


Pentagalloyl Glucose-Targeted Inhibition of P-Glycoprotein and Re-Sensitization of Multidrug-Resistant Leukemic Cells (K562/ADR) to Doxorubicin: In Silico and Functional Studies

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
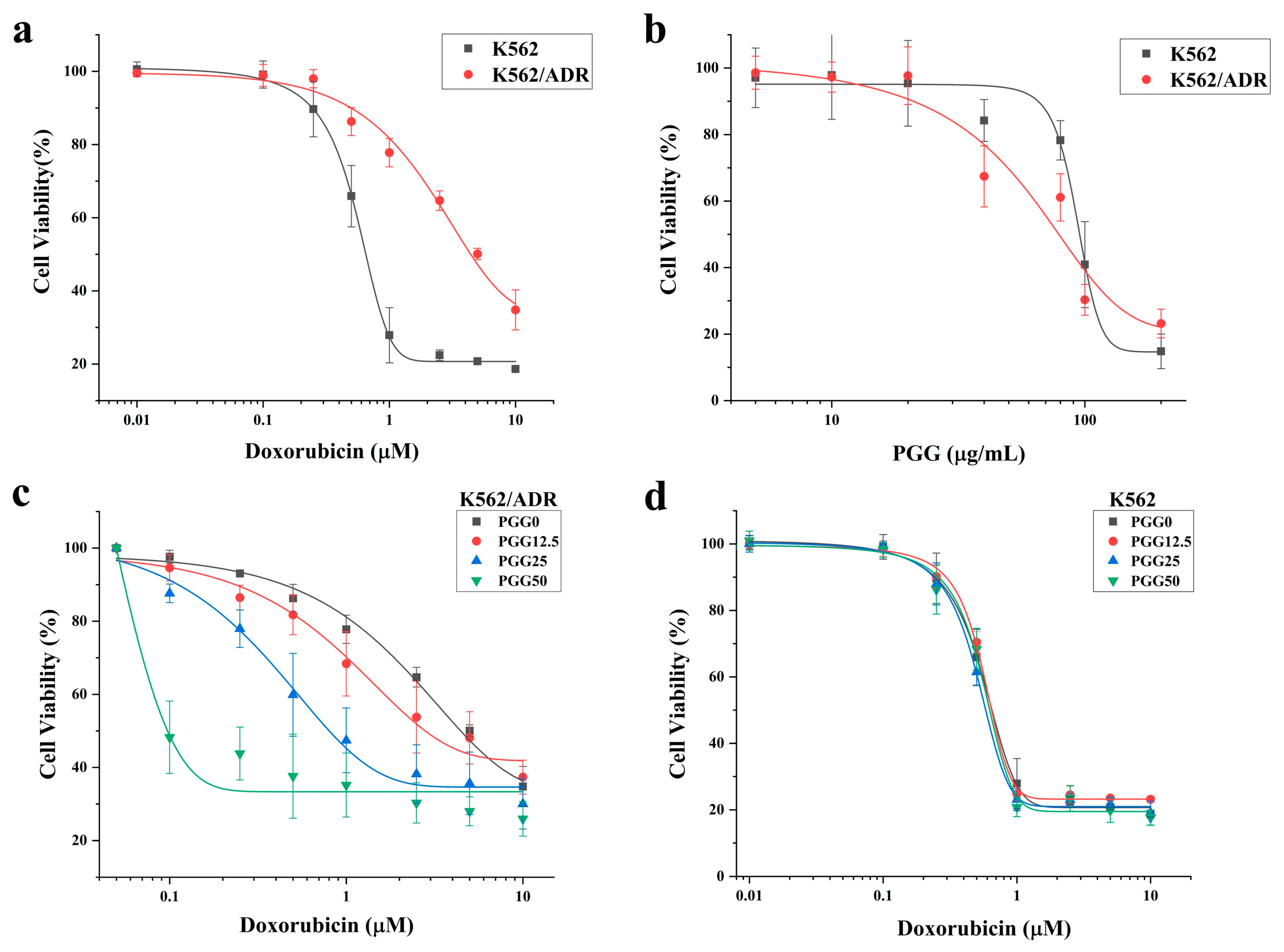
2.1. PGG Enhances the Sensitivity of K562/ADR Cells to DOX
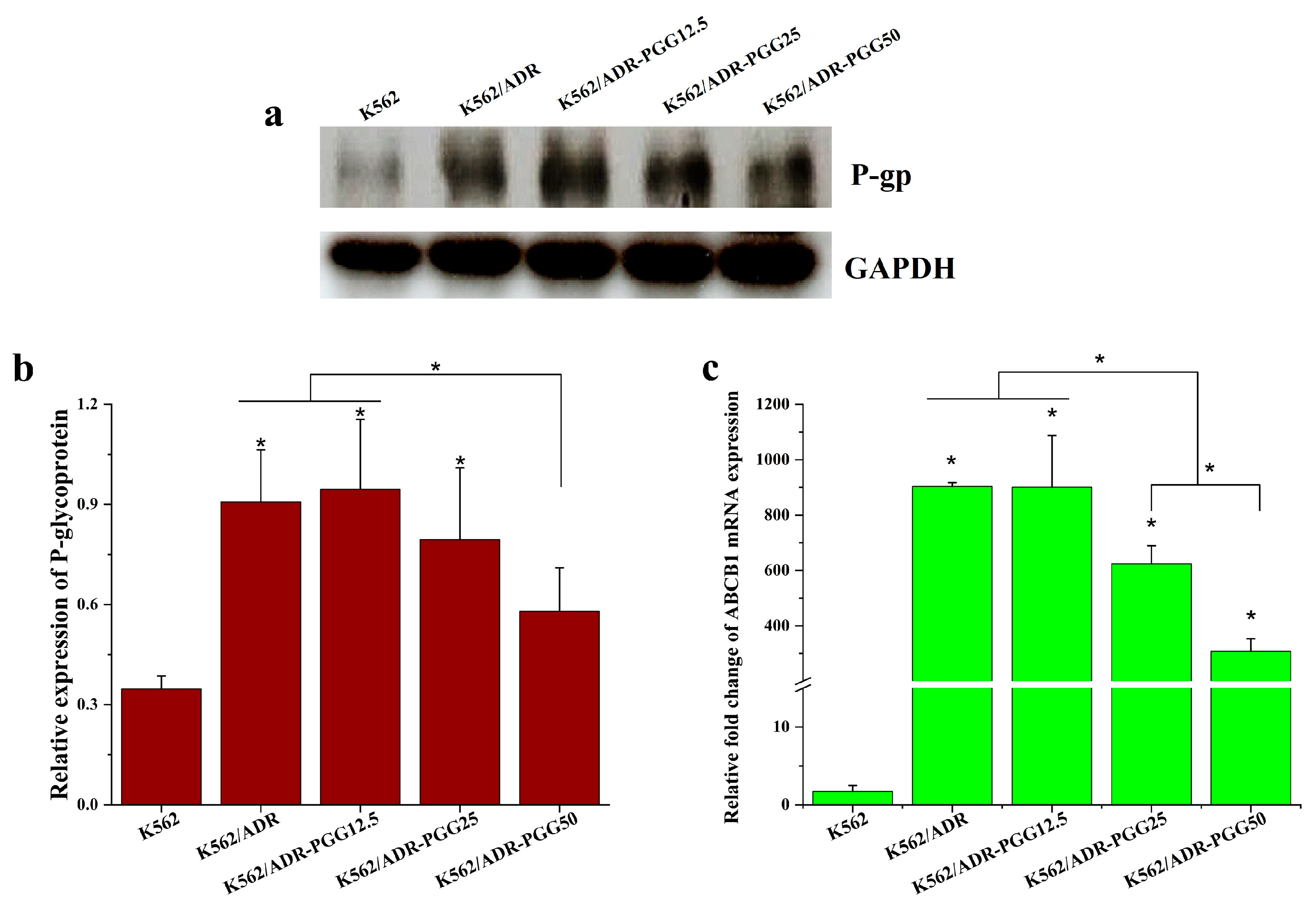
2.2. Effect of PGG on P-gp Protein Expression in K562/ADR Cells
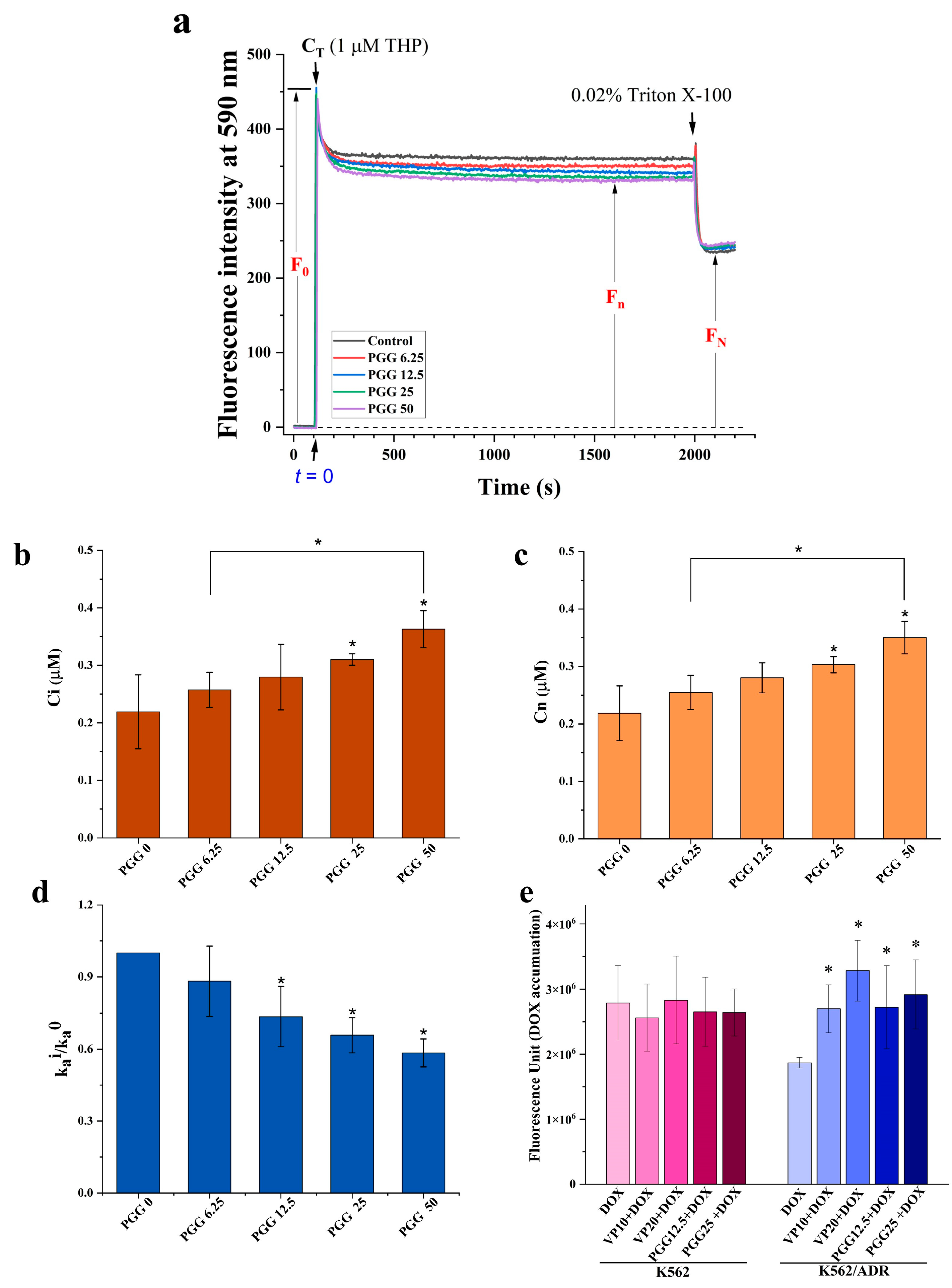
2.3. PGG Can Inhibit the Function of P-gp and Increases the Intracellular Accumulation of DOX
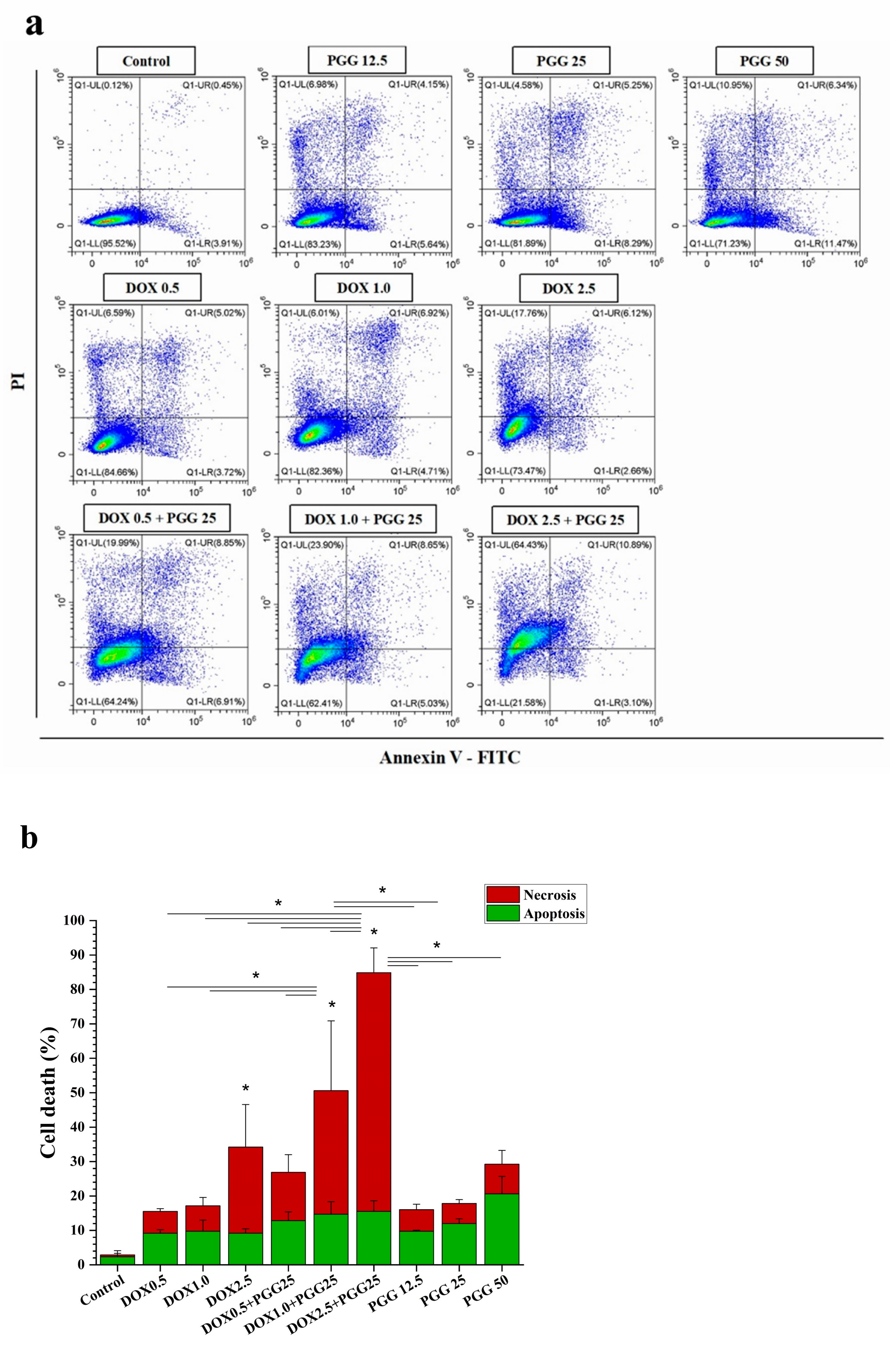
2.4. PGG Augments DOX Treatment in Drug-Resistant K562/ADR Cells
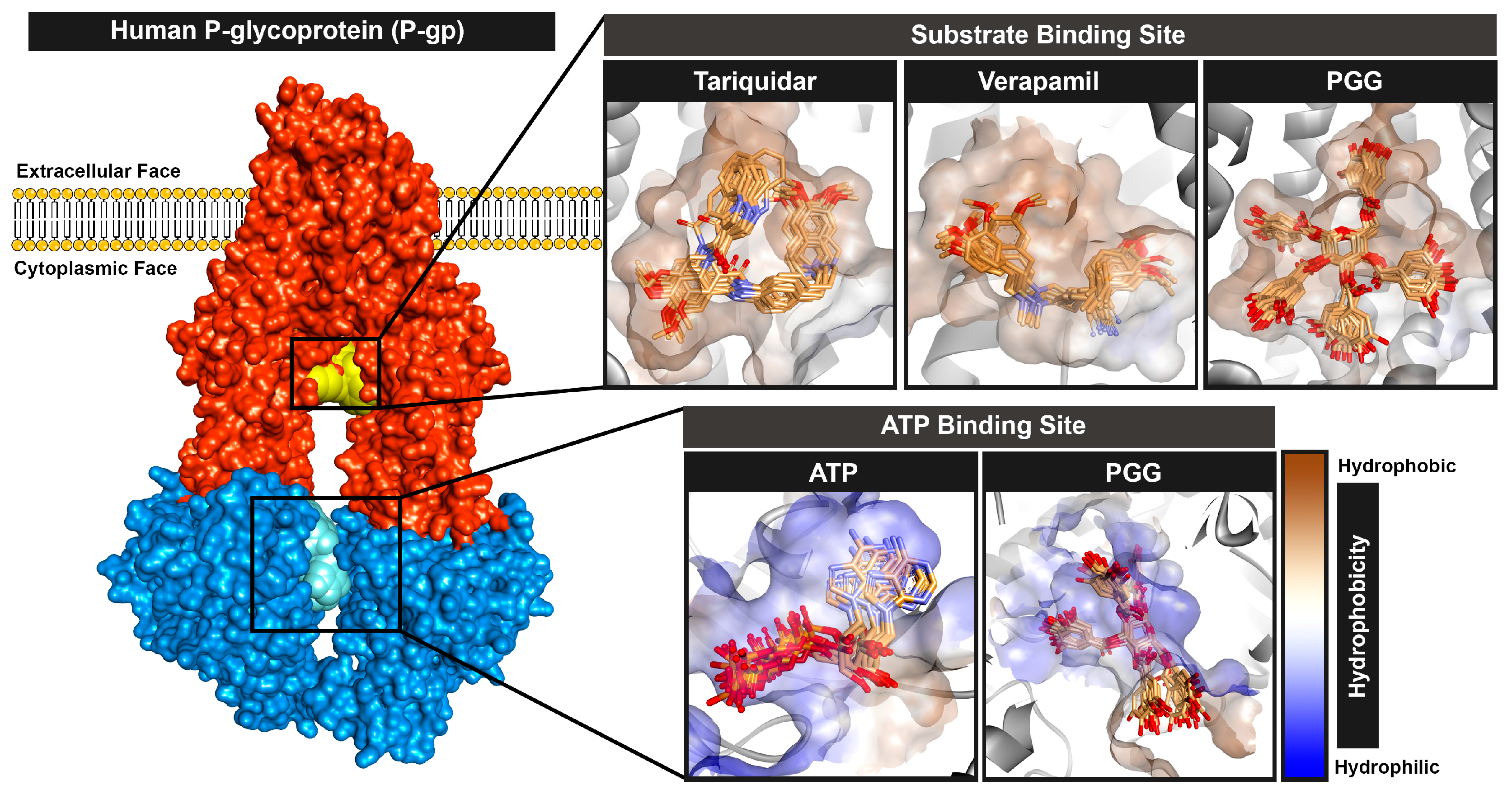
2.5. Molecular Docking
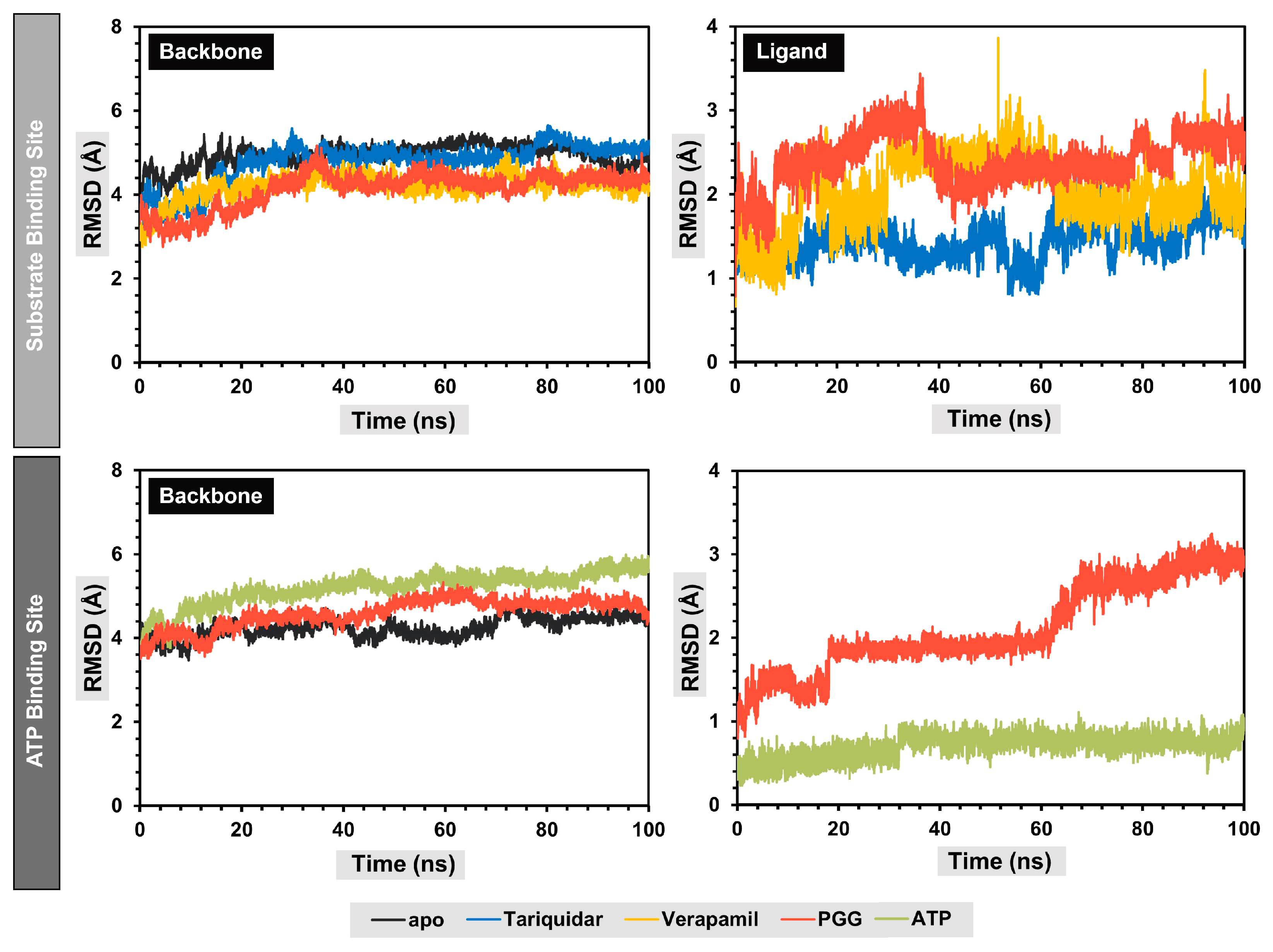
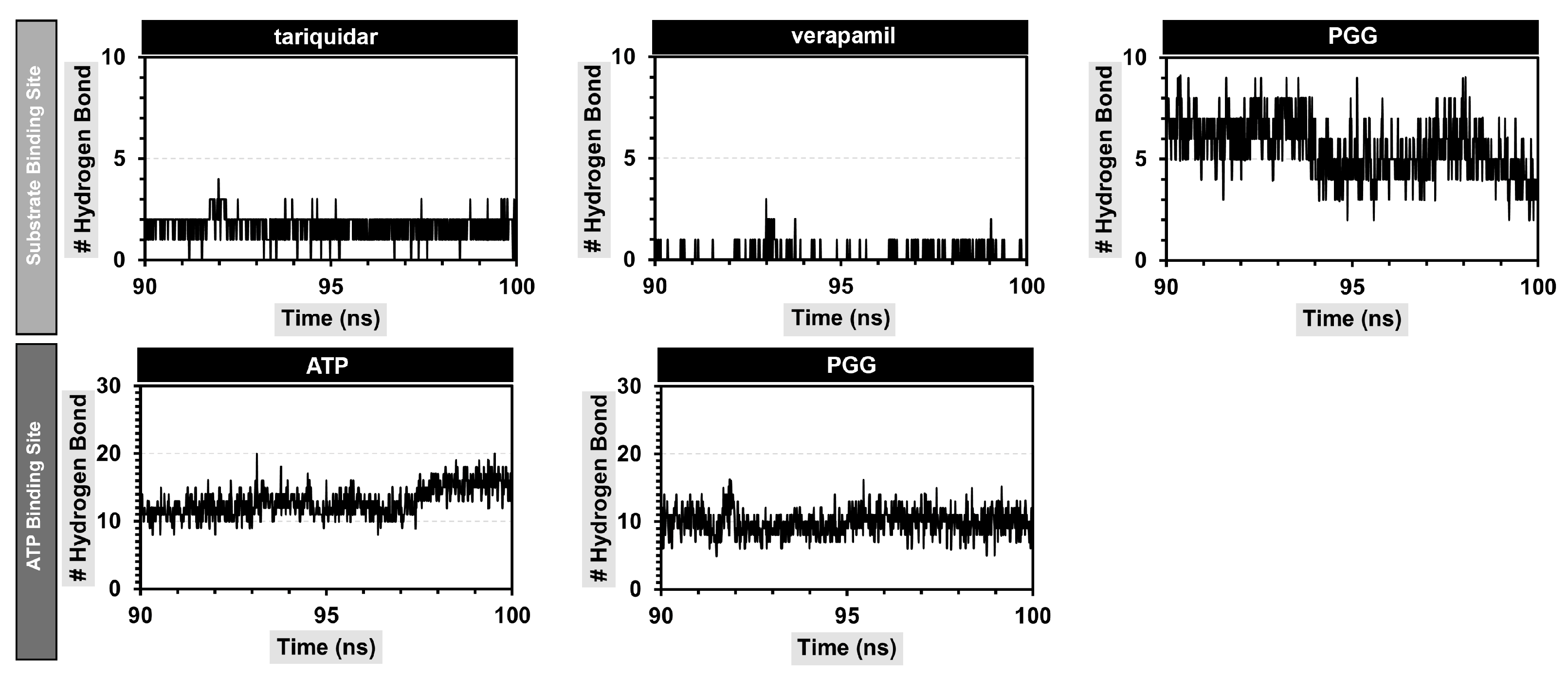
2.6. Molecular Dynamics Simulation
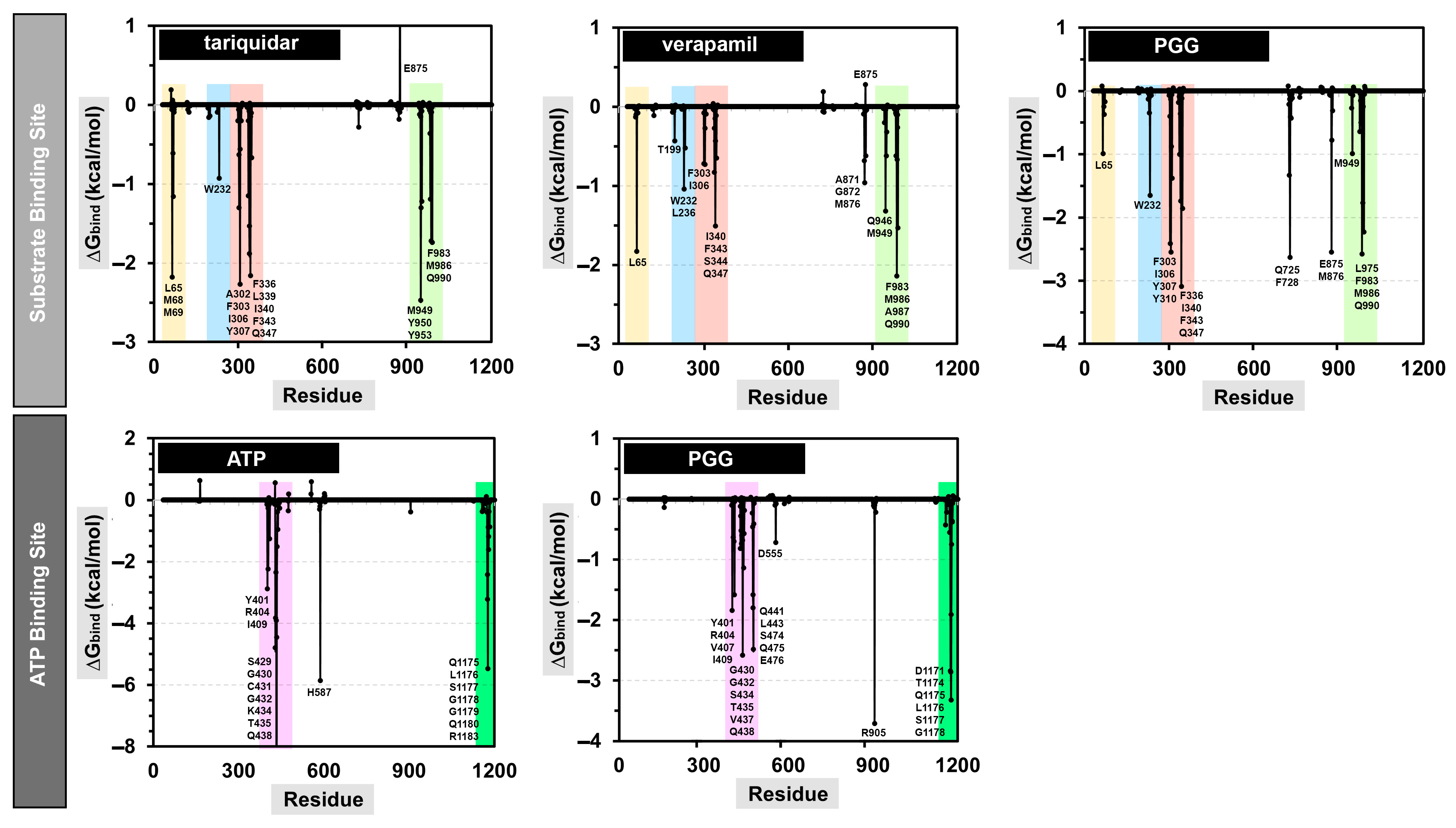
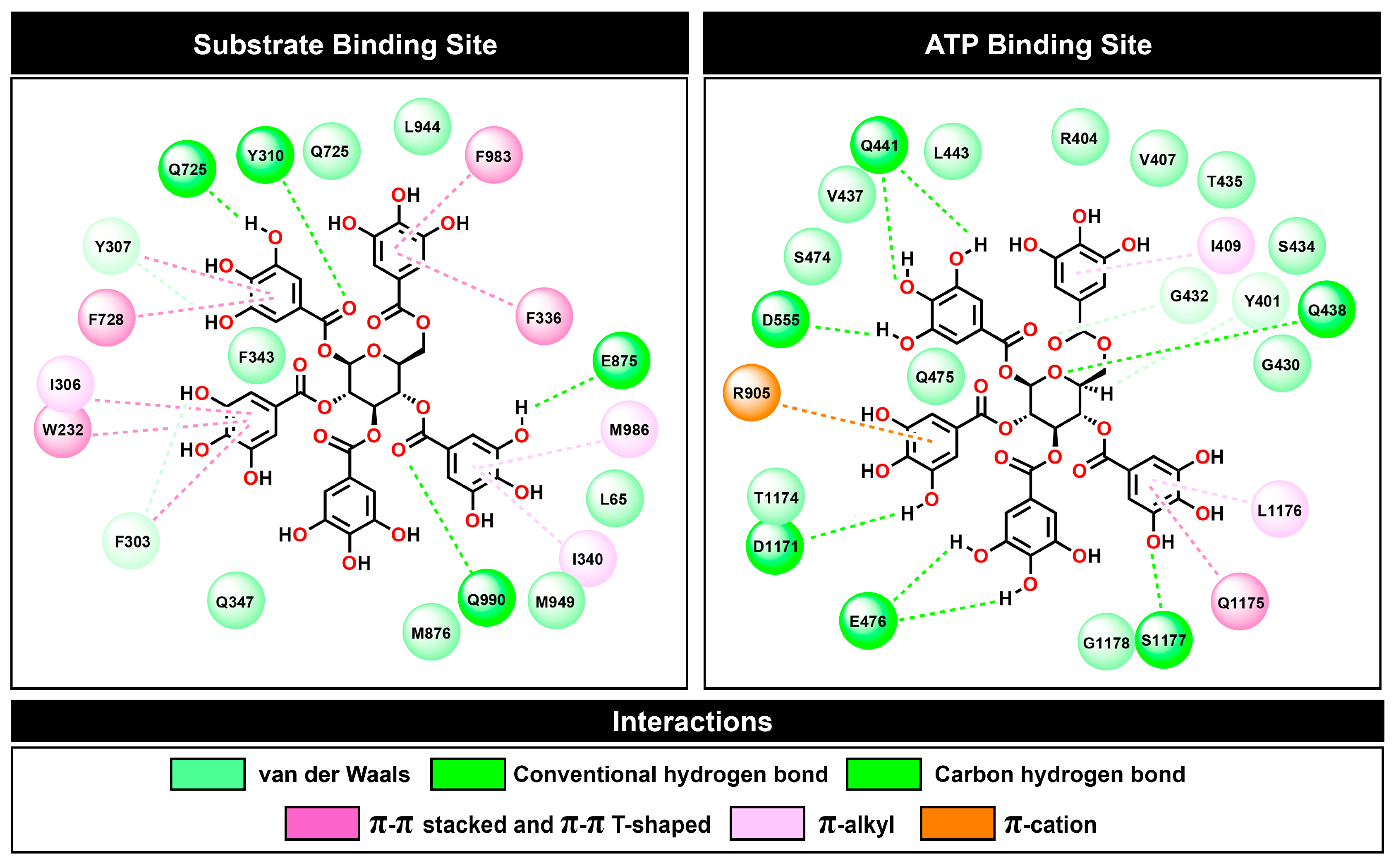
2.7. Calculating the Binding Free Energy
3. Discussion
4. Materials and Methods
4.1. Reagents
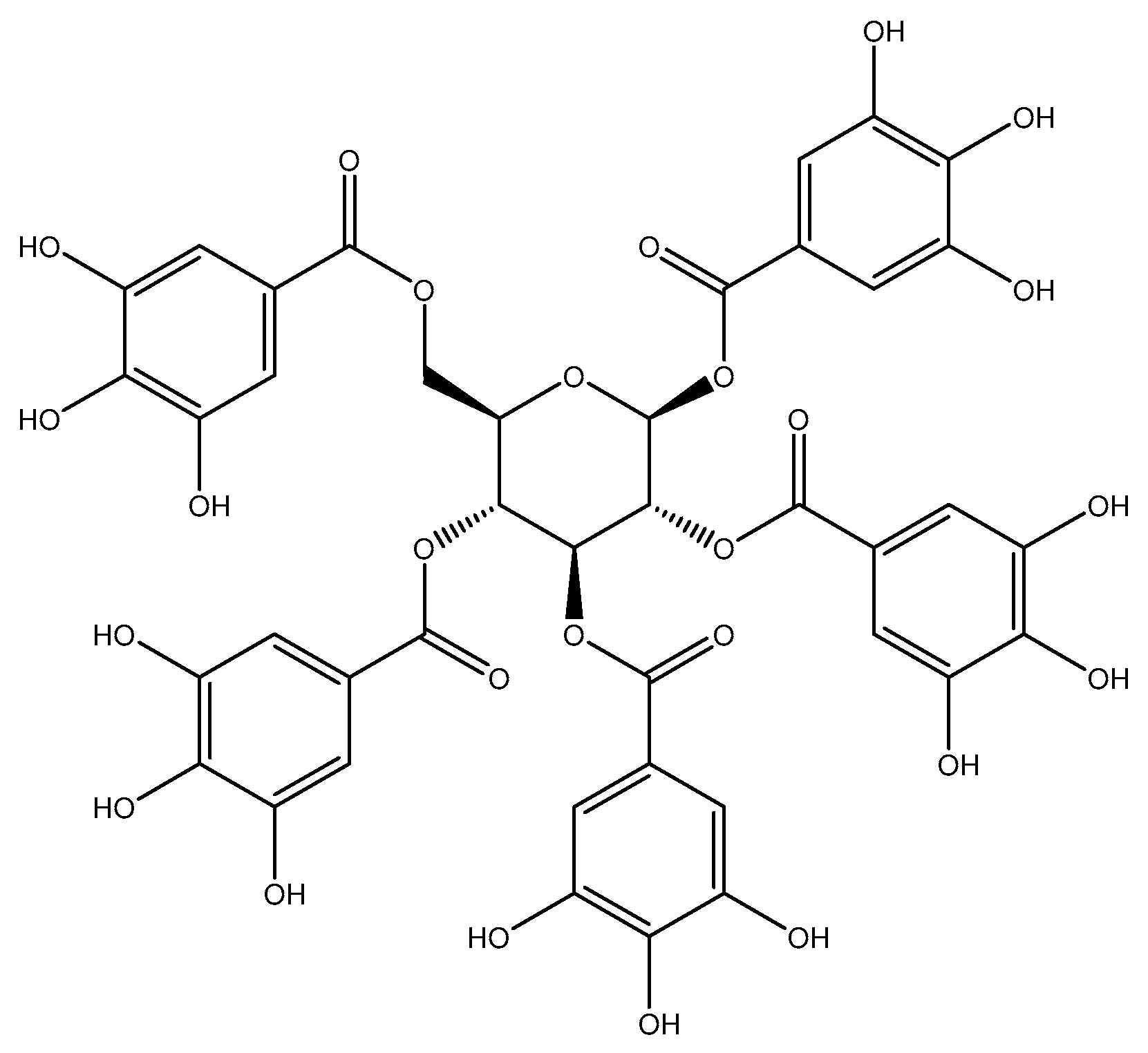
4.2. PGG Isolation and Quantification
4.3. Cell Culture
4.4. Determining the Cytotoxicity and P-Glycoprotein Reversal Effect of PGG Using the CCK-8 Assay
4.5. P-Glycoprotein Expression by Western Blot Analysis
4.6. Real-Time Quantitative PCR
4.7. Kinetic Study of P-gp-Mediated Efflux of THP in K562/ADR Cells Using the Spectrofluorometric Method
4.8. Assay for the Intracellular Accumulation of Doxorubicin
4.9. Apoptosis Assay with Annexin V-FITC/PI Double Staining
4.10. Molecular Docking
4.11. Molecular Dynamics Simulation
4.12. Calculation of Binding Free Energy
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, S.; Gholami, M.H.; Hashemi, F.; Zabolian, A.; Farahani, M.V.; Hushmandi, K.; Zarrabi, A.; Goldman, A.; Ashrafizadeh, M.; Orive, G. Advances in understanding the role of P-gp in doxorubicin resistance: Molecular pathways, therapeutic strategies, and prospects. Drug Discov. Today 2022, 27, 436–455. [Google Scholar] [CrossRef] [PubMed]
- Waghray, D.; Zhang, Q. Inhibit or evade multidrug resistance P-Glycoprotein in cancer treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, S.V.; Kimchi-Sarfaty, C.; Sauna, Z.E.; Gottesman, M.M. P-glycoprotein: From genomics to mechanism. Oncogene 2003, 22, 7468–7485. [Google Scholar] [CrossRef]
- Seelig, A. P-Glycoprotein: One mechanism, many tasks and the consequences for pharmacotherapy of cancers. Front. Oncol. 2020, 10, 576559. [Google Scholar] [CrossRef]
- Lai, J.I.; Tseng, Y.J.; Chen, M.H.; Huang, C.F.; Chang, P.M. Clinical perspective of FDA approved drugs with P-Glycoprotein inhibition activities for potential cancer therapeutics. Front. Oncol. 2020, 10, 561936. [Google Scholar] [CrossRef]
- Bhatia, K.; Bhumika; Das, A. Combinatorial drug therapy in cancer–New insights. Life Sci. 2020, 258, 118134. [Google Scholar] [CrossRef]
- Cháirez-Ramírez, M.H.; de la Cruz-López, K.G.; García-Carrancá, A. Polyphenols as antitumor agents targeting key players in cancer-driving signaling pathways. Front. Pharmacol. 2021, 12, 710304. [Google Scholar] [CrossRef]
- Khan, N.; Afaq, F.; Saleem, M.; Ahmad, N.; Mukhtar, H. Targeting multiple signaling pathways by green tea polyphenol (-)-epigallocatechin-3-gallate. Cancer Res. 2006, 66, 2500–2505. [Google Scholar] [CrossRef]
- Maleki Dana, P.; Sadoughi, F.; Asemi, Z.; Yousefi, B. The role of polyphenols in overcoming cancer drug resistance: A comprehensive review. Cell. Mol. Biol. Lett. 2022, 27, 1. [Google Scholar] [CrossRef]
- Ganesan, M.; Kanimozhi, G.; Pradhapsingh, B.; Khan, H.A.; Alhomida, A.S.; Ekhzaimy, A.; Brindha, G.; Prasad, N.R. Phytochemicals reverse P-glycoprotein mediated multidrug resistance via signal transduction pathways. Biomed. Pharmacother. 2021, 139, 111632. [Google Scholar] [CrossRef] [PubMed]
- Anuchapreeda, S.; Leechanachai, P.; Smith, M.M.; Ambudkar, S.V.; Limtrakul, P.N. Modulation of P-glycoprotein expression and function by curcumin in multidrug-resistant human KB cells. Biochem. Pharmacol. 2002, 64, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Li, J.; Li, Y.; Song, L.; Li, D.; Peng, L.; Wan, Y.; Hua, S. Nanomicelles loaded with doxorubicin and curcumin for alleviating multidrug resistance in lung cancer. Int. J. Nanomed. 2016, 11, 5757–5770. [Google Scholar] [CrossRef] [PubMed]
- Mapoung, S.; Pitchakarn, P.; Yodkeeree, S.; Ovatlarnporn, C.; Sakorn, N.; Limtrakul, P. Chemosensitizing effects of synthetic curcumin analogs on human multi-drug resistance leukemic cells. Chem. Biol. Interact. 2016, 244, 140–148. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Kitagawa, S.; Nabekura, T.; Kamiyama, S. Inhibition of P-glycoprotein function by tea catechins in KB-C2 cells. J. Pharm. Pharmacol. 2004, 56, 1001–1005. [Google Scholar] [CrossRef]
- Kitagawa, S.; Nabekura, T.; Nakamura, Y.; Takahashi, T.; Kashiwada, Y. Inhibition of P-glycoprotein function by tannic acid and pentagalloylglucose. J. Pharm. Pharmacol. 2007, 59, 965–969. [Google Scholar] [CrossRef]
- Kantapan, J.; Paksee, S.; Chawapun, P.; Sangthong, P.; Dechsupa, N. Pentagalloyl glucose- and ethyl gallate-rich extract from Maprang seeds induce apoptosis in MCF-7 breast cancer cells through mitochondria-mediated pathway. Evid.-Based Complement. Altern. Med. 2020, 2020, 5686029. [Google Scholar] [CrossRef]
- Kawk, S.H.; Kang, Y.R.; Kim, Y.H. 1,2,3,4,6-Penta-O-galloyl-β-d-glucose suppresses colon cancer through induction of tumor suppressor. Bioorg. Med. Chem. Lett. 2018, 28, 2117–2123. [Google Scholar] [CrossRef]
- Kim, J.H.; Im, E.; Lee, J.; Lee, H.J.; Sim, D.Y.; Park, J.E.; Ahn, C.-H.; Kwon, H.H.; Shim, B.S.; Kim, B.; et al. Apoptotic and DNA damage effect of 1,2,3,4,6-penta-O-galloyl-beta-D-glucose in cisplatin-resistant non-small lung cancer cells via phosphorylation of H2AX, CHK2 and p53. Cells 2022, 11, 1343. [Google Scholar] [CrossRef]
- Kuo, P.T.; Lin, T.P.; Liu, L.C.; Huang, C.H.; Lin, J.K.; Kao, J.Y.; Way, T.-D. Penta-O-galloyl-beta-D-glucose suppresses prostate cancer bone metastasis by transcriptionally repressing EGF-induced MMP-9 expression. J. Agric. Food Chem. 2009, 57, 3331–3339. [Google Scholar] [CrossRef]
- Kantapan, J.; Paksee, S.; Duangya, A.; Sangthong, P.; Roytrakul, S.; Krobthong, S.; Suttana, W.; Dechsupa, N. A radiosensitizer, gallotannin-rich extract from Bouea macrophylla seeds, inhibits radiation-induced epithelial-mesenchymal transition in breast cancer cells. BMC Complement. Med. Ther. 2021, 21, 189. [Google Scholar] [CrossRef] [PubMed]
- Kantapan, J.; Dechsupa, N.; Tippanya, D.; Nobnop, W.; Chitapanarux, I. Gallotannin from Bouea macrophylla seed extract suppresses cancer stem-like cells and radiosensitizes head and neck cancer. Int. J. Mol. Sci. 2021, 22, 9253. [Google Scholar] [CrossRef] [PubMed]
- Kantapan, J.; Intachai, N.; Khamto, N.; Meepowpan, P.; Sangthong, P.; Wantanajittikul, K.; Dechsupa, N.; Chitapanarux, I. Pentagalloyl glucose and cisplatin combination treatment exhibits a synergistic anticancer effect in 2D and 3D models of head and neck carcinoma. Pharmaceuticals 2022, 15, 830. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.Q.; Zhao, S.; Wang, J.Y.; Zheng, H.C.; Ma, C.M. Inhibitory effects and molecular mechanisms of pentagalloyl glucose in combination with 5-FU on aggressive phenotypes of HepG2 cells. Nat. Prod. Res. 2021, 35, 815–818. [Google Scholar] [CrossRef]
- Knapp, B.; Ospina, L.; Deane, C.M. Avoiding False Positive Conclusions in Molecular Simulation: The Importance of Replicas. J. Chem. Theory Comput. 2018, 14, 6127–6138. [Google Scholar] [CrossRef]
- Kitagawa, S. Inhibitory effects of polyphenols on p-glycoprotein-mediated transport. Biol. Pharm. Bull. 2006, 29, 1–6. [Google Scholar] [CrossRef]
- Gilad, Y.; Gellerman, G.; Lonard, D.M.; O’Malley, B.W. Drug combination in cancer treatment-from cocktails to conjugated combinations. Cancers 2021, 13, 669. [Google Scholar] [CrossRef]
- Goebel, J.; Chmielewski, J.; Hrycyna, C.A. The roles of the human ATP-binding cassette transporters P-glycoprotein and ABCG2 in multidrug resistance in cancer and at endogenous sites: Future opportunities for structure-based drug design of inhibitors. Cancer Drug Resist. 2021, 4, 784–804. [Google Scholar] [CrossRef]
- Khonkarn, R.; Daowtak, K.; Okonogi, S. Chemotherapeutic efficacy enhancement in P-gp-overexpressing cancer cells by flavonoid-loaded polymeric micelles. AAPS PharmSciTech 2020, 21, 121. [Google Scholar] [CrossRef]
- Mankhetkorn, S.; Garnier-Suillerot, A. The ability of verapamil to restore intracellular accumulation of anthracyclines in multidrug resistant cells depends on the kinetics of their uptake. Eur. J. Pharmacol. 1998, 343, 313–321. [Google Scholar] [CrossRef]
- Wen, C.; Dechsupa, N.; Yu, Z.; Zhang, X.; Liang, S.; Lei, X.; Xu, T.; Gao, X.; Hu, Q.; Innuan, P.; et al. Pentagalloyl Glucose: A Review of Anticancer Properties, Molecular Targets, Mechanisms of Action, Pharmacokinetics, and Safety Profile. Molecules 2023, 28, 4856. [Google Scholar] [CrossRef] [PubMed]
- Jiamboonsri, P.; Pithayanukul, P.; Bavovada, R.; Gao, S.; Hu, M. A validated liquid chromatography-tandem mass spectrometry method for the determination of methyl gallate and pentagalloyl glucopyranose: Application to pharmacokinetic studies. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 986–987, 12–17. [Google Scholar] [CrossRef]
- Ma, C.; Zhao, X.; Wang, P.; Jia, P.; Zhao, X.; Xiao, C.; Zheng, X. Metabolite characterization of Penta-O-galloyl-beta-d-glucose in rat biofluids by HPLC-QTOF-MS. Chin. Herb. Med. 2018, 10, 73–79. [Google Scholar] [CrossRef]
- Dechsupa, N.; Kantapan, J.; Tungjai, M.; Intorasoot, S. Maprang “Bouea macrophylla Griffith” seeds: Proximate composition, HPLC fingerprint, and antioxidation, anticancer and antimicrobial properties of ethanolic seed extracts. Heliyon 2019, 5, e02052. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Mankhetkorn, S.; Teodori, E.; Scapecchi, S.; Garnier-Suillerot, A. Study of P-glycoprotein functionality in living resistant K562 cells after photolabeling with a verapamil analogue. Biochem. Pharmacol. 1996, 52, 213–217. [Google Scholar] [CrossRef]
- Reungpatthanaphong, P.; Mankhetkorn, S. Modulation of multidrug resistance by artemisinin, artesunate and dihydroartemisinin in K562/adr and GLC4/adr resistant cell lines. Biol. Pharm. Bull. 2002, 25, 1555–1561. [Google Scholar] [CrossRef]
- Meesungnoen, J.; Jay-Gerin, J.P.; Mankhetkorn, S. Relation between MDR1 mRNA levels, resistance factor, and the efficiency of P-glycoprotein-mediated efflux of pirarubicin in multidrug-resistant K562 sublines. Can. J. Physiol. Pharmacol. 2002, 80, 1054–1063. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA. Discovery Studio Visualizer; Dassault Systèmes: San Diego, CA, USA, 2021. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cisneros, G.A.; Cruzeiro, V.W.D.; et al. Amber 2022; University of California: San Francisco, CA, USA, 2022. [Google Scholar]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the generalized born model suitable for macromolecules. J. Phys. Chem. B 2000, 15, 3712–3720. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | IC50 Mean ± SD a | Resistance Index b | Reversal Index c | |
|---|---|---|---|---|
| K562 | K562/ADR | |||
| PGG | 94.7 ± 3.84 µg/mL | 77.94 ± 7.31 µg/mL | ||
| DOX | 0.64 ± 0.32 µM | 4.54 ± 0.77 µM | 7.1 | 1 |
| DOX+PGG12.5 | 0.65 ± 0.16 µM | 3.30 ± 1.99 µM | 5.2 | 1.4 |
| DOX+PGG25 | 0.60 ± 0.23 µM | 0.95 ± 0.53 µM | 1.5 | 4.7 |
| DOX+PGG50 | 0.63 ± 0.13 µM | 0.22 ± 0.03 µM | 0.3 | 23.7 |
| PGG (μg/mL) | V+ (nM/s) | K+ × 10−9 L/Cell·s | Va (nM/s) | Ka × 10−9 L/Cell·s |
|---|---|---|---|---|
| 0 | 1.60 ± 0.2 | 0.98 ± 0.1 | 0.55 ± 0.03 | 2.9 ± 0.85 |
| 6.25 | 2.10 ± 0.3 | 1.13 ± 0.30 | 0.52 ± 0.02 | 2.3 ± 0.40 |
| 12.5 | 2.38 ± 0.3 a | 1.17 ± 0.05 | 0.49 ± 0.10 | 2.2 ± 0.56 |
| 25 | 2.52 ± 0.2 a | 1.28 ± 0.07 a | 0.42 ± 0.06 a | 2.04 ± 0.8 a |
| 50 | 2.70 ± 0.4 a | 1.36 ± 0.2 a | 0.35 ± 0.11 a | 1.06 ± 0.4 a |
| No. | Hydrogen Bonding a | Distance (Å) b | Occupancy (%) |
|---|---|---|---|
| Substrate binding site | |||
| 1 | E875-(C=O)-O ···· HO-PGG | 2.432 ± 0.789 | 109.09 |
| 2 | Y310-OH ···· O=C-PGG | 2.032 ± 0.284 | 47.65 |
| 3 | Q990-NH2 ···· O=C-PGG | 2.159 ± 0.270 | 34.27 |
| 4 | Q725-C=O ···· HO-PGG | 2.698 ± 1.064 | 33.07 |
| ATP binding site | |||
| 1 | E476-(C=O)-O ···· HO-PGG | 2.230 ± 0.845 | 216.88 |
| 2 | D1171-(C=O)-O ···· HO-PGG | 1.725 ± 0.152 | 111.59 |
| 3 | D555-(C=O)-O ···· HO-PGG | 1.703 ± 0.104 | 105.39 |
| 4 | Q441-NH2 ···· O-PGG | 2.131 ± 0.210 | 96.40 |
| 5 | Q441-C=O ···· HO-PGG | 1.935 ± 0.326 | 65.73 |
| 6 | Q438-NH2 ···· O-PGG | 2.158 ± 0.242 | 51.05 |
| 7 | S1177-NH ···· O-PGG | 2.892 ± 0.434 | 31.87 |
| Compound | Free Energy (kcal/mol) a | |||||
|---|---|---|---|---|---|---|
| Substrate binding site | ||||||
| Verapamil | −57.36 ± 1.51 | −9.45 ± 0.86 | 31.87 ± 0.93 | −8.21 ± 0.18 | 9.28 ± 0.62 | −33.87 ± 1.77 |
| Tariquidar | −76.09 ± 3.37 | −25.31 ± 1.51 | 56.04 ± 1.10 | −10.30 ± 0.48 | 10.02 ± 0.67 | −45.64 ± 4.67 |
| PGG | −85.52 ± 1.67 | −87.62 ± 4.01 | 115.17 ± 6.74 | −14.20 ± 0.23 | 29.10 ± 2.07 | −43.07 ± 4.53 |
| ATP Binding site | ||||||
| PGG | −84.72 ± 3.26 | −65.65 ± 2.84 | 75.80 ± 2.31 | −13.31 ± 0.42 | 16.44 ± 1.35 | −71.45 ± 4.51 |
| ATP | −41.73 ± 0.51 | −519.36 ± 6.30 | 495.84 ± 3.23 | −6.55 ± 0.22 | 50.60 ± 6.81 | −21.21 ± 6.72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dechsupa, N.; Khamto, N.; Chawapun, P.; Siriphong, S.; Innuan, P.; Suwan, A.; Luangsuep, T.; Photilimthana, N.; Maita, W.; Thanacharttanatchaya, R.; et al. Pentagalloyl Glucose-Targeted Inhibition of P-Glycoprotein and Re-Sensitization of Multidrug-Resistant Leukemic Cells (K562/ADR) to Doxorubicin: In Silico and Functional Studies. Pharmaceuticals 2023, 16, 1192. https://doi.org/10.3390/ph16091192
Dechsupa N, Khamto N, Chawapun P, Siriphong S, Innuan P, Suwan A, Luangsuep T, Photilimthana N, Maita W, Thanacharttanatchaya R, et al. Pentagalloyl Glucose-Targeted Inhibition of P-Glycoprotein and Re-Sensitization of Multidrug-Resistant Leukemic Cells (K562/ADR) to Doxorubicin: In Silico and Functional Studies. Pharmaceuticals. 2023; 16(9):1192. https://doi.org/10.3390/ph16091192
Chicago/Turabian StyleDechsupa, Nathupakorn, Nopawit Khamto, Pornthip Chawapun, Sadanon Siriphong, Phattarawadee Innuan, Authaphinya Suwan, Thitiworada Luangsuep, Nichakorn Photilimthana, Witchayaporn Maita, Rossarin Thanacharttanatchaya, and et al. 2023. "Pentagalloyl Glucose-Targeted Inhibition of P-Glycoprotein and Re-Sensitization of Multidrug-Resistant Leukemic Cells (K562/ADR) to Doxorubicin: In Silico and Functional Studies" Pharmaceuticals 16, no. 9: 1192. https://doi.org/10.3390/ph16091192
APA StyleDechsupa, N., Khamto, N., Chawapun, P., Siriphong, S., Innuan, P., Suwan, A., Luangsuep, T., Photilimthana, N., Maita, W., Thanacharttanatchaya, R., Sangthong, P., Meepowpan, P., Udomtanakunchai, C., & Kantapan, J. (2023). Pentagalloyl Glucose-Targeted Inhibition of P-Glycoprotein and Re-Sensitization of Multidrug-Resistant Leukemic Cells (K562/ADR) to Doxorubicin: In Silico and Functional Studies. Pharmaceuticals, 16(9), 1192. https://doi.org/10.3390/ph16091192