1. Introduction
The Kirsten rat sarcoma viral oncogene homolog G12C isoform (KRAS) belongs to the RAS protein family of small guanosine–triphosphate hydrolases (GTPases) [
1]. The KRAS
G12C mutation represents the most prevalent in lung cancer (14%) and is associated with carcinogen exposure, such as cigarette smoke [
2]. The discovery of a druggable pocket on the KRAS protein and the recent development of covalent KRAS
G12C inhibitors with clinical efficacy have been enormous breakthroughs for prolonging the survival of patients with advanced NSCLC [
3,
4,
5]. Sotorasib was the first approved KRAS
G12C inhibitor (KRAS
G12Ci) by the FDA as a monotherapy of second-line treatment for patients with advanced KRAS
G12C mutated NSCLC [
4]. In parallel, adagrasib also showed a response in KRAS
G12C mutant NSCLC patients previously treated with chemotherapy and anti-PD1/L1 [
6]. Despite these new molecules’ benefits, response rates remain below those obtained for other targeted therapies and range around 45% [
2,
7,
8]. In addition, mechanisms of acquired resistance are emerging, including mutations in the KRAS protein, which prevent compound binding or activation of molecular pathways bypassing KRAS signaling [
9,
10,
11].
The Hippo-YAP1 pathway is emerging as an effective bypass mechanism to mitogen-activated protein kinase (MAPK) pathway inhibition [
12,
13]. Hippo-YAP1 acts via the activation of transcriptional coactivator proteins, transcriptional coactivators yes-associated protein 1/transcriptional activator with PDZ-binding motif (YAP1 and TAZ), which interact with transcription factors from the TEAD-family (transcriptional enhanced associate domain 1-4; DNA binding transcription factors) and thereby regulate the transcription of genes required for cell survival and cell proliferation [
14].
Several preclinical studies highlight the role of YAP1/TAZ-TEAD activation as a bypass mechanism to mutant KRAS inactivation. In a KRAS
G12D-dependent mouse pancreatic ductal adenocarcinoma (PDAC) model, YAP1 gene amplification bypasses KRAS inactivation, leading to cancer progression. In a KRAS
G12D-dependent mouse NSCLC model, the knockdown of YAP1 delays tumor regrowth [
15]. In parallel, the loss of the wild-type KRAS allele can promote YAP1 activation, as wt-KRAS favors the cytoplasmic localization of YAP1 [
16].
The crosstalk between KRAS and YAP1 pathways is partly mediated via the src tyrosine kinases (SFKs) family. These kinases modulate KRAS phosphorylation by either signaling on LATS1/2 or by directly acting on YAP1 phosphorylation [
17]. Additional interaction nodes between YAP1 and KRAS pathways that have been suggested include (i) the convergence of both pathways on the activation of the induction of epithelial–mesenchymal transition (EMT) via the transcription factor fos proto-oncogene (FOS), (ii) the KRAS-mediated stabilization of YAP1 via the downregulation of SOCS-box proteins and (iii) the role of YAP1 as an oncogenic KRAS effector in TRP53 mutant pancreas tumor [
15,
18,
19].
Despite the described crosstalks, the direct effects of combining a KRASG12Ci with a TEADi have yet to be investigated. Here, we explore the combination of KRASG12C and YAP1-TEAD inhibition on KRASG12C-NSCLC cell lines. We performed drug combination studies using KRASG12Ci adagrasib (MRTX849) or sotorasib (AMG510) and pan-TEADi K-975 or Sanofi compound A (Cpd A) for the in vivo combination study. We evaluated the effect of this dual inhibition on tumor cell growth and biomarker modulation and characterized the effects of the combination at the transcriptomic, proteomic, and phospho-proteomic levels.
3. Discussion
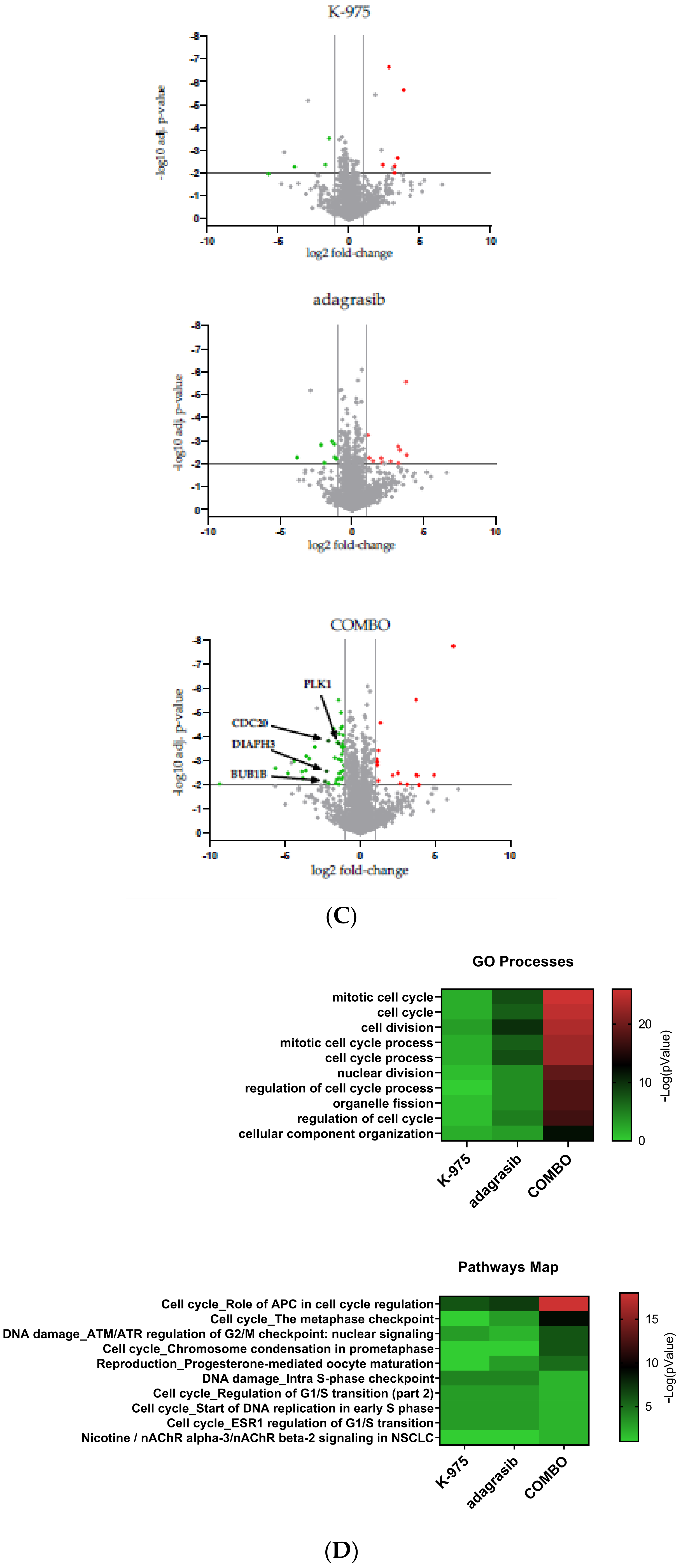
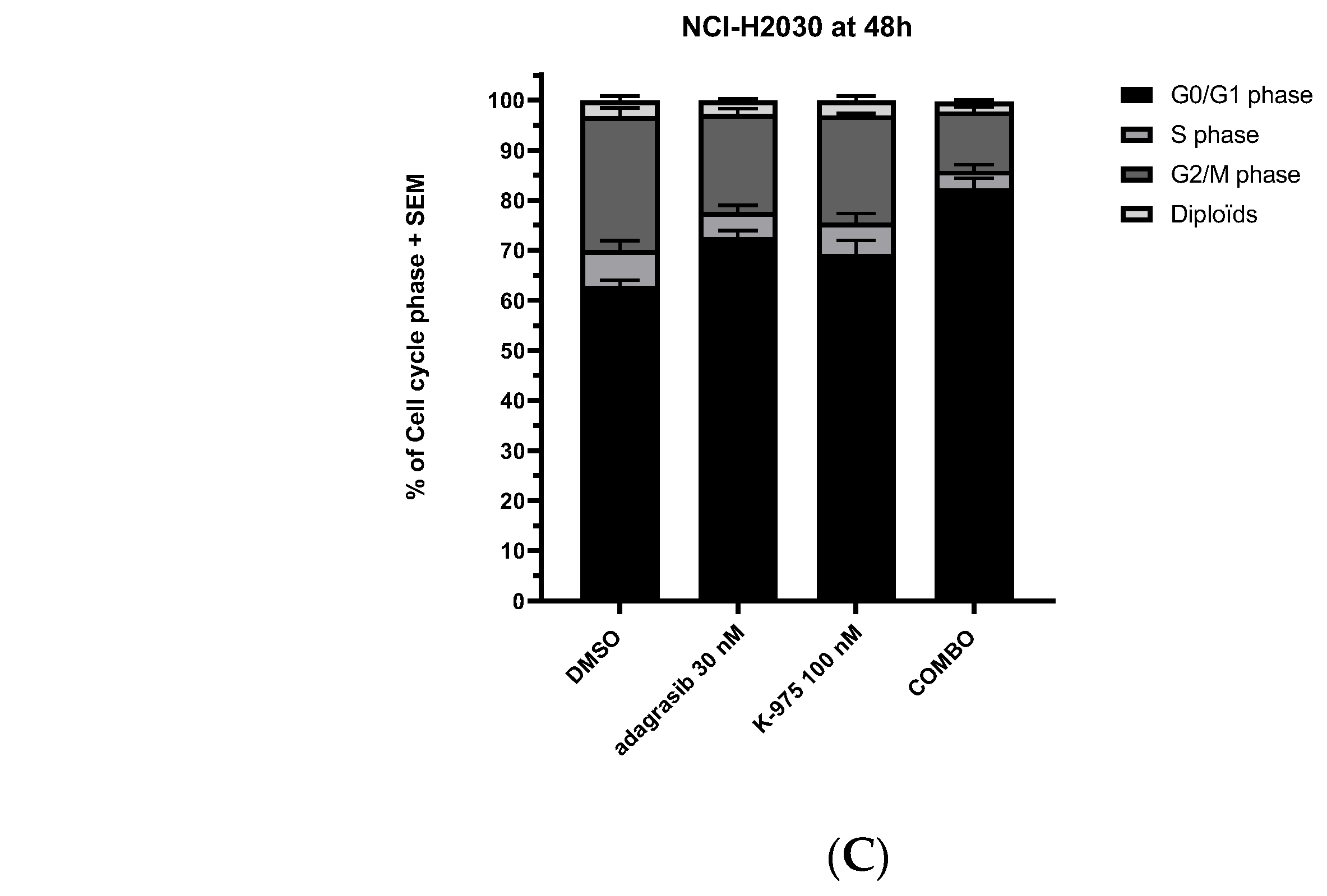
Here, we report an integrative analysis of the effects of combining a KRAS
G12C inhibitor with a YAP1/TAZ-TEAD inhibitor in KRAS
G12C mutant NSCLC cell lines. We show that adding a TEADi potentiates the effect of adagrasib in most cell lines tested. Mechanistically, we demonstrate that the effect of the combination leads to a dual cell cycle arrest characterized by blockage in the G1 and G2/M phases. Previous studies have reported that YAP1-TEAD complexes regulate genes implicated in the cell cycle [
21], including some of the genes we identify in the current study. For example, HELLS and ATAD2 are coactivators that favor the expression of genes implicated in G1/S transition and S phase progression, which we see inhibited in our combination treatment. They drive cell proliferation through the activation of MYC in the case of ATAD2 and E2F in the case of HELLS [
22]. MYC and E2F were the main transcription signature programs downregulated in our KRAS
G12C/TEADi combination.
The other prominent signature modulated by our combination was the G2/M checkpoint. Some data are available on YAP1 activation in the context of resistance to EGFR inhibitor treatment. In EGFR mutant NSCLC cells, the YAP1/FOXM1 axis mediates resistance to EGFR inhibitors by increasing the expression of spindle assembly checkpoint (SAC) proteins [
23]. SAC components play an essential role in the correct attachment of microtubules (MT) of the mitotic spindle to the kinetochore (KT) by inhibiting the formation of the APC/C-CDC20 complex [
24]. APC/C is an E3 ubiquitin ligase necessary for mitotic exit [
25,
26,
27]. SAC proteins inhibit the formation of the APC/C-CDC20 complex when one KT is unattached or is incorrectly attached [
25].
Our studies show that downregulation of the KRAS-YAP1-TEAD pathway also inhibits the expression of essential modulators such as Aurora B and PLK1 kinases at transcript and protein levels. These kinases are known components of SAC that regulate the protein levels of BUB1, BUBR1, and CDC20, which are all implicated in regulating APC/C-CDC20 complex formation. We validated the protein decrease in some of these SAC components in response to the combination treatment.
Based on the literature data and our findings, we propose a model where the combination of KRAS
G12C and TEAD inhibition converges in the downregulation of genes required for cell cycle progression and kinetochore attachment mediated by transcription factors (TFs) such as FOXM1 (
Figure 7). In this model, the combination may, on the one hand, inhibit the transcription of HELLS and ATAD2 via FOXM1, leading to an arrest in G1 [
28]. On the other hand, potential FOXM1 inhibition may decrease the expression of SAC components, inhibiting the formation of the APC/C complex and leading to reduced entry into the mitotic phase.
FOXM1 can modulate the expression of several genes, some of which are implicated in specific checkpoints of cell cycle progression [
29,
30]. Cell cycle analysis showed a reduction of the S/G2/M phases and no changes in late mitosis, a phenotype different from the pure inhibition of SAC components. This observation can be explained by the fact that KRAS
G12Ci and TEADi combination inhibits the SAC, E2F, and MYC transcription programs, resulting in a dual cell cycle effect. Whether this dual cell cycle inhibition event occurs in parallel or whether one precedes the other will need to be investigated. Furthermore, additional evaluations of other cell lines and a detailed genomic study will be required to understand better this phenomenon suggested in the literature [
31,
32] and whether the effect on cell cycle inhibition would be comparable.
4. Materials and Methods
4.1. Cell Lines Culture Conditions
Cell lines were maintained at Sanofi and grown at 37 °C under 5% CO2, except for SW1573, which was at 0% CO2. A short tandem repeat assay authenticated all cell lines at the Microsynth AG (Balgach, Switzerland). PCR using the Venor®GeM kit excluded mycoplasma infection (Biovalley, MB minerva biolabs, Skillman, NJ, USA). HOP 62 (#CRL-11350) RRID: CVCL_1285, NCI-H2030 (#CRL-5914) RRID: CVCL_1517, NCI-H1792 (#CRL-5895) RRID: CVCL_1495, NCI-H23 (#CRL-5800) RRID: CVCL_1547, NCI-H358 (#CRL-5807) RRID: CVCL_1559, NCI-H1373 (#CRL-5866) RRID: CVCL_1465, NCI-H2122 (#CRL-5985) RRID: CVCL_1531, SW1573 (#CRL-2170) RRID: CVCL_1720 were purchased from the American Type Culture Collection (ATCC), while LU99A was purchased from the Japanese Collection of Research Bioresources Cell Bank (JCRB0044) and cultured according to supplier’s recommendation. NCI-H2030 and HOP 62 were stably transduced either with the Incucyte® Cell Cycle Green/Orange Lentivirus reagent (#4809, Sartorius, Göttingen, Germany) or with TEAD Luciferase lentiviral construct (FlashTherapeutics, Toulouse, France) containing a TEAD-responsive synthetic element/region (8xGTIIC region followed by a minimal chicken TNNT2 promoter, driving luciferase expression). All transduced cells were cultured according to the supplier’s recommendations, plus the puromycin or neomycin (#10781691 and #G8168, Sigma, St Louis, MO, USA) to maintain the selected population.
4.2. Luciferase Assay
NCI-H2030 and HOP 62 cells were seeded at 8 × 104 cells/well density in 96-well plates in a complete culture medium and incubated at 37 °C in 5% CO2 for 24 h. Initially dissolved in DMSO, compounds were added to the culture medium at different dilutions with a final amount of 0.1% DMSO. After 24 h of incubation, luciferase activity was detected by a luminescence plate reader (Spark, TECAN Männedorf, canton of Zürich, Switzerland) using Bright-Glo™ Luciferase Assay System (according to the manufacturer’s instruction).
4.3. Compounds Used
We used the TEADi K-975 [
33], the KRAS
G12C inhibitors adagrasib [
34] and sotorasib [
35], the inhibitors for PLK1 (RO-3280) and Aurora B Kinase (SML-0268), and a proprietary compound from Sanofi (Cpd A).
4.4. Compound Combination Assay for A Confirmatory Study
Cell lines were seeded in 384-well plates in a complete culture medium. Cell lines were seeded at 150 cells/well for HOP 62 and 125 cells/well for NCI-H2030. After 24 h at 37 °C and 5% CO2, compounds were added at different dilutions to have a final percentage of 0.2% DMSO in the culture medium. Single compound treatment was performed using a range of fourteen concentrations for adagrasib from 10,000 to 0.006 nM and eighteen concentrations for K-975 from 10,000 to 0.0001 nM. For combination treatments, the same concentrations and doses were used for each inhibitor, leading to 14 × 18 combination points in this large compound combination assay. Cell viability was read after 144 h of treatment using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Madison, WI, USA) reagent with an incubation time of 1 h at RT. Luminescence was detected using the TECAN SPARK plate readers. To characterize the interactions between adagrasib and K-975 on NCI-H2030 and HOP 62 cells, the IC50 of adagrasib alone and with K-975 were estimated from ranges of adagrasib between 0.02 and 10,000 nM alone and mixed with eleven constant concentrations of K-975 between 0.5 and 3333 nM. The ratio R = IC50adagrasib with K-975/IC50adagrasib alone was calculated with its confidence interval with SAS® version 9.4. If R is significantly lower than 1 (i.e., the lower bound of its confidence interval is lower than 1), the IC50 of K-975 with adagrasib is significantly lower than the IC50 of adagrasib alone. Moreover, if K-975 is nonactive, a potentiation can be concluded. Three independent experiments were conducted to obtain six replicates of the wells with single compounds and triplicates of the wells with combinations.
4.5. Compound Combination Assay
Cell lines were seeded in 96-well plates in complete culture medium and cultured at densities allowing for exponential growth: 1250 cells/well (HOP 62), 400 cells/well (NCI-H2030), 500 cells/well (NCI-H1792), 5000 cells/well (NCI-H23), 2500 cells/well (NCI-H353, SW1573), 600 cells/well (NCI-H1373), 1000 cells/well (NCI-H2122), and 1200 cells/well (LU99-A). After 24 h, compounds were added in different dilutions with a final percentage of 0.2% DMSO. Single-agent treatments were performed with a range of ten concentrations per compound (0.3 to 10,000 nM) and a dilution factor of approx. 3x between each of the concentrations. In the combination study, adagrasib was tested at the same range of ten concentrations used in the single-agent studies, while K-975 was added at two fixed doses (100 and 1000 nM). Cell viability was determined after 144 h of compound treatment using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Madison, WI, USA) reagent with incubation of 1 h at room temperature (RT). Luminescence was detected using the TECAN SPARK plate readers. IC50 values were calculated using Excel Fit on three independent experiments.
4.6. Phosphorylated/Total ERK1/2 Detection
HOP 62 and NCI-H2030 cells were seeded at 6000 cells/well in 96-well plates in complete culture medium at 37 °C at 5% CO2. After 24 h, compounds were added at different dilutions to obtain a final percentage of 0.1% DMSO. Single compounds were tested using a range of ten concentrations from 0.0003 μM to 10,000 nM for both inhibitors, adagrasib, and K-975. After 5 and 24 h of compound treatment, the culture medium was removed, and the cells were lysed with the cell extraction buffer (#FNN0011, Invitrogen, Waltham, MA, USA), complemented with Anti-Protease (#04963124001, Roche, Basel, Switzerland) and 1 mM PMSF (#93482, Sigma). Protein extracts were processed according to MSD®, Rockville, Maryland, MULTI-SPOT Assay System (#K15107D) protocol. IC50 values were calculated using Excel Fit on three independent experiments.
4.7. RNA Extraction, HiCAR-Seq Library Preparation, Sequencing, Analysis
RNA was isolated from frozen cell pellets using RNEASY Plus Mini Kit (#74136, Qiagen, Venlo, Netherlands). Then, 50 ng of total RNA was used for HiCARSeq library preparation. HiCARSeq library was prepared as described previously [
36]. HiCARSeq libraries were sequenced along with other samples using NVSEQ 6000 S1 Rgt Kit v1.5 (100 cyc) and NovaSeq 6000 Sequencing System (Illumina, San Diego, CA, USA). NovaSeq 6000 output files were converted into FASTQ files using bcl2fastq and used in downstream analysis. HiCARSeq transcriptome FASTQ files were analyzed on Array studio V10.0 (Omicsoft, Qiagen). Following raw-read QC, the last few bases were trimmed and mapped to the reference genome Human.GRCh38. The read count data were generated using GeneModel RefGene20200204. The resulting data were normalized by the DESeq package, transformed to a log2 value, and used for pathway prediction and one-way ANOVA analyses. For pathway enrichment, GSEA was performed using the Hallmark gene set of Molecular Signatures Database v7.4 [
20]. To estimate the TEAD-dependent transcription rate and to evaluate TEAD inhibitors’ pharmacodynamics, we used a transcriptional signature of TEAD activity and a scoring method described elsewhere [
37].
4.8. Sample Preparation and Liquid Chromatography Mass Spectrometry Analysis (LC/MS)
NCI-H2030 cells were resuspended in cell lysis buffer (#FNN0011, Invitrogen) supplemented with protease and phosphatase inhibitor cocktail (#78446, Thermo Fisher Scientific, Waltham, MA, USA). The mixture was then stirred for 10 min at 70 °C, followed by centrifugation at 10,000 rpm for 10 min. Protein concentration was measured by BCA analysis, and 30 μg of total proteins from cell lysates was diluted in Laemmli buffer and loaded on a stacking gel. The top of the front gel was excised, reduced with DTT, alkylated with iodoacetamide, and in-gel digested with trypsin. Peptides were extracted with 50 mM ammonium bicarbonate and 50% acetonitrile in 0.2% formic acid, dried by evaporation in a speed-vac concentrator, and resuspended in 60 μL of 0.2% formic acid. Samples were spiked with six heavy isotopic AQUA standards peptides at 10 fmol each (Thermo Fisher Scientific). All samples were separated into two aliquots, one analyzed in Data Dependent Acquisition (DDA) mode LC/MS analysis for global proteome differential analysis and the other one subjected to targeted LC/MS analysis for TEAD1, TEAD2, TEAD3, and TEAD4 expression level determination and target occupancy ratio (TOR) measurement. Analyses were performed using a nano-ACQUITY Ultra-Performance LC system (Waters, Milford, MA, USA) coupled to an Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific). LC separation was performed with a trapping column (nano-Acquity Symmetry C18, 100 Å, 5 μm, 180 μm × 20 mm) at 15 μL/min flow rate and an analytical column (nano-Acquity BEH C18, 130 Å, 1.7 μm, 75 μm × 250 mm) directly coupled to the ion source. The mobile phases for LC separation were 0.2% (
v/
v) formic acid in LC-MS grade water (solvent A) and 0.2% (
v/
v) formic acid in acetonitrile (solvent B). Peptides were separated at a 300 nl/min constant flow rate with a linear gradient of 5–85% solvent B for 120 min for global proteome analysis and 35 min for targeted LC/MS. For DDA experiments, a full MS1 survey scan was acquired in the Orbitrap for
m/
z 325–1200. The resolution was set to 120 k at
m/
z 200. Fragmentation was performed in the HCD fragmentation cell (collision energy at 26%), isolating precursor ions in the quadrupole. Target ions previously selected for fragmentation were dynamically excluded for 50 s with a relative mass window of ±10 ppm. TOR experiments were performed as recently described [
37].
4.9. Data Processing for Proteomics Analysis and Pathway Analysis
DDA data were processed with the MaxQuant software (Ver. 1.6.17.0, Max-Planck Institute of Biochemistry, Department of Proteomics and Signal Transduction, Munich, Germany). Database searching was performed against the human FASTA database downloaded from UniProtKB/Swiss-Prot. Interrogation of the databank was based on the following criteria: precursor mass tolerance of 7 ppm, fragment ions mass tolerance of 0.6 Da, and 2 maximum missed cleavages with trypsin as the enzyme. Search parameters for post-translational modifications were variable modifications of
N-acetylation on protein
N-terminal residues, oxidation on methionine residues, and pyro-Glu modification on glutamine residues. The matching between runs was also checked. All the other parameters were the MaxQuant default parameters. Protein intensities were exported from the MaxQuant proteinGroups file. Missing values were replaced by the minimum value of each acquisition. LFQ intensities were transformed into their log2 values. Medians were calculated over the technical replicates. A two-tailed
t test for each peptide was performed on the normalized medians to determine the statistical significance between vehicle and treated sample groups, assuming equal variance. The gene lists generated with
p value < 0.01 and absolute fold change (FC) >2, after analysis of variance of the proteomic levels under various treatments, were submitted to Gene Ontology (GO) process and pathway enrichment analyses using Metacore (
https://portal.genego.com/ (accessed on 18 November 2022)).
4.10. Statistical Analysis of Transcriptomic and Proteomic Levels
Differentially expressed genes were identified by one-way analysis of variance of quantile-normalized signals followed by a Scheffe test for multiple comparisons. The SPSS Statistics version 25 (IBM, Armonk, NY, USA) software was used for all statistical computations. Once this selection was made, the data in the different conditions for proteomic and transcriptomic datasets were compared with the data on the TEAD-responsive elements found in the YAP/TAZ/TEAD Chromatin Immunoprecipitation followed by Sequencing (ChIP-Seq) data [
21] in
Supplementary Table S2. Venn diagrams were drawn by Draw Venn Diagram (
http://ugent.be (accessed on 16 August 2022)).
4.11. Protein Extraction and Quantification
NCI-H2030 cell line was seeded in 6-well plates in a complete medium. After 24 h, compounds were added at different dilutions to obtain a final percentage of 0.2% DMSO. After 24 h of treatment, cells were harvested using TrypLE Express (#12604013, Thermo Fisher Scientific) and washed in 1X PBS. Proteins were extracted by treating with RIPA buffer 1X (#89901, Thermo Fisher Scientific) supplemented by protease and phosphatase inhibitors 1X (#78446, Thermo Fisher Scientific) and DNase Basemuncher (#15417954, Thermo Fischer Scientific). Protein concentrations were determined using bicinchoninic acid method (BCA) (#23225, Thermo Fischer Scientific).
4.12. Capillary Western Immunoassay (Jess)
A total protein assay was conducted for data normalization and comparison on the Jess following the manufacturer’s instructions (ProteinSimple, San Jose, CA, USA) with a 12–230 kDa separation module (SM-W004, ProteinSimple). Target proteins were detected with antibodies from Abcam HAUS6 (ab173281), BUBR1 (ab215351), DIAPH3 (ab245660) and from Cell Signaling Technology (CST), Danvers, MA, USA, BUB1 (#5421) RRID:AB_10691315, CDC20 (#14866) RRID:AB_2715567, PLK1 (#4513) RRID:AB_2167409 and CYR61 (#14479) RRID:AB_2798492. The level was normalized to the total protein for each target protein studied, and then, the inhibition rate was calculated vs. DMSO. Three independent experiments were carried out.
4.13. Fluorescence-Activated Single-Cell Sorting (FACS)
The Guava Cell Cycle Reagent determines, by labeling cellular DNA with propidium iodide (PI), the percentage of cells in G0/G1, S, G2/M, and diploid phases based on DNA content. NCI-H2030 cell line was seeded in 6-well plates in a complete medium for 24 h. Then, compounds were added to cells. After 48 h incubation in compounds, cell cycle staining was realized according to Guava Cell Cycle Reagent Package Insert #4500-0220 (Luminex, Austin, TX, USA). Briefly, after harvesting cells with TrypLE Express (# 12604013, Thermo Fisher Scientific), washed in 1X PBS and counted (Vi-Cell XR, Beckman Coulter, Brea, CA, USA), cells were resuspended in polystyrene tube, and ice-cold 70% ethanol was slowly added. After overnight incubation at −20 °C, the equivalent volume of 150,000 cells was centrifuged at 450× g for 5 min and washed once with 1X PBS. On the cell pellet, 200 μL of Guava Cell Cycle reagent was added to each tube and incubated for 30 min at room temperature in the dark. Quantification of the cell cycle was realized with a flow cytometer from Luminex (Guava easyCyte 6HT-2L) and analyzed by CellCycle GuavaSoft 4.5 software. Recording excitation and emission wavelengths were 488 and 785 nm, respectively. Five thousand events were analyzed for each sample. At least three independent experiments were carried out. For data comparisons and statistical analyses, 2-way ANOVA and Tukey’s multiple comparisons test were used (GraphPad Prism v8). A p value < 0.05 was considered statistically significant.
4.14. Incucyte with Cell-by-Cell Detection
NCI-H2030-CC and NCI-H1792-CC cell lines were seeded at 1500 cells/well in 96-well plates in a complete medium for 24 h. The compounds were added at different dilutions to obtain a final percentage of 0.2% DMSO. Cells were then placed in INCUCYTE S5 (Sartorius, Göttingen, Germany). Photos were taken every 24 h at 10×. Data analysis was performed with a cell-by-cell adherent module with the IncuCyte software v. 2022A, as indicated by the supplier’s recommendations.
4.15. Immunofluorescence (IF)
NCI-H2030 cells were seeded at 5000 cells/well on glass coverslips placed into a 24-well plate. After 24 h, cells were then treated with different compounds. After 24 h of treatment, the media were gently aspirated, briefly rinsed with 1X PBS, and incubated with 600 μL of 4% formaldehyde (4 °C) per well for 20 min. The formaldehyde was aspirated, and the cells were rinsed with 1X PBS before a permeabilization step (Triton 0.1% 15 min at RT) and a saturation step with 1% of BSA, 20 min at RT. Immunofluorescence was performed using mouse anti-Tubulin-Alexa_488 antibody (#ab195887, Abcam, Boston, MA, USA) incubated at 4 °C overnight. Cells were rinsed and stained with DAPI before mounting the coverslips (0.17 mm) on a microscope slide. The observation was performed on the VS-120 Olympus scanner using the extended focus microscopy module to capture the cell volume even though the final image is 2D. A mixed model was performed on log-transformed tubulin density with treatment as fixed factor treatment and area into the well as random factor followed by a Dunnett’s test to adjust for the multiplicity of tests. Statistical analyses were performed using SAS® version 9, USA: 5 770. A p value < 0.05 was considered statistically significant.
4.16. Post Translational Modification (PTM) Scan Analysis
NCI-H2030 cells were seeded in a complete medium at 200,000 cells in 500 cm2. After 72 h, compounds were added to the dishes. After 24 h incubation, cell lysis was realized according to the PTM scan urea protocol provided by CST. This experiment was realized three times for each condition. Samples extracts were sent to CST for PTM scan studies characterized by detecting phosphorylated tyrosine, serine, and threonine. Phosphorylated tyrosine (pY) was detected via immunoprecipitation, and phosphorylated serine and threonine were detected via Immobilized Metal Affinity Chromatography (IMAC). PTM scan data were processed by CST. Log2FC was calculated for each peptide expression, comparing each treatment condition against DMSO-vehicle control. Log2FC values were then normalized by subtracting the log2 ratio for each peptide from the median log2 ratio of all peptides within a treatment condition. Only peptides with normalized log2FC > 1 or < −1 were considered in downstream analysis. A single pooled sample (from three biological replicates) was processed per condition; no statistical tests were performed. PTM scan analysis was performed in R v.3.5.3.
4.17. In Vivo Efficacy Study
Female CB17/lcr-Prkdcscid/lcrIcoCrl mice (6–8 weeks old) were bred at Charles River (Les Oncins, Saint-Germain-Nuelles, France), housed in Sanofi AAALAC-accredited animal facilities, and were provided with irradiated food and filtered water ad libitum. All experiments were carried out following the French law and the European Directive 2010/63/EU for the protection of animals used for scientific purposes and with the approval of the ethics committee #21 (project number APAFIS#5644-2016061311593064.V1). In vivo efficacy study of Cpd A was performed in SCID mice inoculated subcutaneously into the right flank with 3 × 106 NCI-H358 cells mixed with Matrigel. Mice bearing around 200 mm3 subcutaneous NCI-H358 tumors were randomly assigned to 5 groups of 8 mice/group and treated every day for 23 consecutive days with either vehicle, Cpd A 25 mg/kg, adagrasib 10 mg/kg or a combination of Cpd A and adagrasib. Tumor perpendicular diameters were measured twice per week with a caliper, and tumor volume (V) was calculated according to the following equation: V (mm3) = (d2 (mm2) × D (mm))/2, where d is the smallest and D the largest perpendicular tumor diameters. A contrast analysis using Bonferroni–Holm correction for multiplicity following a two-way analysis of variance (ANOVA) { XE “Anova” \f Abbreviation \t “analysis of variance” } with factor treatment and day (repeated) was performed on tumor volume changes from baseline, to compare globally and at each day, all treated groups to the control group and all treated groups with the combination group. A probability of less than 5% (p < 0.05) was considered significant. Statistical analysis was performed using EverStat6 software.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









