
A Bitter Taste Receptor as a Novel Molecular Target on Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma
 ,
,
Abstract
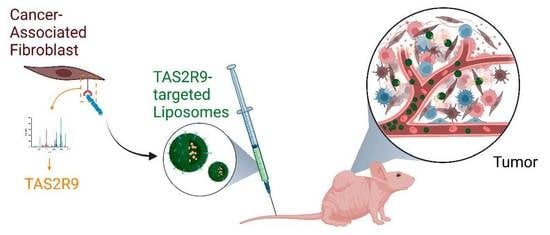
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Identification of TAS2R9 as New CAF-Selective Target
2.2. Validation of TAS2R9 as a CAF Target
2.3. TAS2R9 mRNA Expression Is a Prognostic Indicator in Several Cancers
2.4. Development of a TAS2R9-Targeted Drug Delivery System
2.5. TAS2R9-Targeted Delivery Increases Liposome Accumulation in Tumor of Admix PDAC Model
2.6. TAS2R9-Targeted Liposomal Delivery of a CXCR2 Inhibitor Inhibits Tumor Growth
2.7. Drug-Loaded TAS2R9-Targeted Liposomes Show Target Pathway Engagement and Decrease Cancer Cell Proliferation
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. Cell Culture and Reagents
4.3. Lipids and Peptides for Liposome Preparation
4.4. Phage-Based Pulldown Assay
4.5. TAS2R9 siRNA/shRNA Lentivirus Transduction
4.6. Quantitative PCR
4.7. Phage ELISA
4.8. Western Blot Analysis
4.9. Flow Cytometry with TAS2R9 Primary Antibody
4.10. Immunoflourscent Staining
4.11. Preperation and Characterization of Liposomes
4.12. Liposome Binding Assay
4.13. In Vivo Tumor Studies
4.14. Immunohistochemistry (IHC) Staining and Analysis
4.15. Pharmacokinetics
4.16. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Ushio, J.; Kanno, A.; Ikeda, E.; Ando, K.; Nagai, H.; Miwata, T.; Kawasaki, Y.; Tada, Y.; Yokoyama, K.; Numao, N.; et al. Pancreatic Ductal Adenocarcinoma: Epidemiology and Risk Factors. Diagnostics 2021, 11, 562. [Google Scholar] [CrossRef]
- Leca, J.; Martinez, S.; Lac, S.; Nigri, J.; Secq, V.; Rubis, M.; Bressy, C.; Sergé, A.; Lavaut, M.-N.; Dusetti, N.; et al. Cancer-Associated Fibroblast-Derived Annexin A6+ Extracellular Vesicles Support Pancreatic Cancer Aggressiveness. J. Clin. Investig. 2016, 126, 4140–4156. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Randazzo, O.; Papini, F.; Mantini, G.; Gregori, A.; Parrino, B.; Liu, D.S.K.; Cascioferro, S.; Carbone, D.; Peters, G.J.; Frampton, A.E.; et al. “Open Sesame?”: Biomarker Status of the Human Equilibrative Nucleoside Transporter-1 and Molecular Mechanisms Influencing Its Expression and Activity in the Uptake and Cytotoxicity of Gemcitabine in Pancreatic Cancer. Cancers 2020, 12, 3206. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Zhang, B.; Hu, Q.; Qin, Y.; Xu, W.; Liu, W.; Yu, X.; Xu, J. The Impact of Cancer-Associated Fibroblasts on Major Hallmarks of Pancreatic Cancer. Theranostics 2018, 8, 5072–5087. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal Elements Act to Restrain, Rather than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, R.K.; McDonough, S.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.R.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. A Phase IB/II Randomized Study of MFOLFIRINOX (MFFOX) + Pegylated Recombinant Human Hyaluronidase (PEGPH20) versus MFFOX Alone in Patients with Good Performance Status Metastatic Pancreatic Adenocarcinoma (MPC): SWOG S1313 (NCT #01959139). JCO 2018, 36, 208. [Google Scholar] [CrossRef]
- Catenacci, D.V.T.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. JCO 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic Cancer Stroma: An Update on Therapeutic Targeting Strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Jiang, B.; Zhou, L.; Lu, J.; Wang, Y.; Liu, C.; You, L.; Guo, J. Stroma-Targeting Therapy in Pancreatic Cancer: One Coin With Two Sides? Front. Oncol. 2020, 10, 576399. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Ijichi, H.; Takahashi, R.; Miyabayashi, K.; Fujiwara, H.; Yamada, T.; Kato, H.; Nakatsuka, T.; Tanaka, Y.; Tateishi, K.; et al. Blocking CXCLs–CXCR2 Axis in Tumor–Stromal Interactions Contributes to Survival in a Mouse Model of Pancreatic Ductal Adenocarcinoma through Reduced Cell Invasion/Migration and a Shift of Immune-Inflammatory Microenvironment. Oncogenesis 2019, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [Green Version]
- Brinton, L.T.; Bauknight, D.K.; Dasa, S.S.K.; Kelly, K.A. PHASTpep: Analysis Software for Discovery of Cell-Selective Peptides via Phage Display and Next-Generation Sequencing. PLoS ONE 2016, 11, e0155244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howitt, M.R.; Lavoie, S.; Michaud, M.; Blum, A.M.; Tran, S.V.; Weinstock, J.V.; Gallini, C.A.; Redding, K.; Margolskee, R.F.; Osborne, L.C.; et al. Tuft Cells, Taste-Chemosensory Cells, Orchestrate Parasite Type 2 Immunity in the Gut. Science 2016, 351, 1329–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, D.A.; Wang, W.C.H.; McIlmoyle, E.L.; Robinett, K.S.; Schillinger, R.M.; An, S.S.; Sham, J.S.K.; Liggett, S.B. Bitter Taste Receptors on Airway Smooth Muscle Bronchodilate by Localized Calcium Signaling and Reverse Obstruction. Nat. Med. 2010, 16, 1299–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wente, M.N.; Keane, M.P.; Burdick, M.D.; Friess, H.; Büchler, M.W.; Ceyhan, G.O.; Reber, H.A.; Strieter, R.M.; Hines, O.J. Blockade of the Chemokine Receptor CXCR2 Inhibits Pancreatic Cancer Cell-Induced Angiogenesis. Cancer Lett. 2006, 241, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Bierie, B.; Tada, M.; Mohri, D.; Miyabayashi, K.; Asaoka, Y.; Maeda, S.; et al. Inhibiting Cxcr2 Disrupts Tumor-Stromal Interactions and Improves Survival in a Mouse Model of Pancreatic Ductal Adenocarcinoma. J. Clin. Investig. 2011, 121, 4106–4117. [Google Scholar] [CrossRef]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, Y.; Ohuchida, K.; Otsubo, Y.; Kibe, S.; Takesue, S.; Abe, T.; Iwamoto, C.; Shindo, K.; Moriyama, T.; Nakata, K.; et al. Necroptosis in Pancreatic Cancer Promotes Cancer Cell Migration and Invasion by Release of CXCL5. PLoS ONE 2020, 15, e0228015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purohit, A.; Saxena, S.; Varney, M.; Prajapati, D.R.; Kozel, J.A.; Lazenby, A.; Singh, R.K. Host Cxcr2-Dependent Regulation of Pancreatic Cancer Growth, Angiogenesis, and Metastasis. Am. J. Pathol. 2021, 191, 759–771. [Google Scholar] [CrossRef]
- Hastrup, N.; Khalilieh, S.; Dale, D.C.; Hanson, L.G.; Magnusson, P.; Tzontcheva, A.; Tseng, J.; Huyck, S.; Rosenberg, E.; Krogsgaard, K. The Effects of the CXCR2 Antagonist, MK-7123, on Bone Marrow Functions in Healthy Subjects. Cytokine 2015, 72, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Jurcevic, S.; Humfrey, C.; Uddin, M.; Warrington, S.; Larsson, B.; Keen, C. The Effect of a Selective CXCR2 Antagonist (AZD5069) on Human Blood Neutrophil Count and Innate Immune Functions. Br. J. Clin. Pharmacol. 2015, 80, 1324–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, K.A.; Bardeesy, N.; Anbazhagan, R.; Gurumurthy, S.; Berger, J.; Alencar, H.; Depinho, R.A.; Mahmood, U.; Weissleder, R. Targeted Nanoparticles for Imaging Incipient Pancreatic Ductal Adenocarcinoma. PLoS Med. 2008, 5, e85. [Google Scholar] [CrossRef]
- Peran, I.; Madhavan, S.; Byers, S.W.; McCoy, M.D. Curation of the Pancreatic Ductal Adenocarcinoma Subset of the Cancer Genome Atlas Is Essential for Accurate Conclusions about Survival-Related Molecular Mechanisms. Clin. Cancer Res. 2018, 24, 3813–3819. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liang, Y.; Xu, H.; Zhang, X.; Mao, T.; Cui, J.; Yao, J.; Wang, Y.; Jiao, F.; Xiao, X.; et al. Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Identifies a Novel Fibroblast Subtype Associated with Poor Prognosis but Better Immunotherapy Response. Cell Discov. 2021, 7, 36. [Google Scholar] [CrossRef]
- Chandrashekar, J.; Hoon, M.A.; Ryba, N.J.P.; Zuker, C.S. The Receptors and Cells for Mammalian Taste. Nature 2006, 444, 288–294. [Google Scholar] [CrossRef]
- Bachmanov, A.A.; Beauchamp, G.K. Taste Receptor Genes. Annu. Rev. Nutr. 2007, 27, 389–414. [Google Scholar] [CrossRef] [Green Version]
- Piersma, B.; Hayward, M.-K.; Weaver, V.M. Fibrosis and Cancer: A Strained Relationship. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.R.; Baker, D.; Farren, M.; Pommier, A.; Swann, R.; Wang, X.; Mistry, S.; McDaid, K.; Kendrew, J.; Womack, C.; et al. Tumor Stromal Architecture Can Define the Intrinsic Tumor Response to VEGF-Targeted Therapy. Clin. Cancer Res. 2013, 19, 6943–6956. [Google Scholar] [CrossRef] [Green Version]
- Curry, J.M.; Sprandio, J.; Cognetti, D.; Luginbuhl, A.; Bar-ad, V.; Pribitkin, E.; Tuluc, M. Tumor Microenvironment in Head and Neck Squamous Cell Carcinoma. Semin. Oncol. 2014, 41, 217–234. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein–Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Nagy, Á.; Munkácsy, G.; Győrffy, B. Pancancer Survival Analysis of Cancer Hallmark Genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef]
- He, K.; Tang, M. Safety of Novel Liposomal Drugs for Cancer Treatment: Advances and Prospects. Chem.-Biol. Interact. 2018, 295, 13–19. [Google Scholar] [CrossRef]
- Belfiore, L.; Saunders, D.N.; Ranson, M.; Thurecht, K.J.; Storm, G.; Vine, K.L. Towards Clinical Translation of Ligand-Functionalized Liposomes in Targeted Cancer Therapy: Challenges and Opportunities. J. Control. Release 2018, 277, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Aronson, M.R.; Medina, S.H.; Mitchell, M.J. Peptide Functionalized Liposomes for Receptor Targeted Cancer Therapy. APL Bioeng. 2021, 5, 011501. [Google Scholar] [CrossRef]
- Dasa, S.S.K.; Diakova, G.; Suzuki, R.; Mills, A.M.; Gutknecht, M.F.; Klibanov, A.L.; Slack-Davis, J.K.; Kelly, K.A. Plectin-Targeted Liposomes Enhance the Therapeutic Efficacy of a PARP Inhibitor in the Treatment of Ovarian Cancer. Theranostics 2018, 8, 2782–2798. [Google Scholar] [CrossRef]
- Wei, Y.; Gu, X.; Sun, Y.; Meng, F.; Storm, G.; Zhong, Z. Transferrin-Binding Peptide Functionalized Polymersomes Mediate Targeted Doxorubicin Delivery to Colorectal Cancer in Vivo. J. Control. Release 2020, 319, 407–415. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open Source Software for Digital Pathology Image Analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chari, S.T.; Kelly, K.; Hollingsworth, M.A.; Thayer, S.P.; Ahlquist, D.A.; Andersen, D.K.; Batra, S.K.; Brentnall, T.A.; Canto, M.; Cleeter, D.F.; et al. Early Detection of Sporadic Pancreatic Cancer: Summative Review. Pancreas 2015, 44, 693–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct Populations of Inflammatory Fibroblasts and Myofibroblasts in Pancreatic Cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [Green Version]
- Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers 2020, 12, 1347. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.-Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. JCO 2020, 38, 3185–3194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, L.; Liu, X.; Kammertoens, T.; Blankenstein, T.; Qin, Z. Fibroblast-Specific Protein 1/S100A4–Positive Cells Prevent Carcinoma through Collagen Production and Encapsulation of Carcinogens. Cancer Res. 2013, 73, 2770–2781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, J.N.; Magiera, L.; Kraman, M.; Fearon, D.T. Tumoral Immune Suppression by Macrophages Expressing Fibroblast Activation Protein-Alpha and Heme Oxygenase-1. Cancer Immunol. Res. 2014, 2, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.V.; Rozengurt, N.; Yang, M.; Young, S.H.; Sinnett-Smith, J.; Rozengurt, E. Expression of Bitter Taste Receptors of the T2R Family in the Gastrointestinal Tract and Enteroendocrine STC-1 Cells. Proc. Natl. Acad. Sci. USA 2002, 99, 2392–2397. [Google Scholar] [CrossRef] [Green Version]
- Höfer, D.; Püschel, B.; Drenckhahn, D. Taste Receptor-like Cells in the Rat Gut Identified by Expression of Alpha-Gustducin. Proc. Natl. Acad. Sci. USA 1996, 93, 6631–6634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaik, F.A.; Singh, N.; Arakawa, M.; Duan, K.; Bhullar, R.P.; Chelikani, P. Bitter Taste Receptors: Extraoral Roles in Pathophysiology. Int. J. Biochem. Cell Biol. 2016, 77, 197–204. [Google Scholar] [CrossRef]
- Zehentner, S.; Reiner, A.T.; Grimm, C.; Somoza, V. The Role of Bitter Taste Receptors in Cancer: A Systematic Review. Cancers 2021, 13, 5891. [Google Scholar] [CrossRef]
- Gaida, M.M.; Mayer, C.; Dapunt, U.; Stegmaier, S.; Schirmacher, P.; Wabnitz, G.H.; Hänsch, G.M. Expression of the Bitter Receptor T2R38 in Pancreatic Cancer: Localization in Lipid Droplets and Activation by a Bacteria-Derived Quorum-Sensing Molecule. Oncotarget 2016, 7, 12623–12632. [Google Scholar] [CrossRef] [Green Version]
- Stern, L.; Giese, N.; Hackert, T.; Strobel, O.; Schirmacher, P.; Felix, K.; Gaida, M.M. Overcoming Chemoresistance in Pancreatic Cancer Cells: Role of the Bitter Taste Receptor T2R10. J. Cancer 2018, 9, 711–725. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Zhang, C.-H.; Lifshitz, L.M.; ZhuGe, R. Extraoral Bitter Taste Receptors in Health and Disease. J. Gen. Physiol. 2017, 149, 181–197. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, M.T.; Kemp, S.B.; Salas-Escabillas, D.J.; Zhang, Y.; Steele, N.G.; The, S.; Long, D.; Benitz, S.; Yan, W.; Margolskee, R.F.; et al. The Gustatory Sensory G-Protein GNAT3 Suppresses Pancreatic Cancer Progression in Mice. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 349–369. [Google Scholar] [CrossRef] [PubMed]
- Niethammer, A.G.; Zheng, Z.; Timmer, A.; Lee, T.-L. First-in-Human Evaluation of Oral Denatonium Acetate (ARD-101), a Potential Bitter Taste Receptor Agonist: A Randomized, Double-Blind, Placebo-Controlled Phase 1 Trial in Healthy Adults. Clin. Pharmacol. Drug Dev. 2022, 11, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.-T.; Sun, W.; Zhang, J.-T.; Fan, Y.-Z. Cancer-associated Fibroblast Regulation of Tumor Neo-angiogenesis as a Therapeutic Target in Cancer (Review). Oncol. Lett. 2019, 17, 3055–3065. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, F.; Panneer, N.; Tutino, C.M.; Wu, M.; Skrabal, W.R.; Moskaluk, C.; Kelly, K.A. A Functional Proteomic Method for Biomarker Discovery. PLoS ONE 2011, 6, e22471. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H. Thin-Film Hydration Followed by Extrusion Method for Liposome Preparation. Methods Mol. Biol. 2017, 1522, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.M.; Maxwell, P.J.; Waugh, D.J.J. Rationale and Means to Target Pro-Inflammatory Interleukin-8 (CXCL8) Signaling in Cancer. Pharmaceuticals 2013, 6, 929–959. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, J.; Perez, S.M.; Dasa, S.S.K.; Hall, S.P.; Heckert, D.B.; Murphy, B.P.; Crawford, H.C.; Kelly, K.A.; Brinton, L.T. A Bitter Taste Receptor as a Novel Molecular Target on Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma. Pharmaceuticals 2023, 16, 389. https://doi.org/10.3390/ph16030389
Hung J, Perez SM, Dasa SSK, Hall SP, Heckert DB, Murphy BP, Crawford HC, Kelly KA, Brinton LT. A Bitter Taste Receptor as a Novel Molecular Target on Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma. Pharmaceuticals. 2023; 16(3):389. https://doi.org/10.3390/ph16030389
Chicago/Turabian StyleHung, Jessica, Samantha M. Perez, Siva Sai Krishna Dasa, Sarah P. Hall, Danielle B. Heckert, Brian P. Murphy, Howard C. Crawford, Kimberly A. Kelly, and Lindsey T. Brinton. 2023. "A Bitter Taste Receptor as a Novel Molecular Target on Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma" Pharmaceuticals 16, no. 3: 389. https://doi.org/10.3390/ph16030389
APA StyleHung, J., Perez, S. M., Dasa, S. S. K., Hall, S. P., Heckert, D. B., Murphy, B. P., Crawford, H. C., Kelly, K. A., & Brinton, L. T. (2023). A Bitter Taste Receptor as a Novel Molecular Target on Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma. Pharmaceuticals, 16(3), 389. https://doi.org/10.3390/ph16030389