
The Disruption of NMDAR/TRPM4 Death Signaling with TwinF Interface Inhibitors: A New Pharmacological Principle for Neuroprotection

Abstract
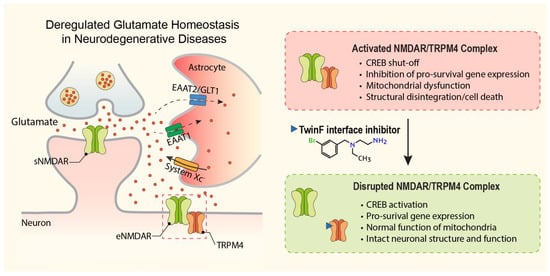
:
1. Introduction
2. Deregulation of Glutamate Homeostasis in Neurodegeneration
3. Glutamate Neurotoxicity and NMDA Receptors
4. Neuroprotectants Targeting Glutamate Neurotoxicity
5. Riluzole
6. Memantine
7. NMDAR Interacting Proteins
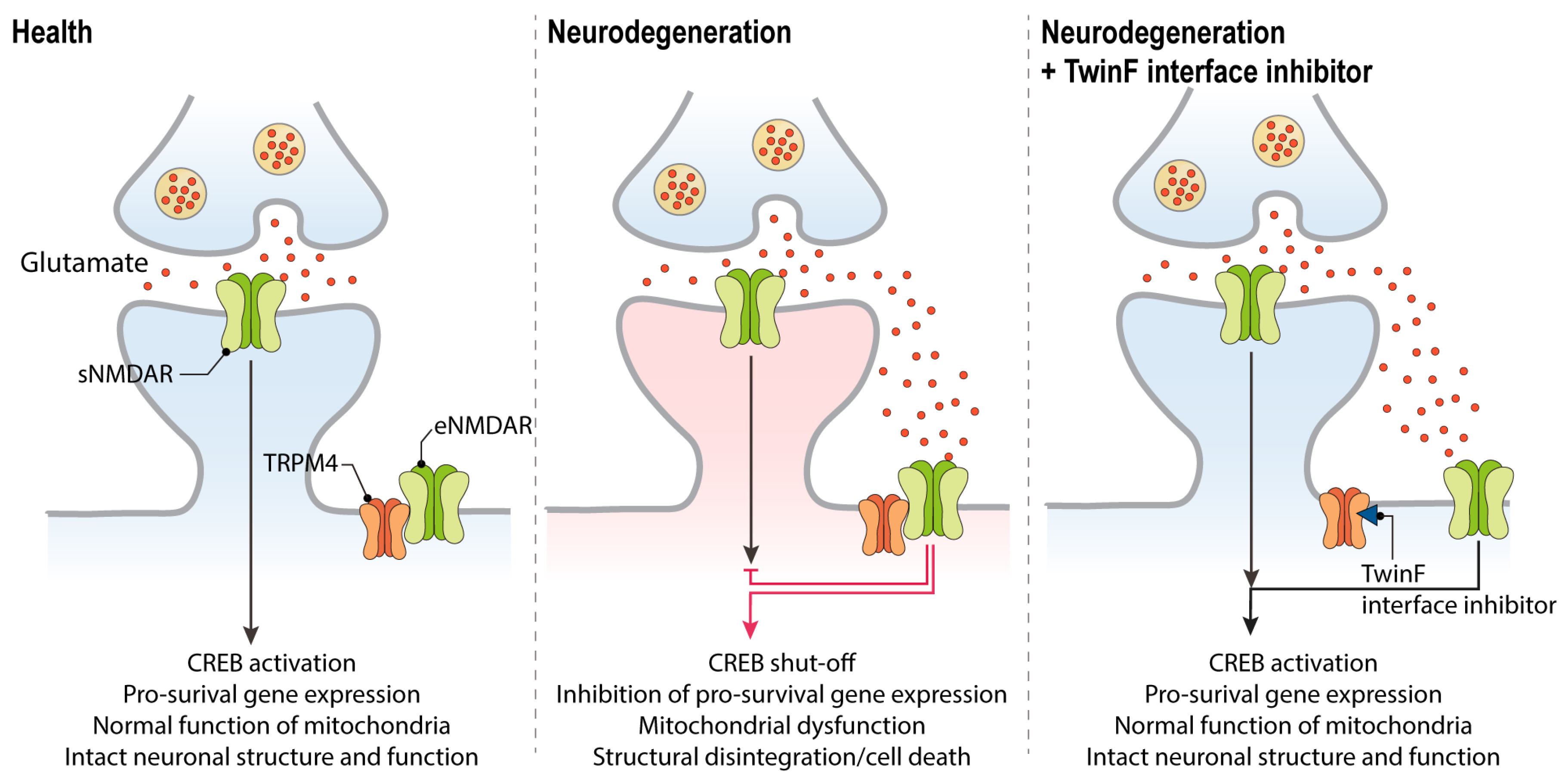
8. The NMDAR/TRPM4 Death Signaling Complex
9. A New Pharmacological Principle in Neuroprotection: Disruption of the NMDAR/TRPM4 Death Signaling Complex
10. TI Inhibitors
11. Wide Range of Possible Therapeutic Applications of TI Inhibitors
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lucas, D.R.; Newhouse, J.P. The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch. Ophthalmol. 1957, 58, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 1969, 164, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Ionic dependence of glutamate neurotoxicity. J. Neurosci. 1987, 7, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef]
- Bading, H. Therapeutic targeting of the pathological triad of extrasynaptic NMDA receptor signaling in neurodegenerations. J. Exp. Med. 2017, 214, 569–578. [Google Scholar] [CrossRef]
- Yan, J.; Bengtson, C.P.; Buchthal, B.; Hagenston, A.M.; Bading, H. Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science 2020, 370, eaay3302. [Google Scholar] [CrossRef]
- Todd, A.C.; Hardingham, G.E. The Regulation of Astrocytic Glutamate Transporters in Health and Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 9607. [Google Scholar] [CrossRef]
- Lin, C.L.; Kong, Q.; Cuny, G.D.; Glicksman, M.A. Glutamate transporter EAAT2: A new target for the treatment of neurodegenerative diseases. Future Med. Chem. 2012, 4, 1689–1700. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef]
- Pitt, D.; Nagelmeier, I.E.; Wilson, H.C.; Raine, C.S. Glutamate uptake by oligodendrocytes: Implications for excitotoxicity in multiple sclerosis. Neurology 2003, 61, 1113–1120. [Google Scholar] [CrossRef]
- Soria, F.N.; Perez-Samartin, A.; Martin, A.; Gona, K.B.; Llop, J.; Szczupak, B.; Chara, J.C.; Matute, C.; Domercq, M. Extrasynaptic glutamate release through cystine/glutamate antiporter contributes to ischemic damage. J. Clin. Investig. 2014, 124, 3645–3655. [Google Scholar] [CrossRef]
- Mead, R.J.; Shan, N.; Reiser, H.J.; Marshall, F.; Shaw, P.J. Amyotrophic lateral sclerosis: A neurodegenerative disorder poised for successful therapeutic translation. Nat. Rev. Drug Discov. 2023, 22, 185–212. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Morris, R.G. NMDA receptors and memory encoding. Neuropharmacology 2013, 74, 32–40. [Google Scholar] [CrossRef]
- Bading, H. Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci. 2013, 14, 593–608. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci 2003, 26, 81–89. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Turski, L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002, 1, 383–386. [Google Scholar] [CrossRef]
- Ogden, K.K.; Traynelis, S.F. New advances in NMDA receptor pharmacology. Trends Pharmacol. Sci. 2011, 32, 726–733. [Google Scholar] [CrossRef]
- Williams, K. Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: Selectivity and mechanisms at recombinant heteromeric receptors. Mol. Pharmacol. 1993, 44, 851–859. [Google Scholar]
- Gotti, B.; Duverger, D.; Bertin, J.; Carter, C.; Dupont, R.; Frost, J.; Gaudilliere, B.; MacKenzie, E.T.; Rousseau, J.; Scatton, B.; et al. Ifenprodil and SL 82.0715 as cerebral anti-ischemic agents. I. Evidence for efficacy in models of focal cerebral ischemia. J. Pharmacol. Exp. Ther. 1988, 247, 1211–1221. [Google Scholar]
- Mishra, V.; Verma, R.; Singh, N.; Raghubir, R. The neuroprotective effects of NMDAR antagonist, ifenprodil and ASIC1a inhibitor, flurbiprofen on post-ischemic cerebral injury. Brain Res. 2011, 1389, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Muir, K.W.; Lees, K.R. Excitatory amino acid antagonists for acute stroke. Cochrane Database Syst. Rev. 2003, 2003, CD001244. [Google Scholar] [CrossRef] [PubMed]
- McCool, B.A.; Lovinger, D.M. Ifenprodil inhibition of the 5-hydroxytryptamine3 receptor. Neuropharmacology 1995, 34, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Karbon, E.W.; Patch, R.J.; Pontecorvo, M.J.; Ferkany, J.W. Ifenprodil potently interacts with [3H](+)-3-PPP-labeled sigma binding sites in guinea pig brain membranes. Eur. J. Pharmacol. 1990, 176, 247–248. [Google Scholar] [CrossRef] [PubMed]
- Church, J.; Fletcher, E.J.; Baxter, K.; MacDonald, J.F. Blockade by ifenprodil of high voltage-activated Ca2+ channels in rat and mouse cultured hippocampal pyramidal neurones: Comparison with N-methyl-D-aspartate receptor antagonist actions. Br. J. Pharmacol. 1994, 113, 499–507. [Google Scholar] [CrossRef]
- Bath, C.P.; Farrell, L.N.; Gilmore, J.; Ward, M.A.; Hicks, C.A.; O’Neill, M.J.; Bleakman, D. The effects of ifenprodil and eliprodil on voltage-dependent Ca2+ channels and in gerbil global cerebral ischaemia. Eur. J. Pharmacol. 1996, 299, 103–112. [Google Scholar] [CrossRef]
- Kundrotiene, J.; Cebers, G.; Wagner, A.; Liljequist, S. The NMDA NR2B subunit-selective receptor antagonist, CP-101,606, enhances the functional recovery the NMDA NR2B subunit-selective receptor and reduces brain damage after cortical compression-induced brain ischemia. J. Neurotrauma 2004, 21, 83–93. [Google Scholar] [CrossRef]
- Tsuchida, E.; Rice, M.; Bullock, R. The neuroprotective effect of the forebrain-selective NMDA antagonist CP101,606 upon focal ischemic brain damage caused by acute subdural hematoma in the rat. J. Neurotrauma 1997, 14, 409–417. [Google Scholar] [CrossRef]
- Di, X.; Bullock, R.; Watson, J.; Fatouros, P.; Chenard, B.; White, F.; Corwin, F. Effect of CP101,606, a novel NR2B subunit antagonist of the N-methyl-D-aspartate receptor, on the volume of ischemic brain damage off cytotoxic brain edema after middle cerebral artery occlusion in the feline brain. Stroke 1997, 28, 2244–2251. [Google Scholar] [CrossRef]
- Merchant, R.E.; Bullock, M.R.; Carmack, C.A.; Shah, A.K.; Wilner, K.D.; Ko, G.; Williams, S.A. A double-blind, placebo-controlled study of the safety, tolerability and pharmacokinetics of CP-101,606 in patients with a mild or moderate traumatic brain injury. Ann. N. Y. Acad. Sci. 1999, 890, 42–50. [Google Scholar] [CrossRef]
- Bullock, M.R.; Merchant, R.E.; Carmack, C.A.; Doppenberg, E.; Shah, A.K.; Wilner, K.D.; Ko, G.; Williams, S.A. An open-label study of CP-101,606 in subjects with a severe traumatic head injury or spontaneous intracerebral hemorrhage. Ann. N. Y. Acad. Sci. 1999, 890, 51–58. [Google Scholar] [CrossRef]
- Machado-Vieira, R.; Henter, I.D.; Zarate, C.A., Jr. New targets for rapid antidepressant action. Prog. Neurobiol. 2017, 152, 21–37. [Google Scholar] [CrossRef]
- Raymond, L.A.; Moshaver, A.; Tingley, W.G.; Huganir, R.L. Glutamate receptor ion channel properties predict vulnerability to cytotoxicity in a transfected nonneuronal cell line. Mol. Cell. Neurosci. 1996, 7, 102–115. [Google Scholar] [CrossRef]
- Anegawa, N.J.; Lynch, D.R.; Verdoorn, T.A.; Pritchett, D.B. Transfection of N-methyl-D-aspartate receptors in a nonneuronal cell line leads to cell death. J. Neurochem. 1995, 64, 2004–2012. [Google Scholar] [CrossRef]
- Zhou, X.; Ding, Q.; Chen, Z.; Yun, H.; Wang, H. Involvement of the GluN2A and GluN2B subunits in synaptic and extrasynaptic N-methyl-D-aspartate receptor function and neuronal excitotoxicity. J. Biol. Chem. 2013, 288, 24151–24159. [Google Scholar] [CrossRef]
- Von Engelhardt, J.; Coserea, I.; Pawlak, V.; Fuchs, E.C.; Kohr, G.; Seeburg, P.H.; Monyer, H. Excitotoxicity in vitro by NR2A- and NR2B-containing NMDA receptors. Neuropharmacology 2007, 53, 10–17. [Google Scholar] [CrossRef]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef]
- Mizoule, J.; Meldrum, B.; Mazadier, M.; Croucher, M.; Ollat, C.; Uzan, A.; Legrand, J.J.; Gueremy, C.; Le Fur, G. 2-Amino-6-trifluoromethoxy benzothiazole, a possible antagonist of excitatory amino acid neurotransmission--I. Anticonvulsant properties. Neuropharmacology 1985, 24, 767–773. [Google Scholar] [CrossRef]
- Benavides, J.; Camelin, J.C.; Mitrani, N.; Flamand, F.; Uzan, A.; Legrand, J.J.; Gueremy, C.; Le Fur, G. 2-Amino-6-trifluoromethoxy benzothiazole, a possible antagonist of excitatory amino acid neurotransmission--II. Biochemical properties. Neuropharmacology 1985, 24, 1085–1092. [Google Scholar] [CrossRef]
- Martin, D.; Thompson, M.A.; Nadler, J.V. The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur. J. Pharmacol. 1993, 250, 473–476. [Google Scholar] [CrossRef]
- Azbill, R.D.; Mu, X.; Springer, J.E. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res. 2000, 871, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, J.; Beal McIlvain, H.; She, Y.; Howland, D.S. Impaired spinal cord glutamate transport capacity and reduced sensitivity to riluzole in a transgenic superoxide dismutase mutant rat model of amyotrophic lateral sclerosis. J. Neurosci. 2003, 23, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, E.; Funicello, M.; Rauen, T.; Gobbi, M.; Mennini, T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur. J. Pharmacol. 2008, 578, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Debono, M.W.; Le Guern, J.; Canton, T.; Doble, A.; Pradier, L. Inhibition by riluzole of electrophysiological responses mediated by rat kainate and NMDA receptors expressed in Xenopus oocytes. Eur. J. Pharmacol. 1993, 235, 283–289. [Google Scholar] [CrossRef]
- Deflorio, C.; Palma, E.; Conti, L.; Roseti, C.; Manteca, A.; Giacomelli, E.; Catalano, M.; Limatola, C.; Inghilleri, M.; Grassi, F. Riluzole blocks human muscle acetylcholine receptors. J. Physiol. 2012, 590, 2519–2528. [Google Scholar] [CrossRef]
- Palma, E.; Inghilleri, M.; Conti, L.; Deflorio, C.; Frasca, V.; Manteca, A.; Pichiorri, F.; Roseti, C.; Torchia, G.; Limatola, C.; et al. Physiological characterization of human muscle acetylcholine receptors from ALS patients. Proc. Natl. Acad. Sci. USA 2011, 108, 20184–20188. [Google Scholar] [CrossRef]
- Song, J.H.; Huang, C.S.; Nagata, K.; Yeh, J.Z.; Narahashi, T. Differential action of riluzole on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels. J. Pharmacol. Exp. Ther. 1997, 282, 707–714. [Google Scholar]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Miller, R.G.; Bouchard, J.P.; Duquette, P.; Eisen, A.; Gelinas, D.; Harati, Y.; Munsat, T.L.; Powe, L.; Rothstein, J.; Salzman, P.; et al. Clinical trials of riluzole in patients with ALS. ALS/Riluzole Study Group-II. Neurology 1996, 47, S86–S92; discussion S90–S92. [Google Scholar] [CrossRef]
- Lacomblez, L.; Bensimon, G.; Leigh, P.N.; Guillet, P.; Meininger, V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 1996, 347, 1425–1431. [Google Scholar] [CrossRef]
- Schiefer, J.; Landwehrmeyer, G.B.; Luesse, H.G.; Sprunken, A.; Puls, C.; Milkereit, A.; Milkereit, E.; Kosinski, C.M. Riluzole prolongs survival time and alters nuclear inclusion formation in a transgenic mouse model of Huntington’s disease. Mov. Disord. 2002, 17, 748–757. [Google Scholar] [CrossRef]
- Landwehrmeyer, G.B.; Dubois, B.; de Yebenes, J.G.; Kremer, B.; Gaus, W.; Kraus, P.H.; Przuntek, H.; Dib, M.; Doble, A.; Fischer, W.; et al. Riluzole in Huntington’s disease: A 3-year, randomized controlled study. Ann. Neurol. 2007, 62, 262–272. [Google Scholar] [CrossRef]
- Barneoud, P.; Mazadier, M.; Miquet, J.M.; Parmentier, S.; Dubedat, P.; Doble, A.; Boireau, A. Neuroprotective effects of riluzole on a model of Parkinson’s disease in the rat. Neuroscience 1996, 74, 971–983. [Google Scholar] [CrossRef]
- Carbone, M.; Duty, S.; Rattray, M. Riluzole neuroprotection in a Parkinson’s disease model involves suppression of reactive astrocytosis but not GLT-1 regulation. BMC Neurosci. 2012, 13, 38. [Google Scholar] [CrossRef]
- Bensimon, G.; Ludolph, A.; Agid, Y.; Vidailhet, M.; Payan, C.; Leigh, P.N.; Group, N.S. Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: The NNIPPS study. Brain 2009, 132, 156–171. [Google Scholar] [CrossRef]
- Hunsberger, H.C.; Weitzner, D.S.; Rudy, C.C.; Hickman, J.E.; Libell, E.M.; Speer, R.R.; Gerhardt, G.A.; Reed, M.N. Riluzole rescues glutamate alterations, cognitive deficits, and tau pathology associated with P301L tau expression. J. Neurochem. 2015, 135, 381–394. [Google Scholar] [CrossRef]
- Okamoto, M.; Gray, J.D.; Larson, C.S.; Kazim, S.F.; Soya, H.; McEwen, B.S.; Pereira, A.C. Riluzole reduces amyloid beta pathology, improves memory, and restores gene expression changes in a transgenic mouse model of early-onset Alzheimer’s disease. Transl. Psychiatry 2018, 8, 153. [Google Scholar] [CrossRef]
- Lesuis, S.L.; Kaplick, P.M.; Lucassen, P.J.; Krugers, H.J. Treatment with the glutamate modulator riluzole prevents early life stress-induced cognitive deficits and impairments in synaptic plasticity in APPswe/PS1dE9 mice. Neuropharmacology 2019, 150, 175–183. [Google Scholar] [CrossRef]
- Hascup, K.N.; Findley, C.A.; Britz, J.; Esperant-Hilaire, N.; Broderick, S.O.; Delfino, K.; Tischkau, S.; Bartke, A.; Hascup, E.R. Riluzole attenuates glutamatergic tone and cognitive decline in AbetaPP/PS1 mice. J. Neurochem. 2021, 156, 513–523. [Google Scholar] [CrossRef]
- Saba, K.; Patel, A.B. Riluzole restores memory and brain energy metabolism in AbetaPP-PS1 mouse model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2022, 610, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.C.; Gray, J.D.; Kogan, J.F.; Davidson, R.L.; Rubin, T.G.; Okamoto, M.; Morrison, J.H.; McEwen, B.S. Age and Alzheimer’s disease gene expression profiles reversed by the glutamate modulator riluzole. Mol. Psychiatry 2017, 22, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.C.; Mao, X.; Dowd, K.; Tsakanikas, D.; Jiang, C.S.; Meuser, C.; Andrews, R.D.; Lukic, A.S.; Lee, J.; Hampilos, N.; et al. Riluzole, a glutamate modulator, slows cerebral glucose metabolism decline in patients with Alzheimer’s disease. Brain 2021, 144, 3742–3755. [Google Scholar] [CrossRef] [PubMed]
- Mottet, I.; Demeure, R.; Rataud, J.; Lucas, M.; Wahl, F.; Warscotte, V.; Thiran, J.P.; Goudemant, J.F.; Maldague, B.; Maloteaux, J.M.; et al. Effects of riluzole on the evolution of focal cerebral ischemia: A magnetic resonance imaging study. Magn. Reson. Mater. Phys. Biol. Med. 1997, 5, 185–191. [Google Scholar] [CrossRef]
- Heurteaux, C.; Laigle, C.; Blondeau, N.; Jarretou, G.; Lazdunski, M. Alpha-linolenic acid and riluzole treatment confer cerebral protection and improve survival after focal brain ischemia. Neuroscience 2006, 137, 241–251. [Google Scholar] [CrossRef]
- Pirhan, D.; Yuksel, N.; Emre, E.; Cengiz, A.; Kursat Yildiz, D. Riluzole- and Resveratrol-Induced Delay of Retinal Ganglion Cell Death in an Experimental Model of Glaucoma. Curr. Eye Res. 2016, 41, 59–69. [Google Scholar] [CrossRef]
- Verma, S.K.; Arora, I.; Javed, K.; Akhtar, M.; Samim, M. Enhancement in the Neuroprotective Power of Riluzole Against Cerebral Ischemia Using a Brain Targeted Drug Delivery Vehicle. ACS Appl. Mater. Interfaces 2016, 8, 19716–19723. [Google Scholar] [CrossRef]
- Gerzon, K.; Krumkalns, E.V.; Brindle, R.L.; Marshall, F.J.; Root, M.A. The Adamantyl Group in Medicinal Agents. I. Hypoglycemic N-Arylsulfonyl-N’-Adamantylureas. J. Med. Chem. 1963, 6, 760–763. [Google Scholar] [CrossRef]
- Bormann, J. Memantine is a potent blocker of N-methyl-D-aspartate (NMDA) receptor channels. Eur. J. Pharmacol. 1989, 166, 591–592. [Google Scholar] [CrossRef]
- Parsons, C.G.; Danysz, W.; Quack, G. Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist--a review of preclinical data. Neuropharmacology 1999, 38, 735–767. [Google Scholar] [CrossRef]
- Seeman, P.; Caruso, C.; Lasaga, M. Memantine agonist action at dopamine D2High receptors. Synapse 2008, 62, 149–153. [Google Scholar] [CrossRef]
- Aracava, Y.; Pereira, E.F.; Maelicke, A.; Albuquerque, E.X. Memantine blocks alpha7* nicotinic acetylcholine receptors more potently than n-methyl-D-aspartate receptors in rat hippocampal neurons. J. Pharmacol. Exp. Ther. 2005, 312, 1195–1205. [Google Scholar] [CrossRef]
- Peeters, M.; Romieu, P.; Maurice, T.; Su, T.P.; Maloteaux, J.M.; Hermans, E. Involvement of the sigma 1 receptor in the modulation of dopaminergic transmission by amantadine. Eur. J. Neurosci. 2004, 19, 2212–2220. [Google Scholar] [CrossRef]
- Rammes, G.; Rupprecht, R.; Ferrari, U.; Zieglgansberger, W.; Parsons, C.G. The N-methyl-D-aspartate receptor channel blockers memantine, MRZ 2/579 and other amino-alkyl-cyclohexanes antagonise 5-HT(3) receptor currents in cultured HEK-293 and N1E-115 cell systems in a non-competitive manner. Neurosci. Lett. 2001, 306, 81–84. [Google Scholar] [CrossRef]
- Reiser, G.; Binmoller, F.J.; Koch, R. Memantine (1-amino-3,5-dimethyladamantane) blocks the serotonin-induced depolarization response in a neuronal cell line. Brain Res. 1988, 443, 338–344. [Google Scholar] [CrossRef]
- Kilpatrick, G.J.; Tilbrook, G.S. Memantine. Merz. Curr. Opin. Investig. Drugs 2002, 3, 798–806. [Google Scholar]
- Reisberg, B.; Doody, R.; Stoffler, A.; Schmitt, F.; Ferris, S.; Mobius, H.J.; Memantine Study Group. Memantine in moderate-to-severe Alzheimer’s disease. N. Engl. J. Med. 2003, 348, 1333–1341. [Google Scholar] [CrossRef]
- Cummings, J.L.; Schneider, E.; Tariot, P.N.; Graham, S.M.; Memantine MEM-MD-02 Study Group. Behavioral effects of memantine in Alzheimer disease patients receiving donepezil treatment. Neurology 2006, 67, 57–63. [Google Scholar] [CrossRef]
- Tariot, P.N.; Farlow, M.R.; Grossberg, G.T.; Graham, S.M.; McDonald, S.; Gergel, I.; Memantine Study Group. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: A randomized controlled trial. JAMA 2004, 291, 317–324. [Google Scholar] [CrossRef]
- Howard, R.; McShane, R.; Lindesay, J.; Ritchie, C.; Baldwin, A.; Barber, R.; Burns, A.; Dening, T.; Findlay, D.; Holmes, C.; et al. Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N. Engl. J. Med. 2012, 366, 893–903. [Google Scholar] [CrossRef]
- Milnerwood, A.J.; Gladding, C.M.; Pouladi, M.A.; Kaufman, A.M.; Hines, R.M.; Boyd, J.D.; Ko, R.W.; Vasuta, O.C.; Graham, R.K.; Hayden, M.R.; et al. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron 2010, 65, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Beister, A.; Kraus, P.; Kuhn, W.; Dose, M.; Weindl, A.; Gerlach, M. The N-methyl-D-aspartate antagonist memantine retards progression of Huntington’s disease. J. Neural Transmission. Suppl. 2004, 68, 117–122. [Google Scholar] [CrossRef]
- Ondo, W.G.; Mejia, N.I.; Hunter, C.B. A pilot study of the clinical efficacy and safety of memantine for Huntington’s disease. Park. Relat. Disord. 2007, 13, 453–454. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.; Guo, L.; Cordeiro, M.F. Neuroprotection in glaucoma: Drug-based approaches. Optom. Vis. Sci. 2008, 85, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Hare, W.; WoldeMussie, E.; Lai, R.; Ton, H.; Ruiz, G.; Feldmann, B.; Wijono, M.; Chun, T.; Wheeler, L. Efficacy and safety of memantine, an NMDA-type open-channel blocker, for reduction of retinal injury associated with experimental glaucoma in rat and monkey. Surv. Ophthalmol. 2001, 45 (Suppl. 3), S284–S289; discussion S286–S295. [Google Scholar] [CrossRef]
- Sanchez-Lopez, E.; Egea, M.A.; Davis, B.M.; Guo, L.; Espina, M.; Silva, A.M.; Calpena, A.C.; Souto, E.M.B.; Ravindran, N.; Ettcheto, M.; et al. Memantine-Loaded PEGylated Biodegradable Nanoparticles for the Treatment of Glaucoma. Small 2018, 14, 1701808. [Google Scholar] [CrossRef]
- Weinreb, R.N.; Liebmann, J.M.; Cioffi, G.A.; Goldberg, I.; Brandt, J.D.; Johnson, C.A.; Zangwill, L.M.; Schneider, S.; Badger, H.; Bejanian, M. Oral Memantine for the Treatment of Glaucoma: Design and Results of 2 Randomized, Placebo-Controlled, Phase 3 Studies. Ophthalmology 2018, 125, 1874–1885. [Google Scholar] [CrossRef]
- Howell, G.R.; Libby, R.T.; Jakobs, T.C.; Smith, R.S.; Phalan, F.C.; Barter, J.W.; Barbay, J.M.; Marchant, J.K.; Mahesh, N.; Porciatti, V.; et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J. Cell Biol. 2007, 179, 1523–1537. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, D. Memantine prolongs survival in an amyotrophic lateral sclerosis mouse model. Eur. J. Neurosci. 2005, 22, 2376–2380. [Google Scholar] [CrossRef]
- De Carvalho, M.; Pinto, S.; Costa, J.; Evangelista, T.; Ohana, B.; Pinto, A. A randomized, placebo-controlled trial of memantine for functional disability in amyotrophic lateral sclerosis. Amyotroph. Lateral. Scler. 2010, 11, 456–460. [Google Scholar] [CrossRef]
- Villoslada, P.; Arrondo, G.; Sepulcre, J.; Alegre, M.; Artieda, J. Memantine induces reversible neurologic impairment in patients with MS. Neurology 2009, 72, 1630–1633. [Google Scholar] [CrossRef]
- Rosas, H.D.; Koroshetz, W.J.; Jenkins, B.G.; Chen, Y.I.; Hayden, D.L.; Beal, M.F.; Cudkowicz, M.E. Riluzole therapy in Huntington’s disease (HD). Mov. Disord. 1999, 14, 326–330. [Google Scholar] [CrossRef]
- Seppi, K.; Mueller, J.; Bodner, T.; Brandauer, E.; Benke, T.; Weirich-Schwaiger, H.; Poewe, W.; Wenning, G.K. Riluzole in Huntington’s disease (HD): An open label study with one year follow up. J. Neurol. 2001, 248, 866–869. [Google Scholar] [CrossRef]
- Huntington Study, G. Dosage effects of riluzole in Huntington’s disease: A multicenter placebo-controlled study. Neurology 2003, 61, 1551–1556. [Google Scholar] [CrossRef]
- Chataway, J.; De Angelis, F.; Connick, P.; Parker, R.A.; Plantone, D.; Doshi, A.; John, N.; Stutters, J.; MacManus, D.; Prados Carrasco, F.; et al. Efficacy of three neuroprotective drugs in secondary progressive multiple sclerosis (MS-SMART): A phase 2b, multiarm, double-blind, randomised placebo-controlled trial. Lancet Neurol. 2020, 19, 214–225. [Google Scholar] [CrossRef]
- Husi, H.; Ward, M.A.; Choudhary, J.S.; Blackstock, W.P.; Grant, S.G. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 2000, 3, 661–669. [Google Scholar] [CrossRef]
- Kornau, H.C.; Schenker, L.T.; Kennedy, M.B.; Seeburg, P.H. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 1995, 269, 1737–1740. [Google Scholar] [CrossRef]
- Sattler, R.; Xiong, Z.; Lu, W.Y.; Hafner, M.; MacDonald, J.F.; Tymianski, M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 1999, 284, 1845–1848. [Google Scholar] [CrossRef]
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.T.; Salter, M.W.; Tymianski, M. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science 2002, 298, 846–850. [Google Scholar] [CrossRef]
- Cook, D.J.; Teves, L.; Tymianski, M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature 2012, 483, 213–217. [Google Scholar] [CrossRef]
- Tu, W.; Xu, X.; Peng, L.; Zhong, X.; Zhang, W.; Soundarapandian, M.M.; Balel, C.; Wang, M.; Jia, N.; Zhang, W.; et al. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell 2010, 140, 222–234. [Google Scholar] [CrossRef] [PubMed]
- McQueen, J.; Ryan, T.J.; McKay, S.; Marwick, K.; Baxter, P.; Carpanini, S.M.; Wishart, T.M.; Gillingwater, T.H.; Manson, J.C.; Wyllie, D.J.A.; et al. Pro-death NMDA receptor signaling is promoted by the GluN2B C-terminus independently of Dapk1. eLife 2017, 6, e17161. [Google Scholar] [CrossRef] [PubMed]
- Zong, P.; Feng, J.; Yue, Z.; Li, Y.; Wu, G.; Sun, B.; He, Y.; Miller, B.; Yu, A.S.; Su, Z.; et al. Functional coupling of TRPM2 and extrasynaptic NMDARs exacerbates excitotoxicity in ischemic brain injury. Neuron 2022, 110, 1944–1958.e8. [Google Scholar] [CrossRef] [PubMed]
- Jones, S. A new villain in neuronal death. Science 2020, 370, 168–169. [Google Scholar] [CrossRef]
- Launay, P.; Fleig, A.; Perraud, A.L.; Scharenberg, A.M.; Penner, R.; Kinet, J.P. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 2002, 109, 397–407. [Google Scholar] [CrossRef]
- Fliegert, R.; Glassmeier, G.; Schmid, F.; Cornils, K.; Genisyuerek, S.; Harneit, A.; Schwarz, J.R.; Guse, A.H. Modulation of Ca2+ entry and plasma membrane potential by human TRPM4b. FEBS J. 2007, 274, 704–713. [Google Scholar] [CrossRef]
- Schattling, B.; Steinbach, K.; Thies, E.; Kruse, M.; Menigoz, A.; Ufer, F.; Flockerzi, V.; Bruck, W.; Pongs, O.; Vennekens, R.; et al. TRPM4 cation channel mediates axonal and neuronal degeneration in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2012, 18, 1805–1811. [Google Scholar] [CrossRef]
- Kurland, D.B.; Tosun, C.; Pampori, A.; Karimy, J.K.; Caffes, N.M.; Gerzanich, V.; Simard, J.M. Glibenclamide for the treatment of acute CNS injury. Pharmaceuticals 2013, 6, 1287–1303. [Google Scholar] [CrossRef]
- Tosun, C.; Kurland, D.B.; Mehta, R.; Castellani, R.J.; deJong, J.L.; Kwon, M.S.; Woo, S.K.; Gerzanich, V.; Simard, J.M. Inhibition of the Sur1-Trpm4 channel reduces neuroinflammation and cognitive impairment in subarachnoid hemorrhage. Stroke 2013, 44, 3522–3528. [Google Scholar] [CrossRef]
- Khanna, A.; Walcott, B.P.; Kahle, K.T.; Simard, J.M. Effect of glibenclamide on the prevention of secondary brain injury following ischemic stroke in humans. Neurosurg. Focus 2014, 36, E11. [Google Scholar] [CrossRef]
- Loh, K.P.; Ng, G.; Yu, C.Y.; Fhu, C.K.; Yu, D.; Vennekens, R.; Nilius, B.; Soong, T.W.; Liao, P. TRPM4 inhibition promotes angiogenesis after ischemic stroke. Pflugers Arch. 2014, 466, 563–576. [Google Scholar] [CrossRef]
- Simard, J.M.; Sheth, K.N.; Kimberly, W.T.; Stern, B.J.; del Zoppo, G.J.; Jacobson, S.; Gerzanich, V. Glibenclamide in cerebral ischemia and stroke. Neurocrit. Care 2014, 20, 319–333. [Google Scholar] [CrossRef]
- Sheth, K.N.; Simard, J.M.; Elm, J.; Kronenberg, G.; Kunte, H.; Kimberly, W.T. Human Data Supporting Glyburide in Ischemic Stroke. Acta Neurochir. Suppl. 2016, 121, 13–18. [Google Scholar] [CrossRef]
- Jiang, B.; Li, L.; Chen, Q.; Tao, Y.; Yang, L.; Zhang, B.; Zhang, J.H.; Feng, H.; Chen, Z.; Tang, J.; et al. Role of Glibenclamide in Brain Injury After Intracerebral Hemorrhage. Transl. Stroke Res. 2017, 8, 183–193. [Google Scholar] [CrossRef]
- Zhang, S.J.; Zou, M.; Lu, L.; Lau, D.; Ditzel, D.A.; Delucinge-Vivier, C.; Aso, Y.; Descombes, P.; Bading, H. Nuclear calcium signaling controls expression of a large gene pool: Identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet. 2009, 5, e1000604. [Google Scholar] [CrossRef]
- Inquimbert, P.; Moll, M.; Latremoliere, A.; Tong, C.K.; Whang, J.; Sheehan, G.F.; Smith, B.M.; Korb, E.; Athie, M.C.P.; Babaniyi, O.; et al. NMDA Receptor Activation Underlies the Loss of Spinal Dorsal Horn Neurons and the Transition to Persistent Pain after Peripheral Nerve Injury. Cell Rep. 2018, 23, 2678–2689. [Google Scholar] [CrossRef]
- Trudler, D.; Sanz-Blasco, S.; Eisele, Y.S.; Ghatak, S.; Bodhinathan, K.; Akhtar, M.W.; Lynch, W.P.; Pina-Crespo, J.C.; Talantova, M.; Kelly, J.W.; et al. alpha-Synuclein Oligomers Induce Glutamate Release from Astrocytes and Excessive Extrasynaptic NMDAR Activity in Neurons, Thus Contributing to Synapse Loss. J. Neurosci. 2021, 41, 2264–2273. [Google Scholar] [CrossRef]
- Takahashi, M.; Billups, B.; Rossi, D.; Sarantis, M.; Hamann, M.; Attwell, D. The role of glutamate transporters in glutamate homeostasis in the brain. J. Exp. Biol. 1997, 200, 401–409. [Google Scholar] [CrossRef]
- Rossi, D.J.; Oshima, T.; Attwell, D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 2000, 403, 316–321. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Patients Complete/Total | Details | Evaluation (Primary Endpoint) | Dose | Phase | Outcome | Ref. | |
|---|---|---|---|---|---|---|---|---|
| RILUZOLE | ALS | 155/155 | Randomized, placebo-controlled, double-blind, 1-year trial | Survival and Norris scales | 100 mg o.d. | N.D. | Positive | [49] |
| ALS | 959/959 | Randomized, placebo-controlled, double-blind, 12- or 18-month trial | Survival | 50, 100, or 200 mg. o.d. | N.D. | Positive | [51] | |
| AD | 42/50 | Randomized, placebo-controlled, double-blind, 6-month trial | Cerebral glucose metabolism and NAA | 50 mg b.i.d. | Phase 2 NCT01703117 | Positive | [63] | |
| HD | 8/8 | Open-label, 6-week trial | UHDRS | 50 mg b.i.d. | N.D. | Negative | [92] | |
| HD | 7/9 | Open-label, 12-month trial | UHDRS | 50 mg b.i.d. | N.D. | Benefits at 3 months, but no longer at 12 months | [93] | |
| HD | 56/63 | Randomized, placebo-controlled, double-blind, 8-week trial | UHDRS | 100 or 200 mg o.d. | N.D. | Negative | [94] | |
| HD | 379/537 | Randomized, placebo-controlled, double-blind, 3-year trial | UHDRS | 50 mg b.i.d. | Phase 3 NCT00277602 | Negative | [53] | |
| PPS | 760/767 | Randomized, placebo-controlled, double-blind, 3-year trial | Survival | 50–200 mg o.d. | Phase 3 NCT00211224 | Negative | [56] | |
| MS | 198/223 | Randomized, placebo-controlled, double-blind, 96-week trial | Change in brain volume | 50 mg o.d. until week 4, then b.i.d. | Phase 2b NCT01910259 | Negative | [95] | |
| MEMANTINE | AD (moderate to severe) | 181/252 | Randomized, placebo-controlled, double-blind, 28-week trial | CIBIC-Plus ADCS-ADLsev | 20 mg o.d. | N.D. | Positive | [77] |
| Glaucoma | 1877/2298 | Randomized, placebo-controlled, double-blind, 2-year trials | Visual field progression | 10 and 20 mg o.d. | Phase 3 NCT00141882 NCT00168350 | Negative | [87] | |
| HD | 9/12 | Open-label, 3-month pilot study | UHDRS | 5–20 mg o.d. | N.D. | Positive/negative | [83] | |
| HD | 27/27 | Open-label, 2-year pilot study | Clinical assessment | 30 mg *, o.d. | N.D. | Positive | [82] | |
| ALS | 50/63 | Randomized, placebo-controlled, double-blind, 12-month trial | change in ALSFRS | 20 mg o.d. | Phase 2/3 NCT00353665 | Negative | [90] | |
| MS | 19/50 | Randomized, placebo-controlled, double-blind, 12-month trial | Verbal memory | 30 mg o.d. | NCT00638833 | Neurologic symptoms worsened | [91] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, J.; Bading, H. The Disruption of NMDAR/TRPM4 Death Signaling with TwinF Interface Inhibitors: A New Pharmacological Principle for Neuroprotection. Pharmaceuticals 2023, 16, 1085. https://doi.org/10.3390/ph16081085
Yan J, Bading H. The Disruption of NMDAR/TRPM4 Death Signaling with TwinF Interface Inhibitors: A New Pharmacological Principle for Neuroprotection. Pharmaceuticals. 2023; 16(8):1085. https://doi.org/10.3390/ph16081085
Chicago/Turabian StyleYan, Jing, and Hilmar Bading. 2023. "The Disruption of NMDAR/TRPM4 Death Signaling with TwinF Interface Inhibitors: A New Pharmacological Principle for Neuroprotection" Pharmaceuticals 16, no. 8: 1085. https://doi.org/10.3390/ph16081085
APA StyleYan, J., & Bading, H. (2023). The Disruption of NMDAR/TRPM4 Death Signaling with TwinF Interface Inhibitors: A New Pharmacological Principle for Neuroprotection. Pharmaceuticals, 16(8), 1085. https://doi.org/10.3390/ph16081085