
Exploring the Antitubercular Activity of Anthranilic Acid Derivatives: From MabA (FabG1) Inhibition to Intrabacterial Acidification

 , , , , , , ,
, , , , , , ,
Abstract
:
1. Introduction
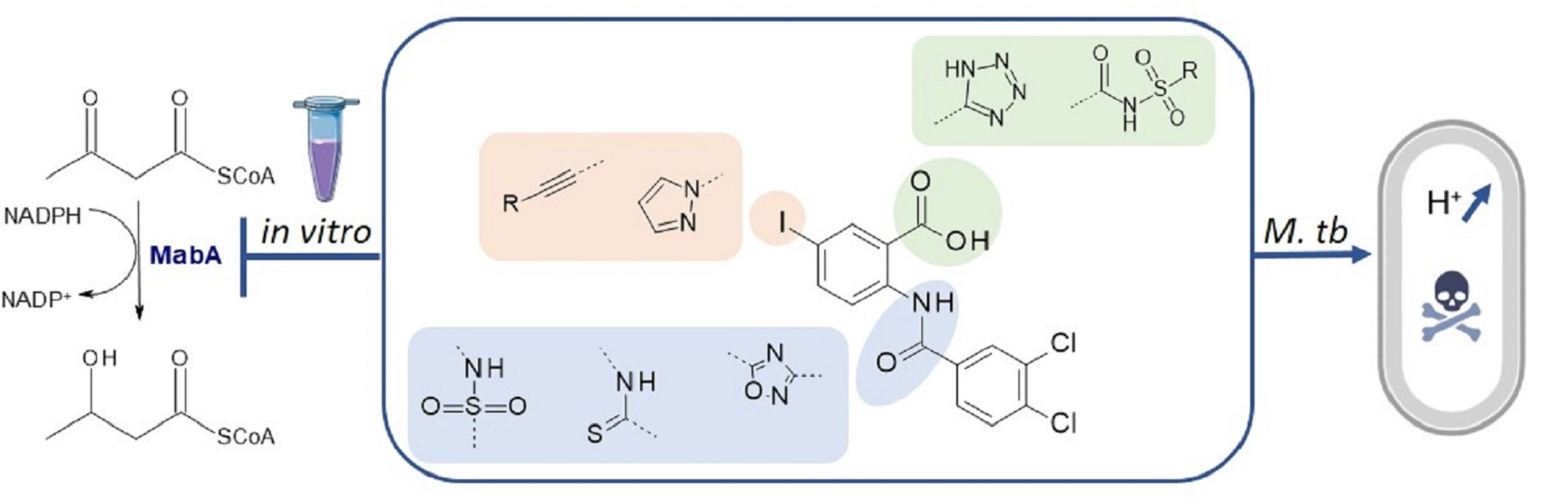
2. Results and Discussion
2.1. Chemistry
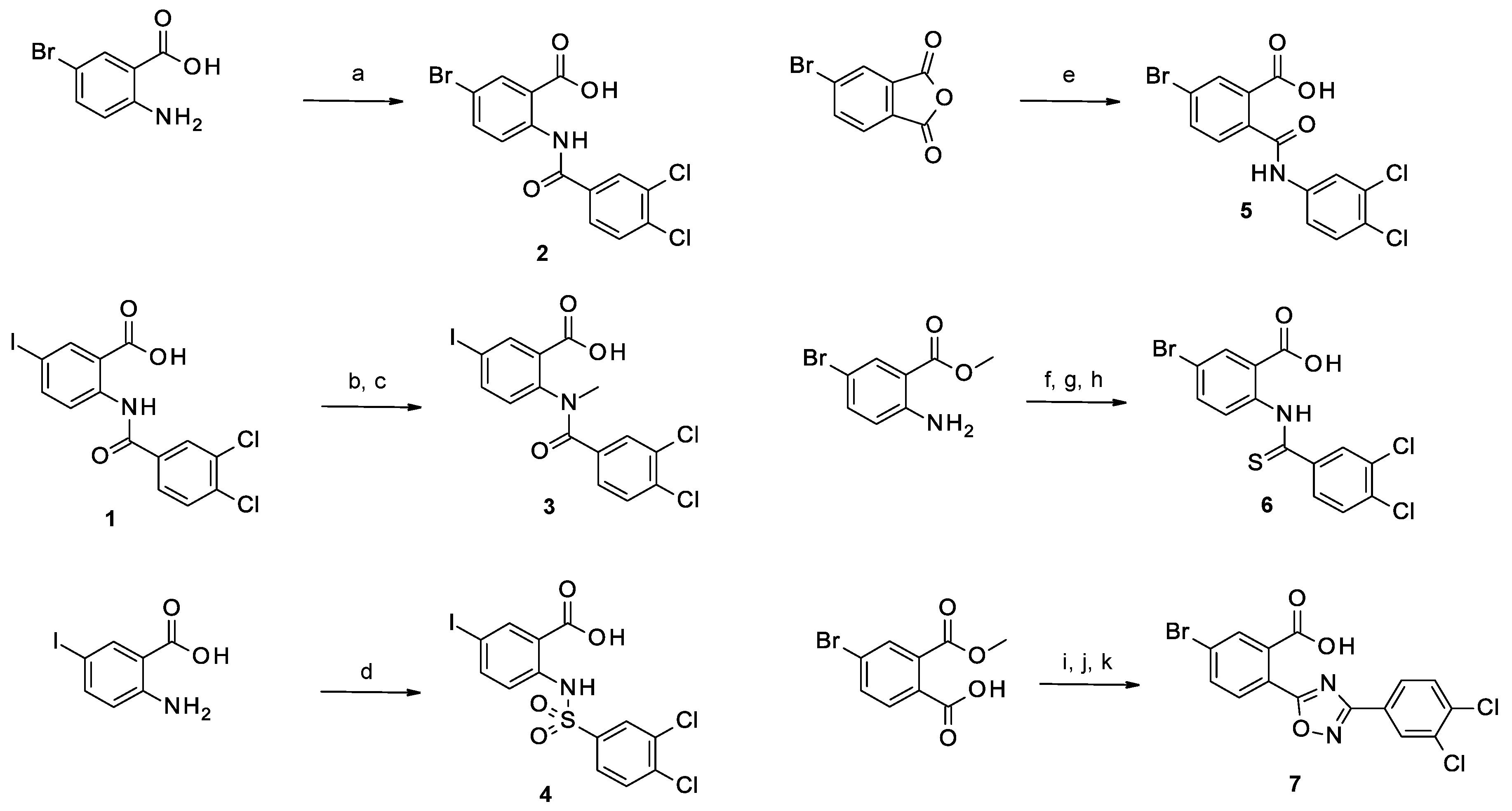
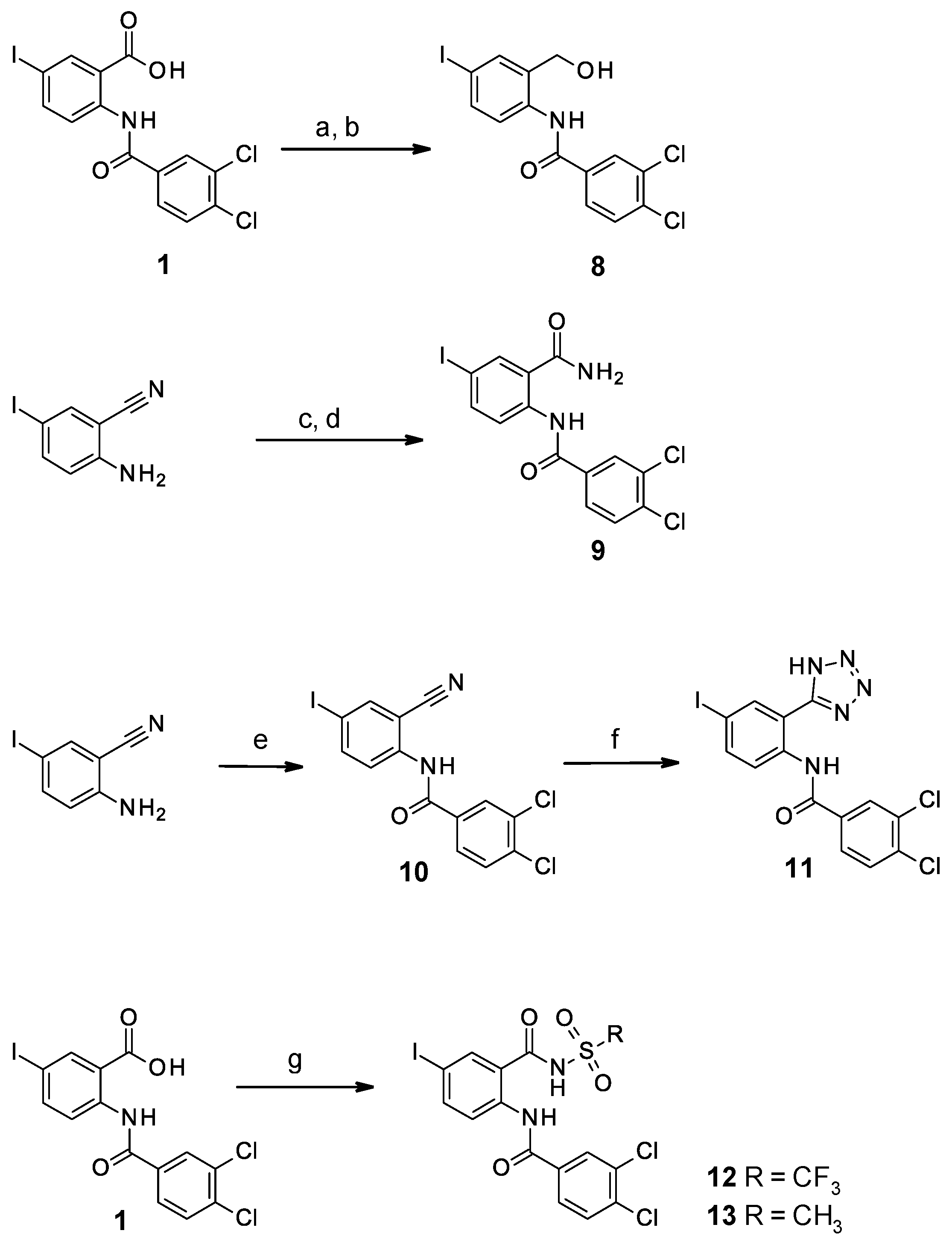
2.1.1. Study of the Modification of the Amide Bond
2.1.2. Study of the Modification of the Carboxylic Acid Moiety
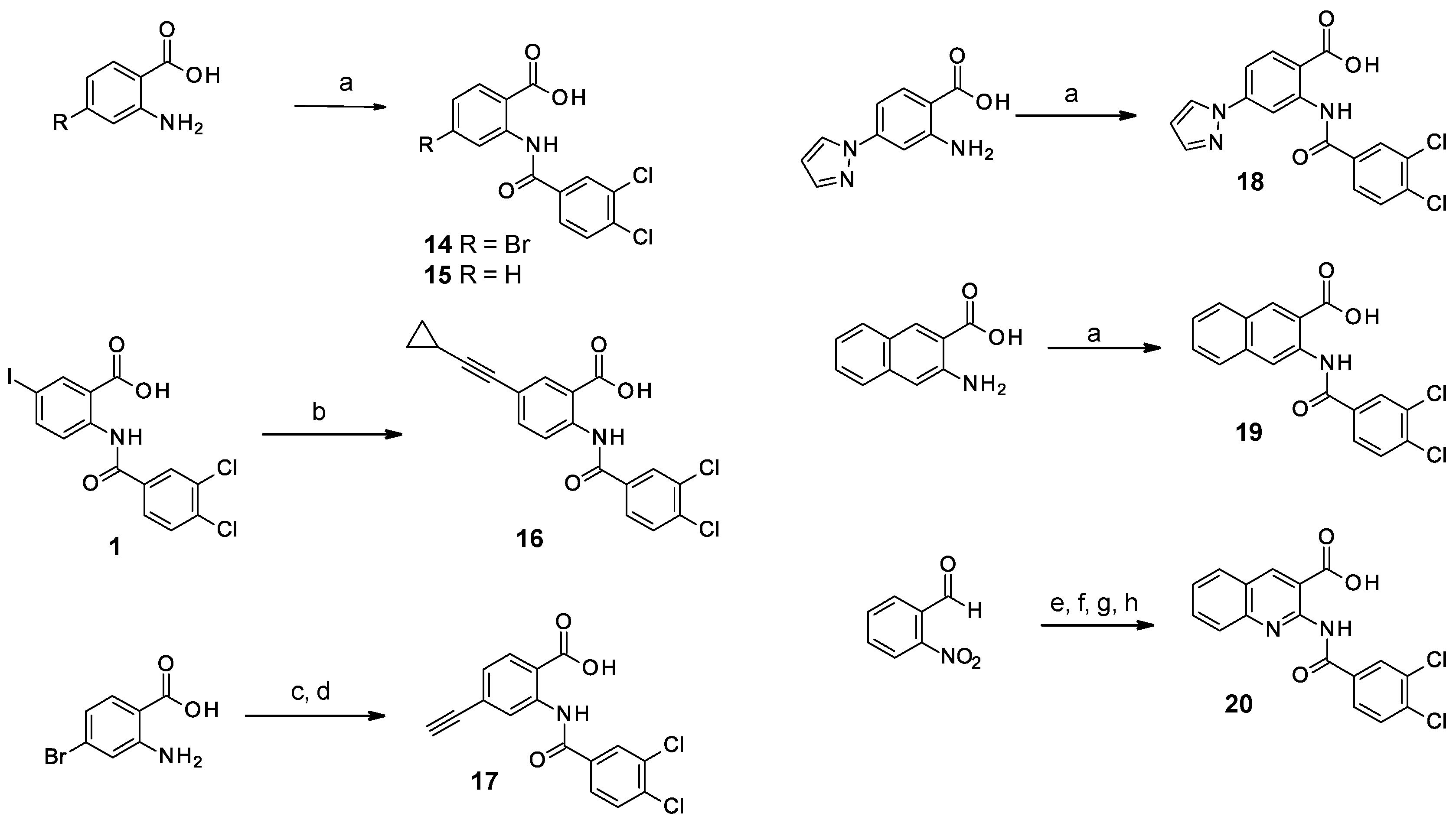
2.1.3. Study of the Modification of the Phenyl Ring
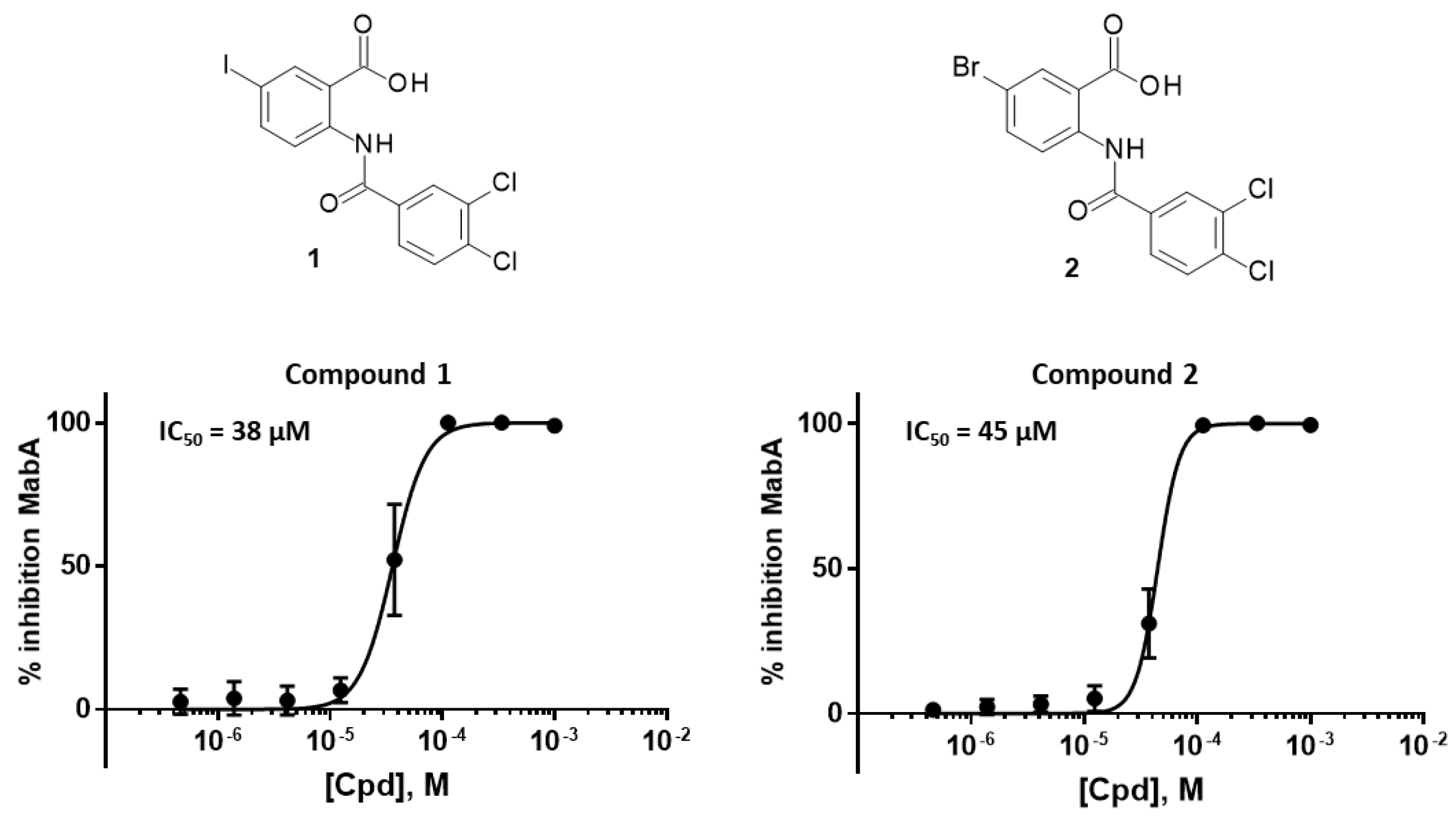
2.2. Biological Results
2.2.1. Modifications of the Amide Bond
2.2.2. Modifications of the Carboxylic Acid Moiety
2.2.3. Modifications of the Phenyl Ring
2.2.4. Evaluation of the Interaction of the Fluorinated Compound 12 with MabA by NMR
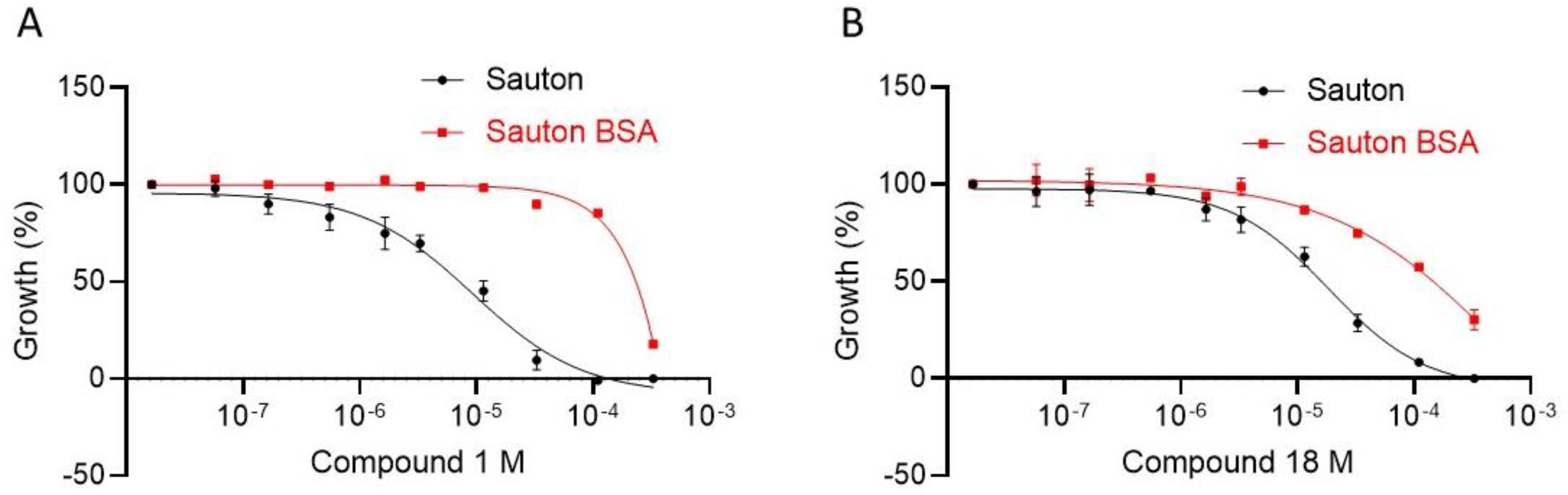
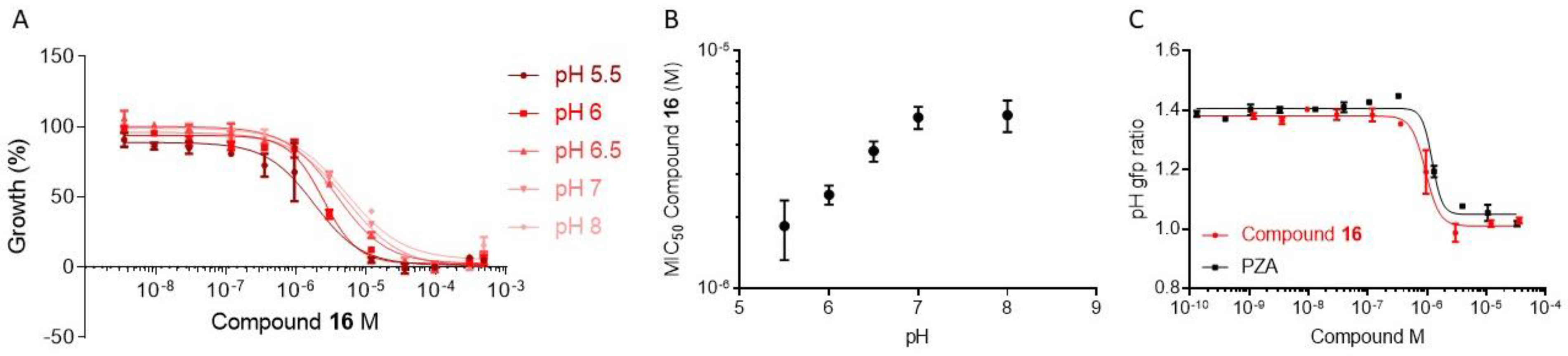
2.2.5. Evaluation of Physico-Chemical Properties and Antimycobacterial Activity
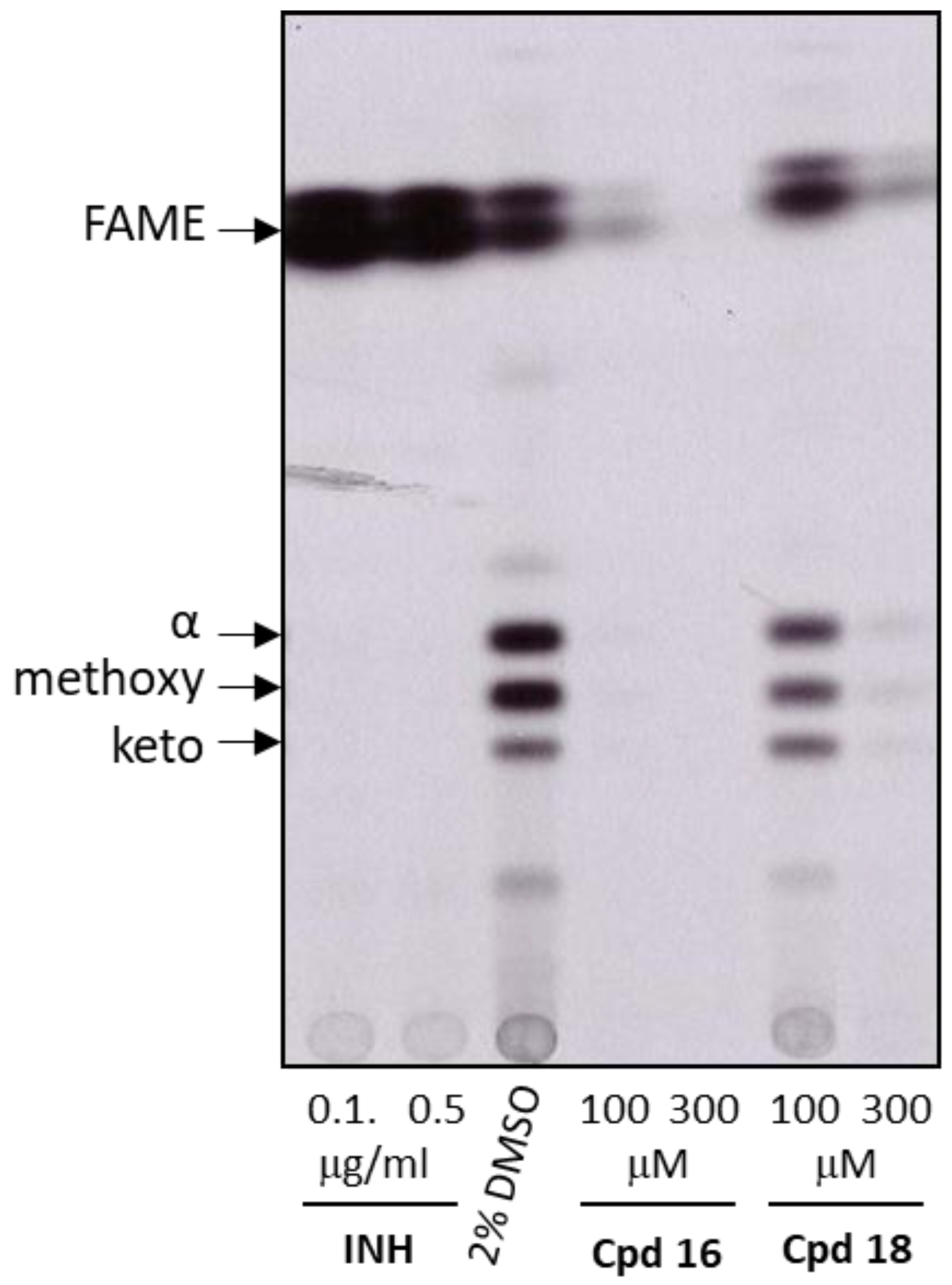
2.2.6. Analysis of Mycolic Acids Inhibition in M. tuberculosis
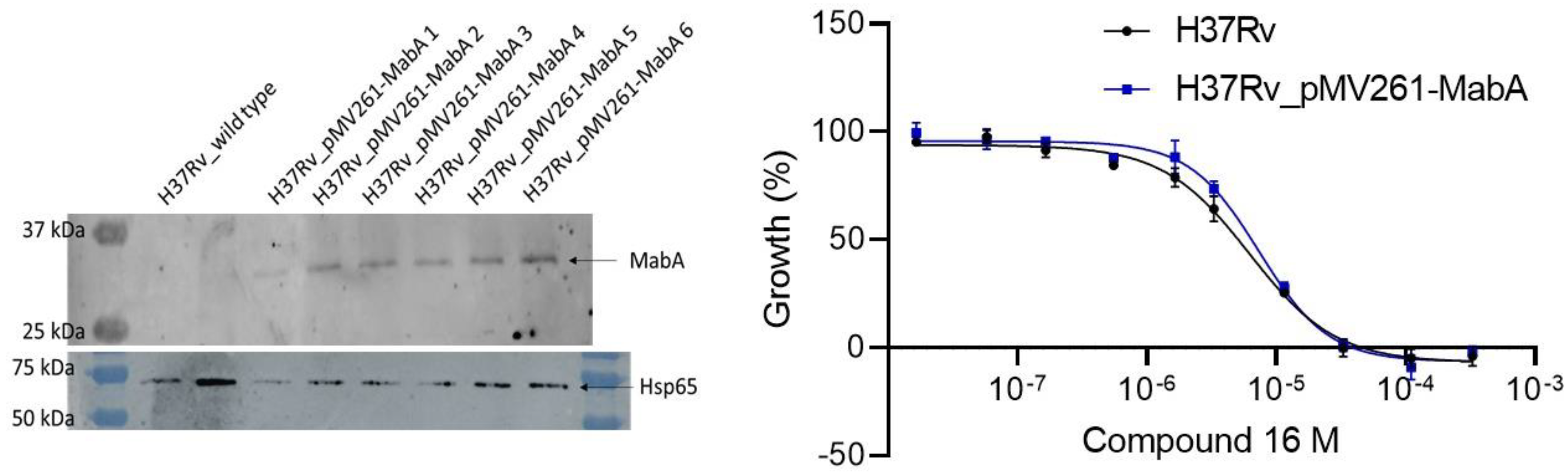
2.2.7. Exploration of the Mechanism of Action of Inhibitors in Bacteria
3. Materials and Methods
3.1. Chemistry
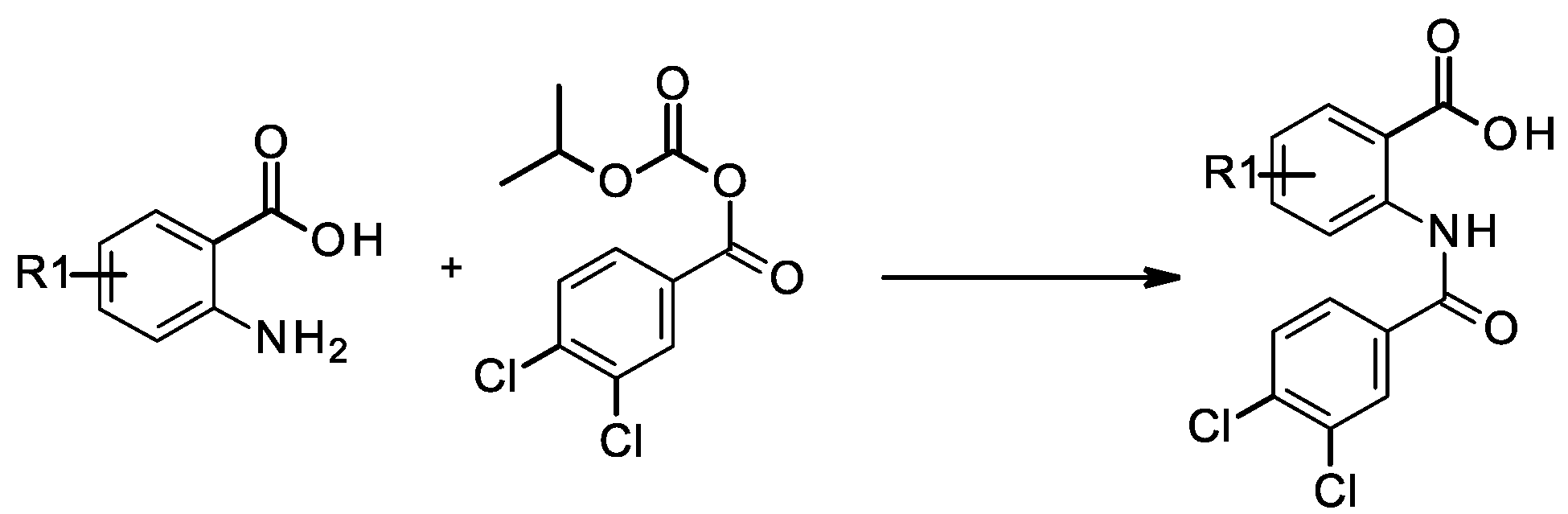
3.1.1. General Method for the Coupling of Anilines with Isopropoxycarbonyl 3,4-dichlorobenzoate
5-bromo-2-[(3,4-dichlorobenzoyl)amino]benzoic Acid (2)
4-bromo-2-[(3,4-dichlorobenzoyl)amino]benzoic Acid (14)
2-[(3,4-dichlorobenzoyl)amino]benzoic Acid (15)
2-[(3,4-dichlorobenzoyl)amino]-4-pyrazol-1-yl-benzoic Acid (18)
3-[(3,4-dichlorobenzoyl)amino]naphthalene-2-carboxylic Acid (19)
3.1.2. Synthesis of 2-[(3,4-dichlorobenzoyl)-methyl-amino]-5-iodo-benzoic Acid (3)
3.1.3. Synthesis of 2-[(3,4-dichlorophenyl)sulfonylamino]-5-iodo-benzoic Acid (4)
3.1.4. Synthesis of 5-bromo-2-[(3,4-dichlorophenyl)carbamoyl]benzoic Acid (5)
3.1.5. Synthesis of 5-bromo-2-[(3,4-dichlorobenzenecarbothioyl)amino]benzoic Acid (6)
Methyl 5-bromo-2-[(3,4-dichlorobenzoyl)amino]benzoate (Intermediate 6a)
Methyl 5-bromo-2-[(3,4-dichlorobenzenecarbothioyl)amino]benzoate (Intermediate 6b)
5-bromo-2-[(3,4-dichlorobenzenecarbothioyl)amino]benzoic Acid (6)
3.1.6. Synthesis of 5-bromo-2-[3-(3,4-dichlorophenyl)-1,2,4-oxadiazol-5-yl]benzoic Acid (7)
3,4-dichloro-N′-hydroxy-benzamidine (Intermediate 7a)
5-bromoisobenzofuran-1,3-dione (Intermediate 7b)
4-bromo-2-methoxycarbonyl-benzoic Acid (Intermediate 7c)
Methyl 5-bromo-2-chlorocarbonyl-benzoate (Intermediate 7d)
Methyl 5-bromo-2-[3-(3,4-dichlorophenyl)-1,2,4-oxadiazol-5-yl]benzoate (Intermediate 7e)
5-bromo-2-[3-(3,4-dichlorophenyl)-1,2,4-oxadiazol-5-yl]benzoic Acid (7)
3.1.7. Synthesis of 3,4-dichloro-N-[2-(hydroxymethyl)-4-iodo-phenyl]benzamide (8)
(2,5-dioxopyrrolidin-1-yl)-2-[(3,4-dichlorobenzoyl)amino]-5-iodo-benzoate (Intermediate 8a)
3,4-dichloro-N-[2-(hydroxymethyl)-4-iodo-phenyl]benzamide (8)
3.1.8. Synthesis of N-(2-carbamoyl-4-iodo-phenyl)-3,4-dichloro-benzamide (9)
2-amino-5-iodobenzamide (Intermediate 9a)
N-(2-carbamoyl-4-iodo-phenyl)-3,4-dichloro-benzamide (9)
3.1.9. 3,4-dichloro-N-(2-cyano-4-iodo-phenyl)benzamide (10)
3.1.10. 3,4-dichloro-N-[4-iodo-2-(1H-tetrazol-5-yl)phenyl]benzamide (11)
3.1.11. 3,4-dichloro-N-[4-iodo-2-(trifluoromethylsulfonylcarbamoyl)phenyl]benzamide (12)
3.1.12. 3,4-dichloro-N-[4-iodo-2-(methylsulfonylcarbamoyl)phenyl]benzamide (13)
3.1.13. 5-(2-cyclopropylethynyl)-2-[(3,4-dichlorobenzoyl)amino]benzoic Acid (16)
3.1.14. Synthesis of 2-[(3,4-dichlorobenzoyl)amino]-4-ethynyl-benzoic Acid (17)
2-amino-4-(2-trimethylsilylethynyl)benzoic Acid (Intermediate 17a)
2-[(3,4-dichlorobenzoyl)amino]-4-ethynyl-benzoic Acid (17)
3.1.15. Synthesis of 2-[(3,4-dichlorobenzoyl)amino]quinoline-3-carboxylic Acid (20)
Ethyl (Z)-2-cyano-3-(2-nitrophenyl)prop-2-enoate (Intermediate 20a)
Ethyl 2-aminoquinoline-3-carboxylate (Intermediate 20b)
Ethyl 2-[bis(3,4-dichlorobenzoyl)amino]quinoline-3-carboxylate (Intermediate 20c)
2-[(3,4-dichlorobenzoyl)amino]quinoline-3-carboxylic Acid (20)
3.2. Biology
3.2.1. MabA Expression and Purification
3.2.2. MabA Enzymatic Assay
Dose-Response Experiments
LC-MS/MS Analysis
3.2.3. Ligand-Observed NMR Experiments
3.2.4. MIC Determination
3.2.5. RNA-seq
3.2.6. Measurement of Intrabacterial pH
3.2.7. Western Blot
3.2.8. Metabolic Labelling with 14C Acetate and Mycolic Acids Analysis
3.3. Physico-Chemical Properties
3.3.1. LogD
3.3.2. Solubility
3.3.3. Plasma Protein Binding
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- MacNeil, A.; Glaziou, P.; Sismanidis, C.; Date, A.; Maloney, S.; Floyd, K. Global Epidemiology of Tuberculosis and Progress Toward Meeting Global Targets—Worldwide, 2018. Morb. Mortal. Wkly. Rep. 2020, 69, 281–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Global Tuberculosis Report; World Health Organization: Geneva, Switzerland, 2022; ISBN 978-92-4-006172-9. [Google Scholar]
- Cox, E.; Laessig, K. FDA Approval of Bedaquiline—The Benefit–Risk Balance for Drug-Resistant Tuberculosis. N. Engl. J. Med. 2014, 371, 689–691. [Google Scholar] [CrossRef] [Green Version]
- Ryan, N.J.; Lo, J.H. Delamanid: First Global Approval. Drugs 2014, 74, 1041–1045. [Google Scholar] [CrossRef]
- Keam, S.J. Pretomanid: First Approval. Drugs 2019, 79, 1797–1803. [Google Scholar] [CrossRef]
- Torres, N.M.C.; Rodríguez, J.J.Q.; Andrade, P.S.P.; Arriaga, M.B.; Netto, E.M. Factors Predictive of the Success of Tuberculosis Treatment: A Systematic Review with Meta-Analysis. PLoS ONE 2019, 14, e0226507. [Google Scholar] [CrossRef] [Green Version]
- Dulberger, C.L.; Rubin, E.J.; Boutte, C.C. The Mycobacterial Cell Envelope—A Moving Target. Nat. Rev. Microbiol. 2020, 18, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, K.A.; Besra, G.S. Synthesis and Recycling of the Mycobacterial Cell Envelope. Curr. Opin. Microbiol. 2021, 60, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Marrakchi, H.; Lanéelle, M.-A.; Daffé, M. Mycolic Acids: Structures, Biosynthesis, and Beyond. Chem. Biol. 2014, 21, 67–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batt, S.M.; Minnikin, D.E.; Besra, G.S. The Thick Waxy Coat of Mycobacteria, a Protective Layer against Antibiotics and the Host’s Immune System. Biochem. J. 2020, 477, 1983–2006. [Google Scholar] [CrossRef]
- Bhatt, A.; Molle, V.; Besra, G.S.; Jacobs, W.R.; Kremer, L. The Mycobacterium Tuberculosis FAS-II Condensing Enzymes: Their Role in Mycolic Acid Biosynthesis, Acid-Fastness, Pathogenesis and in Future Drug Development. Mol. Microbiol. 2007, 64, 1442–1454. [Google Scholar] [CrossRef]
- Parish, T.; Roberts, G.; Laval, F.; Schaeffer, M.; Daffé, M.; Duncan, K. Functional Complementation of the Essential Gene FabG1 of Mycobacterium Tuberculosis by Mycobacterium Smegmatis FabG but Not Escherichia Coli FabG. J. Bacteriol. 2007, 189, 3721–3728. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.K.; Bhatt, A.; Singh, A.; Saparia, E.; Evans, A.F.; Besra, G.S. Identification of the Dehydratase Component of the Mycobacterial Mycolic Acid-Synthesizing Fatty Acid Synthase-II Complex. Microbiology 2007, 153, 4166–4173. [Google Scholar] [CrossRef] [Green Version]
- Vilchèze, C.; Morbidoni, H.R.; Weisbrod, T.R.; Iwamoto, H.; Kuo, M.; Sacchettini, J.C.; Jacobs, W.R. Inactivation of the InhA-Encoded Fatty Acid Synthase II (FASII) Enoyl-Acyl Carrier Protein Reductase Induces Accumulation of the FASI End Products and Cell Lysis of Mycobacterium Smegmatis. J. Bacteriol. 2000, 182, 4059–4067. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, A.; Kremer, L.; Dai, A.Z.; Sacchettini, J.C.; Jacobs, W.R. Conditional Depletion of KasA, a Key Enzyme of Mycolic Acid Biosynthesis, Leads to Mycobacterial Cell Lysis. J. Bacteriol. 2005, 187, 7596–7606. [Google Scholar] [CrossRef] [Green Version]
- North, E.J.; Jackson, M.; Lee, R.E. New Approaches to Target the Mycolic Acid Biosynthesis Pathway for the Development of Tuberculosis Therapeutics. Curr. Pharm. Des. 2014, 20, 4357–4378. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Dubnau, E.; Quemard, A.; Balasubramanian, V.; Um, K.S.; Wilson, T.; Collins, D.; de Lisle, G.; Jacobs, W.R. InhA, a Gene Encoding a Target for Isoniazid and Ethionamide in Mycobacterium Tuberculosis. Science 1994, 263, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Grzegorzewicz, A.E.; Korduláková, J.; Jones, V.; Born, S.E.M.; Belardinelli, J.M.; Vaquié, A.; Gundi, V.A.K.B.; Madacki, J.; Slama, N.; Laval, F.; et al. A Common Mechanism of Inhibition of the Mycobacterium Tuberculosis Mycolic Acid Biosynthetic Pathway by Isoxyl and Thiacetazone. J. Biol. Chem. 2012, 287, 38434–38441. [Google Scholar] [CrossRef] [Green Version]
- Kremer, L.; Douglas, J.D.; Baulard, A.R.; Morehouse, C.; Guy, M.R.; Alland, D.; Dover, L.G.; Lakey, J.H.; Jacobs, W.R.; Brennan, P.J.; et al. Thiolactomycin and Related Analogues as Novel Anti-Mycobacterial Agents Targeting KasA and KasB Condensing Enzymes in Mycobacterium Tuberculosis. J. Biol. Chem. 2000, 275, 16857–16864. [Google Scholar] [CrossRef] [Green Version]
- Faïon, L.; Djaout, K.; Frita, R.; Pintiala, C.; Cantrelle, F.X.; Moune, M.; Vandeputte, A.; Bourbiaux, K.; Piveteau, C.; Herledan, A.; et al. Discovery of the First Mycobacterium Tuberculosis MabA (FabG1) Inhibitors through a Fragment-Based Screening. Eur. J. Med. Chem. 2020, 200, 112440. [Google Scholar] [CrossRef]
- Marrakchi, H.; Ducasse, S.; Labesse, G.; Montrozier, H.; Margeat, E.; Emorine, L.; Charpentier, X.; Daffé, M.; Quémard, A. MabA (FabG1), a Mycobacterium Tuberculosis Protein Involved in the Long-Chain Fatty Acid Elongation System FAS-II. Microbiology 2002, 148, 951–960. [Google Scholar] [CrossRef] [Green Version]
- Reed, C.W.; Washecheck, J.P.; Quitlag, M.C.; Jenkins, M.T.; Rodriguez, A.L.; Engers, D.W.; Blobaum, A.L.; Jeffrey Conn, P.; Niswender, C.M.; Lindsley, C.W. Surveying Heterocycles as Amide Bioisosteres within a Series of MGlu 7 NAMs: Discovery of VU6019278. Bioorg. Med. Chem. Lett. 2019, 29, 1211–1214. [Google Scholar] [CrossRef]
- Claffey, M.M.; Helal, C.J.; Verhoest, P.R.; Kang, Z.; Fors, K.S.; Jung, S.; Zhong, J.; Bundesmann, M.W.; Hou, X.; Lui, S.; et al. Application of Structure-Based Drug Design and Parallel Chemistry to Identify Selective, Brain Penetrant, in Vivo Active Phosphodiesterase 9A Inhibitors. J. Med. Chem. 2012, 55, 9055–9068. [Google Scholar] [CrossRef]
- Wilcken, R.; Zimmermann, M.O.; Bauer, M.R.; Rutherford, T.J.; Fersht, A.R.; Joerger, A.C.; Boeckler, F.M. Experimental and Theoretical Evaluation of the Ethynyl Moiety as a Halogen Bioisostere. ACS Chem. Biol. 2015, 10, 2725–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchholz, C.R.; Pomerantz, W.C.K. 19 F NMR Viewed through Two Different Lenses: Ligand-Observed and Protein-Observed 19 F NMR Applications for Fragment-Based Drug Discovery. RSC Chem. Biol. 2021, 2, 1312–1330. [Google Scholar] [CrossRef] [PubMed]
- Mureddu, L.G.; Vuister, G.W. Fragment-Based Drug Discovery by NMR. Where Are the Successes and Where Can It Be Improved? Front. Mol. Biosci. 2022, 9, 834453. [Google Scholar] [CrossRef] [PubMed]
- Freiberg, C.; Brötz-Oesterhelt, H.; Labischinski, H. The Impact of Transcriptome and Proteome Analyses on Antibiotic Drug Discovery. Curr. Opin. Microbiol. 2004, 7, 451–459. [Google Scholar] [CrossRef]
- Denkin, S.; Byrne, S.; Jie, C.; Zhang, Y. Gene Expression Profiling Analysis of Mycobacterium Tuberculosis Genes in Response to Salicylate. Arch. Microbiol. 2005, 184, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H.; Sun, Z. Susceptibility of Mycobacterium Tuberculosis to Weak Acids. J. Antimicrob. Chemother. 2003, 52, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Vandal, O.H.; Pierini, L.M.; Schnappinger, D.; Nathan, C.F.; Ehrt, S. A Membrane Protein Preserves Intrabacterial PH in Intraphagosomal Mycobacterium Tuberculosis. Nat. Med. 2008, 14, 849–854. [Google Scholar] [CrossRef]
- Ducasse-Cabanot, S.; Cohen-Gonsaud, M.; Marrakchi, H.; Nguyen, M.; Zerbib, D.; Bernadou, J.; Daffé, M.; Labesse, G.; Quémard, A. In Vitro Inhibition of the Mycobacterium Tuberculosis β-Ketoacyl-Acyl Carrier Protein Reductase MabA by Isoniazid. Antimicrob. Agents Chemother. 2004, 48, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.; Claparols, C.; Bernadou, J.; Meunier, B. A Fast and Efficient Metal-Mediated Oxidation of Isoniazid and Identification of Isoniazid-NAD(H) Adducts. ChemBioChem 2001, 2, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Veyron-Churlet, R.; Guerrini, O.; Mourey, L.; Daffé, M.; Zerbib, D. Protein-Protein Interactions within the Fatty Acid Synthase-II System of Mycobacterium Tuberculosis Are Essential for Mycobacterial Viability. Mol. Microbiol. 2004, 54, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Phetsuksiri, B.; Baulard, A.R.; Cooper, A.M.; Minnikin, D.E.; Douglas, J.D.; Besra, G.S.; Brennan, P.J. Antimycobacterial Activities of Isoxyl and New Derivatives through the Inhibition of Mycolic Acid Synthesis. Antimicrob. Agents Chemother. 1999, 43, 1042–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Cpd | X | -Y- | IC50 (µM) |
| 1 | I | -NH-CO- | 38 ± 6 |
| 3 | I | -N(CH3)-CO- | >200 |
| 4 | I | -NH-SO2- | 40 |
| 2 | Br | -NH-CO- | 45 |
| 5 | Br | -CO-NH- | 268 |
| 6 | Br | -NH-CS- | 39 |
| 7 | Br |  | 102 |
 | ||
|---|---|---|
| Cpd | R | IC50 (µM) |
| 1 | -COOH | 38 ± 6 |
| 8 | -CH2-OH | >333 |
| 9 | -CONH2 | >500 |
| 10 | -C≡N | >1000 |
| 11 |  | 24 |
| 12 | -CO-NH-SO2-CF3 | 35 |
| 13 | -CO-NH-SO2-CH3 | 28 |
 | ||
|---|---|---|
| Cpd | R | IC50 (µM) |
| 1 |  | 38 |
| 2 |  | 45 |
| 14 |  | 34 |
| 15 |  | >1000 |
| 16 |  | 60 |
| 17 |  | 77 |
| 18 |  | 23 |
| 19 |  | 33 |
| 20 |  | 76 |
| Cpd | Structure | IC50 (µM) | MIC90 (µM) | Solubility (µM) | LogD7.4 | PPB (%) |
|---|---|---|---|---|---|---|
| 1 |  | 38 | 100 | >200 | 3.401 | 99.86 |
| 2 |  | 45 | 100 | 181 | 2.998 | - |
| 12 |  | 35 | 300 | 180 | 4.102 | 99.98 |
| 16 |  | 60 | 100 | >200 | - | 99.94 |
| 18 |  | 23 | 300 | 149 | 2.537 | >99.99 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faïon, L.; Djaout, K.; Pintiala, C.; Piveteau, C.; Leroux, F.; Biela, A.; Slupek, S.; Antoine, R.; Záhorszká, M.; Cantrelle, F.-X.; et al. Exploring the Antitubercular Activity of Anthranilic Acid Derivatives: From MabA (FabG1) Inhibition to Intrabacterial Acidification. Pharmaceuticals 2023, 16, 335. https://doi.org/10.3390/ph16030335
Faïon L, Djaout K, Pintiala C, Piveteau C, Leroux F, Biela A, Slupek S, Antoine R, Záhorszká M, Cantrelle F-X, et al. Exploring the Antitubercular Activity of Anthranilic Acid Derivatives: From MabA (FabG1) Inhibition to Intrabacterial Acidification. Pharmaceuticals. 2023; 16(3):335. https://doi.org/10.3390/ph16030335
Chicago/Turabian StyleFaïon, Léo, Kamel Djaout, Catalin Pintiala, Catherine Piveteau, Florence Leroux, Alexandre Biela, Stéphanie Slupek, Rudy Antoine, Monika Záhorszká, Francois-Xavier Cantrelle, and et al. 2023. "Exploring the Antitubercular Activity of Anthranilic Acid Derivatives: From MabA (FabG1) Inhibition to Intrabacterial Acidification" Pharmaceuticals 16, no. 3: 335. https://doi.org/10.3390/ph16030335
APA StyleFaïon, L., Djaout, K., Pintiala, C., Piveteau, C., Leroux, F., Biela, A., Slupek, S., Antoine, R., Záhorszká, M., Cantrelle, F. -X., Hanoulle, X., Korduláková, J., Deprez, B., Willand, N., Baulard, A. R., & Flipo, M. (2023). Exploring the Antitubercular Activity of Anthranilic Acid Derivatives: From MabA (FabG1) Inhibition to Intrabacterial Acidification. Pharmaceuticals, 16(3), 335. https://doi.org/10.3390/ph16030335