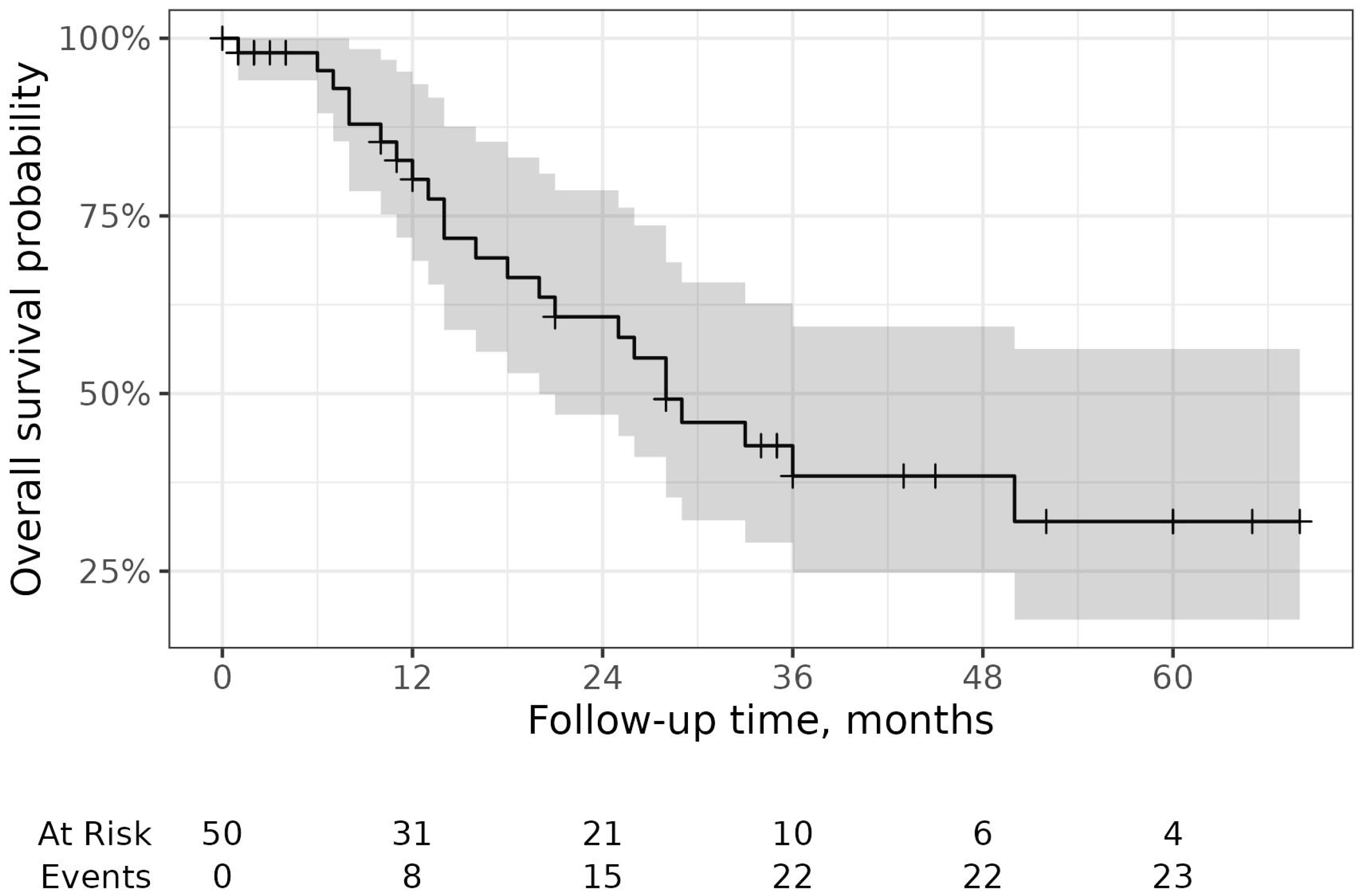
2. Results
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Data Collection
4.3. Assessment of Imaging Involvement
4.4. Assessment of Pulmonary Involvement
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Serra López-Matencio, J.M.; Gómez, M.; Vicente-Rabaneda, E.F.; González-Gay, M.A.; Ancochea, J.; Castañeda, S. Pharmacological Interactions of Nintedanib and Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis in Times of COVID-19 Pandemic. Pharmaceuticals 2021, 14, 819. [Google Scholar] [CrossRef]
- Ruaro, B.; Cerinic, M.M.; Salton, F.; Baratella, E.; Confalonieri, M.; Hughes, M. Editorial: Pulmonary Fibrosis: One Manifestation, Various Diseases. Front. Pharmacol. 2022, 13, 1027332. [Google Scholar] [CrossRef]
- Cameli, P.; Alonzi, V.; d’Alessandro, M.; Bergantini, L.; Pordon, E.; Guerrieri, M.; Refini, R.M.; Sestini, P.; Bargagli, E. The Effectiveness of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis, Familial Pulmonary Fibrosis and Progressive Fibrosing Interstitial Lung Diseases: A Real-World Study. Biomedicines 2022, 10, 1973. [Google Scholar] [CrossRef]
- Hostettler, K.E.; Zhong, J.; Papakonstantinou, E.; Karakiulakis, G.; Tamm, M.; Seidel, P.; Sun, Q.; Mandal, J.; Lardinois, D.; Lambers, C.; et al. Anti-Fibrotic Effects of Nintedanib in Lung Fibroblasts Derived from Patients with Idiopathic Pulmonary Fibrosis. Respir. Res. 2014, 15, 157. [Google Scholar] [CrossRef]
- Distler, O.; Highland, K.B.; Gahlemann, M.; Azuma, A.; Fischer, A.; Mayes, M.D.; Raghu, G.; Sauter, W.; Girard, M.; Alves, M.; et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N. Engl. J. Med. 2019, 380, 2518–2528. [Google Scholar] [CrossRef]
- Denton, C.P.; Goh, N.S.; Humphries, S.M.; Maher, T.M.; Spiera, R.; Devaraj, A.; Ho, L.; Stock, C.; Erhardt, E.; Alves, M.; et al. Extent of Fibrosis and Lung Function Decline in Patients with Systemic Sclerosis and Interstitial Lung Disease: Data from the SENSCIS Trial. Rheumatology 2022, keac535. [Google Scholar] [CrossRef]
- Allanore, Y.; Vonk, M.C.; Distler, O.; Azuma, A.; Mayes, M.D.; Gahlemann, M.; James, A.; Kohlbrenner, V.; Alves, M.; Khanna, D.; et al. Continued Treatment with Nintedanib in Patients with Systemic Sclerosis-Associated Interstitial Lung Disease: Data from SENSCIS-ON. Ann. Rheum. Dis. 2022, 81, 1722–1729. [Google Scholar] [CrossRef]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple Angiokinase Inhibitor with Sustained Receptor Blockade and Good Antitumor Efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef] [Green Version]
- Confalonieri, P.; Volpe, M.C.; Jacob, J.; Maiocchi, S.; Salton, F.; Ruaro, B.; Confalonieri, M.; Braga, L. Regeneration or Repair? The Role of Alveolar Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF). Cells 2022, 11, 2095. [Google Scholar] [CrossRef]
- Wells, A.U.; Flaherty, K.R.; Brown, K.K.; Inoue, Y.; Devaraj, A.; Richeldi, L.; Moua, T.; Crestani, B.; Wuyts, W.A.; Stowasser, S.; et al. Nintedanib in Patients with Progressive Fibrosing Interstitial Lung Diseases-Subgroup Analyses by Interstitial Lung Disease Diagnosis in the INBUILD Trial: A Randomised, Double-Blind, Placebo-Controlled, Parallel-Group Trial. Lancet Respir. Med. 2020, 8, 453–460. [Google Scholar] [CrossRef]
- Zhu, H.; Xia, M.-M.; Tong, K.-H.; Duan, W.-B. Nintedanib Induces the Autophagy-Dependent Death of Gastric Cancer Cells by Inhibiting the STAT3/Beclin1 Pathway. Dig Dis. Sci. 2022. [Google Scholar] [CrossRef]
- Shukla, S.K.; Nguyen, V.; Goyal, M.; Gupta, V. Cationically Modified Inhalable Nintedanib Niosomes: Enhancing Therapeutic Activity against Non-Small-Cell Lung Cancer. Nanomedicine 2022, 17, 935–958. [Google Scholar] [CrossRef]
- Baldini, C.; Danlos, F.-X.; Varga, A.; Texier, M.; Halse, H.; Mouraud, S.; Cassard, L.; Champiat, S.; Signolle, N.; Vuagnat, P.; et al. Safety, Recommended Dose, Efficacy and Immune Correlates for Nintedanib in Combination with Pembrolizumab in Patients with Advanced Cancers. J. Exp. Clin. Cancer Res. 2022, 41, 217. [Google Scholar] [CrossRef]
- Richeldi, L.; Costabel, U.; Selman, M.; Kim, D.S.; Hansell, D.M.; Nicholson, A.G.; Brown, K.K.; Flaherty, K.R.; Noble, P.W.; Raghu, G.; et al. Efficacy of a Tyrosine Kinase Inhibitor in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2011, 365, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- OFEV (Nintedanib) Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/Ofev (accessed on 1 December 2022).
- Lamb, Y.N. Nintedanib: A Review in Fibrotic Interstitial Lung Diseases. Drugs 2021, 81, 575–586. [Google Scholar] [CrossRef]
- Chen, C.-H.; Lin, H.-C.; Wang, Y.-H.; Wang, C.-Y.; Lin, Y.S.; Lai, C.-C. The Safety of Nintedanib for the Treatment of Interstitial Lung Disease: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. PLoS ONE 2021, 16, e0251636. [Google Scholar] [CrossRef]
- Crestani, B.; Huggins, J.T.; Kaye, M.; Costabel, U.; Glaspole, I.; Ogura, T.; Song, J.W.; Stansen, W.; Quaresma, M.; Stowasser, S.; et al. Long-Term Safety and Tolerability of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis: Results from the Open-Label Extension Study, INPULSIS-ON. Lancet Respir. Med. 2019, 7, 60–68. [Google Scholar] [CrossRef]
- Noth, I.; Oelberg, D.; Kaul, M.; Conoscenti, C.S.; Raghu, G. Safety and Tolerability of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis in the USA. Eur. Respir. J. 2018, 52, 1702106. [Google Scholar] [CrossRef] [Green Version]
- Antoniou, K.; Markopoulou, K.; Tzouvelekis, A.; Trachalaki, A.; Vasarmidi, E.; Organtzis, J.; Tzilas, V.; Bouros, E.; Kounti, G.; Rampiadou, C.; et al. Efficacy and Safety of Nintedanib in a Greek Multicentre Idiopathic Pulmonary Fibrosis Registry: A Retrospective, Observational, Cohort Study. ERJ Open Res. 2020, 6, 00172–02019. [Google Scholar] [CrossRef] [Green Version]
- Fournier, D.; Jouneau, S.; Bouzillé, G.; Polard, E.; Osmont, M.-N.; Scailteux, L.-M. Real-World Safety Profiles of Pirfenidone and Nintedanib in Idiopathic Pulmonary Fibrosis Patients. Pulm. Pharmacol. Ther. 2022, 76, 102149. [Google Scholar] [CrossRef]
- Pitre, T.; Mah, J.; Helmeczi, W.; Khalid, M.F.; Cui, S.; Zhang, M.; Husnudinov, R.; Su, J.; Banfield, L.; Guy, B.; et al. Medical Treatments for Idiopathic Pulmonary Fibrosis: A Systematic Review and Network Meta-Analysis. Thorax 2022, 77, 1243–1250. [Google Scholar] [CrossRef]
- Caro, F.; Buendía-Roldán, I.; Noriega-Aguirre, L.; Alberti, M.L.; Amaral, A.; Arbo, G.; Auteri, S.; Bermúdez, A.; Curbelo, P.; de Jesús Díaz Verduzco, M.; et al. Latin American Registry of Idiopathic Pulmonary Fibrosis (REFIPI): Clinical Characteristics, Evolution and Treatment. Arch. Bronconeumol. 2022, 58, 794–801. [Google Scholar] [CrossRef]
- Mondoni, M.; Alfano, F.; Varone, F.; Muscato, G.; Conti, C.; Saderi, L.; Chiesa, A.; Di Marco, F.; Vancheri, C.; Richeldi, L.; et al. Observational, Multicenter Study on the Efficacy, Tolerability, and Safety of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis Older than 80 Years. Respir. Int. Rev. Thorac. Dis. 2022, 1, 9. [Google Scholar] [CrossRef]
- Kaunisto, J.; Salomaa, E.R.; Hodgson, U.; Kaarteenaho, R.; Kankaanranta, H.; Koli, K.; Vahlberg, T.; Myllärniemi, M. Demographics and survival of patients with idiopathic pulmonary fibrosis in the FinnishIPF registry. ERJ Open Res. 2019, 5, 00170–02018. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Albera, C.; Bradford, W.Z.; Costabel, U.; du Bois, R.M.; Leff, J.A.; Nathan, S.D.; Sahn, S.A.; Valeyre, D.; Noble, P.W. All-cause mortality rate in patients with idiopathic pulmonary fibrosis. Implications for the design and execution of clinical trials. Am. J. Respir. Crit. Care Med. 2014, 189, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Marshall, D.C.; Salciccioli, J.D.; Shea, B.S.; Akuthota, P. Trends in mortality from idiopathic pulmonary fibrosis in the European Union: An observational study of the WHO mortality database from 2001–2013. Eur. Respir. J. 2018, 51, 1701603. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.istat.it/it/files/2017/12/cap01.pdf (accessed on 1 December 2022).
- Sulli, A.; Ruaro, B.; Cutolo, M. Evaluation of blood perfusion by laser speckle contrast analysis in different areas of hands and face in patients with systemic sclerosis. Ann Rheum Dis. 2014, 73, 2059–2061. [Google Scholar] [CrossRef]
- Bernero, E.; Sulli, A.; Ferrari, G.; Ravera, F.; Pizzorni, C.; Ruaro, B.; Zampogna, G.; Alessandri, E.; Cutolo, M. Prospective capillaroscopy-based study on transition from primary to secondary Raynaud’s phenomenon: Preliminary results. Reumatismo 2013, 65, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Ruaro, B.; Confalonieri, M.; Matucci-Cerinic, M.; Salton, F.; Confalonieri, P.; Santagiuliana, M.; Citton, G.M.; Baratella, E.; Bruni, C. The Treatment of Lung Involvement in Systemic Sclerosis. Pharmaceuticals 2021, 14, 154. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
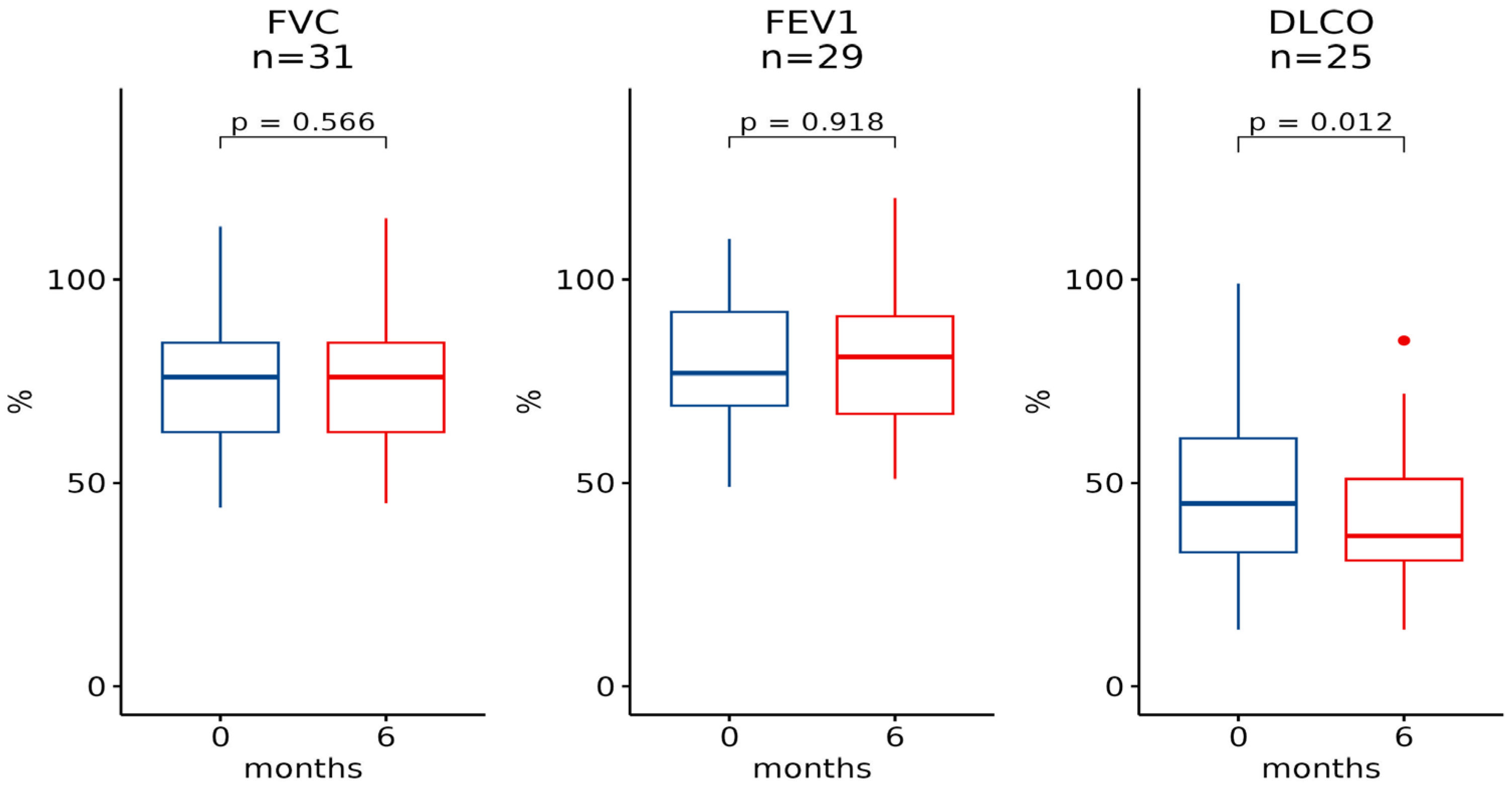
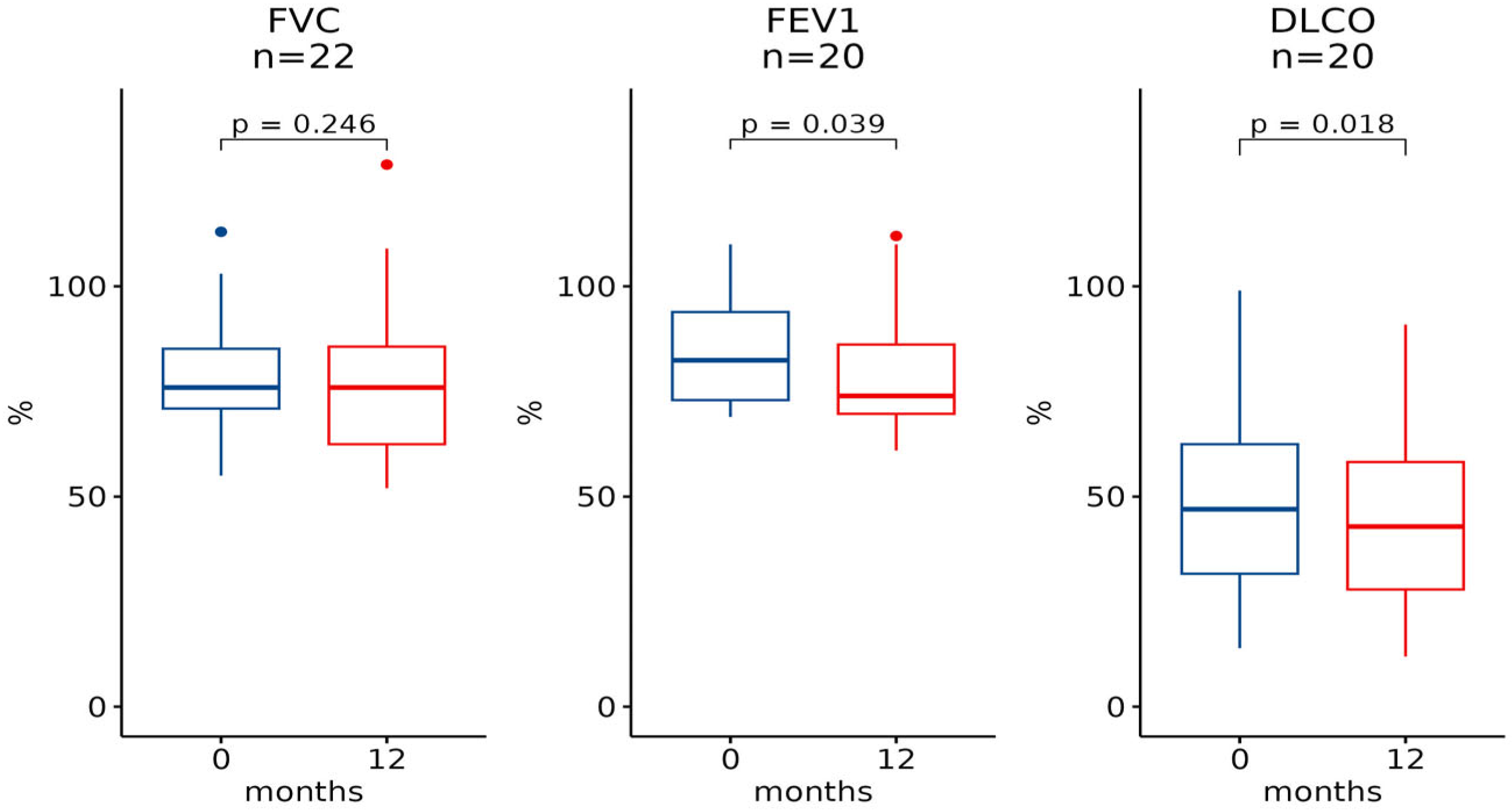
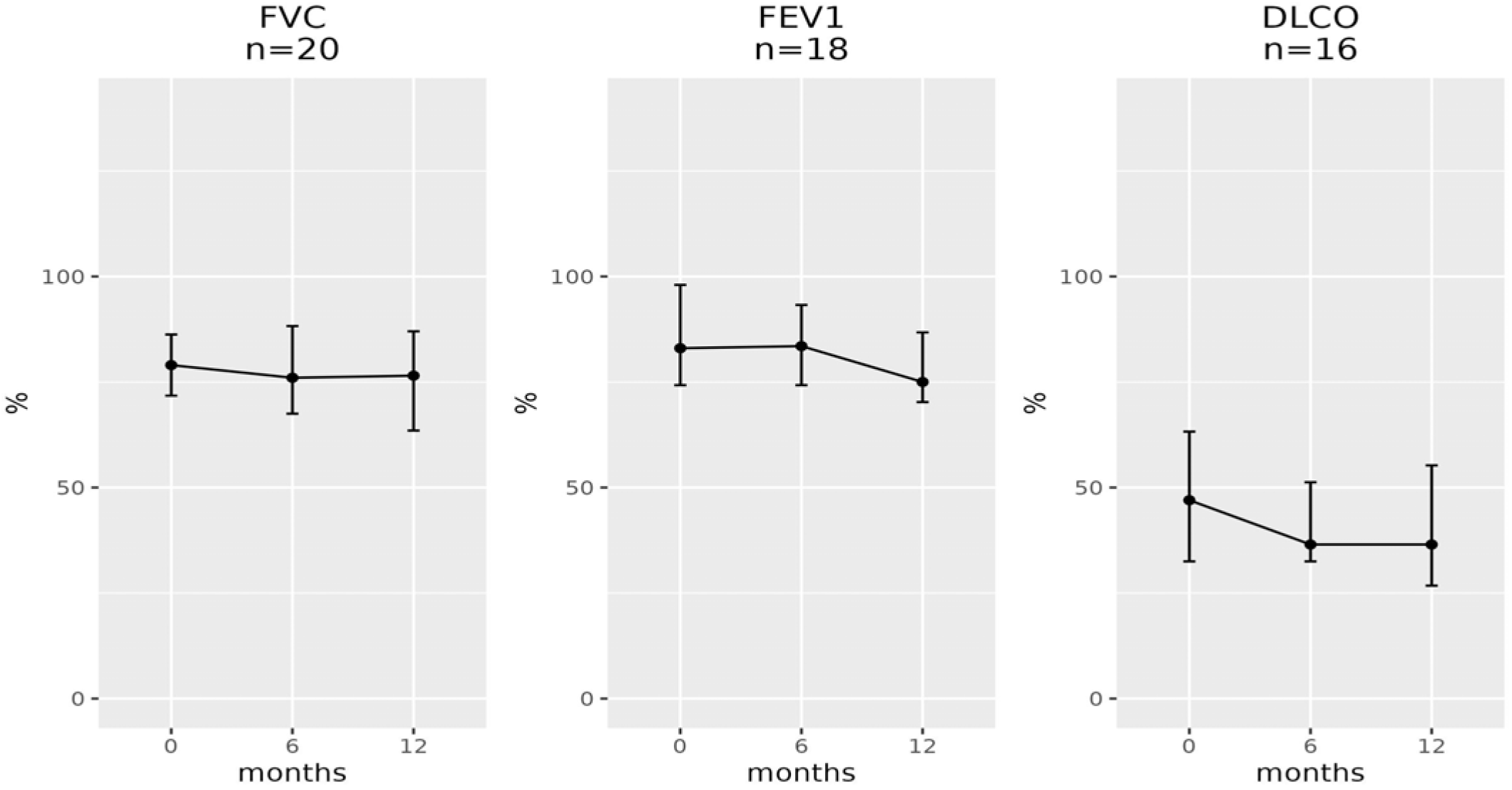
| Baseline [Median (IQR)] | 6 Months [Median (IQR)] | n | p-Value | Baseline [Median (IQR)] | 12 Months [Median (IQR)] | n | p-Value |
|---|---|---|---|---|---|---|---|
| FVC | |||||||
| 76.0 (22.0) | 76.0 (22.0) | 31 | 0.6 | 76.0 (14.2) | 76.0 (23.2) | 22 | 0.2 |
| FEV1 | |||||||
| 77.0 (23.0) | 81.0 (24.0) | 29 | >0.9 | 82.5 (21.0) | 74.0 (16.5) | 20 | 0.039 |
| DLCO | |||||||
| 45.0 (28.0) | 37.0 (20.0) | 25 | 0.012 | 47.0 (30.8) | 43.0 (30.2) | 20 | 0.018 |
| FVC | |||||
| 6 months | 12 months | ||||
| Responder (n = 25) | Non-responder (n = 6) | p-value | Responder (n = 19) | Non-responder (n = 3) | p-value |
| 10 (40%) | 2 (33%) | >0.9 | 7 (39%) | 2 (67%) | 0.6 |
| DLCO | |||||
| 6 months | 12 months | ||||
| Responder (n = 12) | Non-responder (n = 13) | p-value | Responder (n = 15) | Non-responder (n = 5) | p-value |
| 4 (33%) | 6 (46%) | 0.7 | 4 (29%) | 4 (80%) | 0.1 |
| Clinical and demographic characteristics | 56 patients |
| Females, n (%) | 19 (34%) |
| Mean onset age (years), mean ± SD | 71 ± 11 years |
| Mean age at baseline (years), mean ± SD | 74 ± 9 years |
| Smokers (active or former) | 34 (62%) |
| Autoantibodies | |
| Antinuclear antibodies (ANA) | 7 (12%) |
| Anti-Ro52, n (%) | 3 (5%) |
| Organ involvement | |
| COPD (n, %) | 3 (5%) |
| Cancer (n, %) | 8 (15%) |
| Hypertension (n, %) | 36 (64%) |
| Coronary heart disease (n, %) | 11 (19%) |
| Diabetes mellitus (n, %) | 22 (39%) |
| Rheumatic diseases (n, %) | 1 (2%) |
| Previous episodes of venous thromboembolism (n, %) | 11 (19%) |
| Cardiac comorbidities (n, %) | 31 (55%) |
| Concurrent Therapies | |
| Mycophenolate mofetil, n (%) | 3 (5%) |
| Azathioprine, n (%) | 1 (2%) |
| Hydroxychloroquine, n (%) | 2 (4%) |
| Rituximab, n (%) | 1 (2%) |
| Steroid high dose (i.e., >15 mg/die), n (%) | 12 (21%) |
| Steroid low dose (i.e., <15 mg/die), n (%) | 8 (15%) |
| Antihypertensive drugs, n (%) | 36 (64%) |
| Pump inhibitor drugs, n (%) | 38 (59%) |
| Anticoagulant drugs, n (%) | 14 (25%) |
| Warfarin, n (%) | 11 (20%) |
| Apixaban and Rivaroxaban | 3 (5%) |
| Baseline HRCT | 56 patients |
| NSIP, n (%) | 11 (20%) |
| UIP, n (%) | 45 (80%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruaro, B.; Gandin, I.; Pozzan, R.; Tavano, S.; Bozzi, C.; Hughes, M.; Kodric, M.; Cifaldi, R.; Lerda, S.; Confalonieri, M.; et al. Nintedanib in Idiopathic Pulmonary Fibrosis: Tolerability and Safety in a Real Life Experience in a Single Centre in Patients also Treated with Oral Anticoagulant Therapy. Pharmaceuticals 2023, 16, 307. https://doi.org/10.3390/ph16020307
Ruaro B, Gandin I, Pozzan R, Tavano S, Bozzi C, Hughes M, Kodric M, Cifaldi R, Lerda S, Confalonieri M, et al. Nintedanib in Idiopathic Pulmonary Fibrosis: Tolerability and Safety in a Real Life Experience in a Single Centre in Patients also Treated with Oral Anticoagulant Therapy. Pharmaceuticals. 2023; 16(2):307. https://doi.org/10.3390/ph16020307
Chicago/Turabian StyleRuaro, Barbara, Ilaria Gandin, Riccardo Pozzan, Stefano Tavano, Chiara Bozzi, Michael Hughes, Metka Kodric, Rossella Cifaldi, Selene Lerda, Marco Confalonieri, and et al. 2023. "Nintedanib in Idiopathic Pulmonary Fibrosis: Tolerability and Safety in a Real Life Experience in a Single Centre in Patients also Treated with Oral Anticoagulant Therapy" Pharmaceuticals 16, no. 2: 307. https://doi.org/10.3390/ph16020307
APA StyleRuaro, B., Gandin, I., Pozzan, R., Tavano, S., Bozzi, C., Hughes, M., Kodric, M., Cifaldi, R., Lerda, S., Confalonieri, M., Baratella, E., Confalonieri, P., & Salton, F. (2023). Nintedanib in Idiopathic Pulmonary Fibrosis: Tolerability and Safety in a Real Life Experience in a Single Centre in Patients also Treated with Oral Anticoagulant Therapy. Pharmaceuticals, 16(2), 307. https://doi.org/10.3390/ph16020307