1. Introduction
Olfactory receptors (ORs), part of the G protein-coupled receptor (GPCR) superfamily, are primarily expressed in the cilia of olfactory sensory neurons where olfactory perception is initiated upon odorant binding to ORs. These receptors are also ectopically expressed in almost all tissues including liver, lung, sperm, colon, and epithelial cells [
1,
2]. These ectopic expressions of ORs underscore their regulatory roles in biological and physiological processes, such as sperm chemotaxis, energy metabolism, wound healing, and hair growth [
3,
4,
5,
6]. Currently, only a limited number of ligands for ORs have been identified [
7], and a mere 20–30% of human ORs have been deorphanized and finding a potent ligand for human OR remains a challenge. OR2H2 is one such deorphanized human olfactory receptor [
8], with aldehyde 13-13 as its ligand. OR2H2, ectopically expressed in human myoblasts, is activated by aldehyde 13-13 and influences skeletal muscle differentiation by inhibiting myoblast fusion [
9].
ORs are also present in human genital organs. GTEx analysis (
https://gtexportal.org) indicates that OR2H2 is found in vaginal tissue, though its exact function remains elusive. The human vaginal epithelium is a multi-layered, non-keratinized squamous epithelium without glands that envelops the inner surface of the vagina.
Impaired function of the vaginal epithelium, especially in older women, can compromise vaginal health, increasing the risk of vaginal atrophy due to thinning of the vagina’s inner layer [
10]. Both autophagy function and senescence progression are instrumental in the onset of vaginal atrophy. Many studies have delved into the induction of autophagy in human genital cells to preserve vaginal health. For instance, Shao et al. discussed how the Chinese traditional herb prescription JieZe-1 enhanced autophagy by inhibiting the PI3K/Akt/mTOR signaling axis in VK2/E6E7 cells [
11]. Yang et al. demonstrated that inducing autophagy in VK2/E6E7 cells bolstered the barrier function of the vaginal epithelium [
12,
13]. A decline in autophagy activity is often linked to senescence. As autophagy facilitates the removal of impaired cellular components, it can prevent the accumulation of cellular damage leading to senescence, making it a potential protective mechanism against premature aging.
AMP-activated protein kinase (AMPK), a central regulator of energy metabolism, stands among the regulators that induce autophagy, hence promoting cellular homeostasis and senescence protection. AMPK propels autophagy through various means. For example, AMPK triggers inhibitory phosphorylation on TSC1/2 and Raptor, which inhibit autophagy and phosphorylates ULK1, initiating autophagy [
14]. Concurrently, AMPK activation is known to curb cellular senescence via several pathways. AMPK-dependent phosphorylation of p27
kip1 enhances p27
kip1 stability, which in turn reduces cell-senescence indicators, such as beta-galactosidase, P16
INK4A, and P21 [
15]. Furthermore, AMPK can inhibit senescence by activating sirtuin 1 (SIRT1) [
16], an anti-aging protein entangled in numerous signaling routes, including cell senescence [
17].
In this study, we explored the function of OR2H2, the most predominant OR in VK2/E6E7 cells. Results from phosphokinase array experiments revealed that aldehyde 13-13 robustly initiated AMPK phosphorylation. Consequently, we postulated that OR2H2 might bolster cellular health through the regulation of autophagy and senescence in VK2/E6E7 cells.
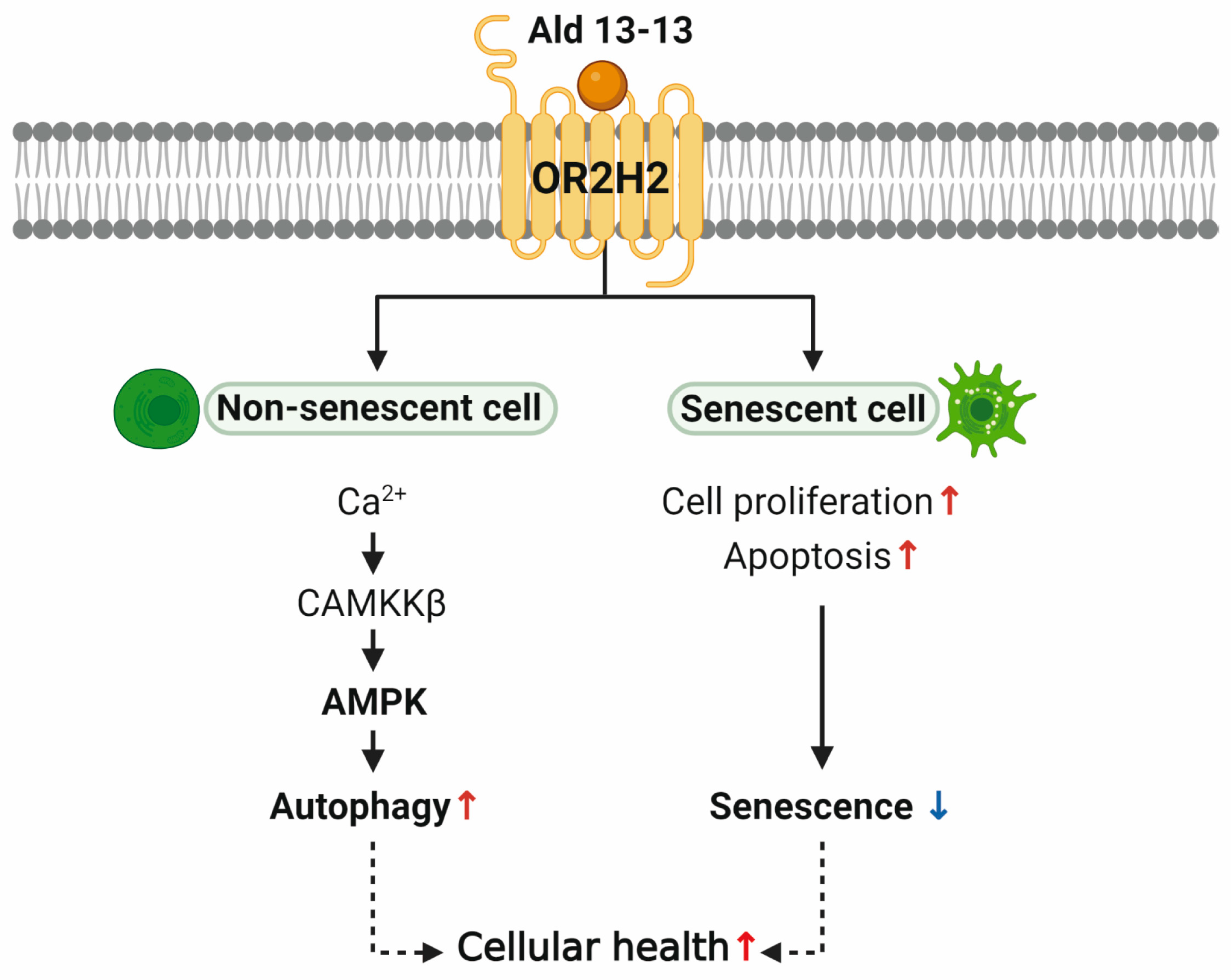
In our findings, we highlight the functionality of ectopic OR expression in VK2/E6E7 human vaginal epithelial cells. OR2H2 was profusely expressed in VK2/E6E7 cells, and its activation by its ligand, aldehyde 13-13, initiated intracellular calcium signaling and CAMKKβ–AMPK–autophagy pathways. Activating OR2H2 revitalized cell proliferation and curtailed primary markers of cellular senescence in H2O2-induced senescent cells. OR2H2 activation also exhibited a senolytic effect by inducing apoptosis in these cells. In sum, our findings suggest that OR2H2 activation augments the autophagy pathway and impedes cellular aging via AMPK activation, proposing OR2H2 as a potential molecular target to enhance cellular health and combat aging.
3. Discussion
Ectopic olfactory receptors perform varied functions across diverse organs and tissues [
5,
26,
27]. Given that genital organs are consistently exposed to odorants, ectopic olfactory receptors might influence intracellular signaling pathways and associated physiological processes in these organs. In this study, we delved into the role of OR2H2 in VK2/E6E7 human vaginal epithelial cells.
Currently, three extant studies discuss the functionality of ectopic OR2H2, pointing to its tissue-specific roles. In human myoblasts, OR2H2 elevates the intracellular calcium concentration and diminishes myoblast fusion via Phosphoinositide 3-kinase signaling [
9]. In the thyroid gland, OR2H2 expression decreases in malignant cells, compared to surrounding healthy tissues. Aldehyde 13-13 inhibits the proliferation of thyroid cancer cells [
28]. The single nucleotide polymorphism of OR2H2, rs123388, is linked to celiac disease in patients from Indian and Dutch populations [
29]. This polymorphism, along with five others, is associated with immune functions and Toll-like receptor signaling pathways [
29]. Such findings indicate the significance of OR2H2 in gut and immune cells. Furthermore, we discerned a new function of OR2H2 in VK2/E6E7 cells. OR2H2 activation by aldehyde 13-13 energized the Ca
2+–CAMKKβ–AMPK–autophagy signaling pathway, curbing cellular senescence via a senolytic effect (
Figure 9).
Aldehyde 13-13 is a potent OR2H2 ligand [
9]. Our findings also underscore its ability to robustly activate OR2H2 in VK2/E6E7 cells, thereby triggering intracellular Ca
2+ elevations. Various effectors modulate intracellular Ca
2+ levels, including adenylyl cyclase, which can escalate Ca
2+ levels [
30], and phospholipase C (PLC), which controls the IP
3-DAG-Ca
2+ signaling route [
31]. Sarco/endoplasmic reticulum calcium ATPase (SERCA) channels Ca
2+ from the cytosol to the sarcoplasmic reticulum lumen [
32], and cyclic nucleotide-gated (CNG) channels facilitate Na
+, K
+, and Ca
2+ ion flow [
33]. Aldehyde 13-13, in our observations, amplified the intracellular Ca
2+ concentration, an effect that was diminished by multiple calcium inhibitors like adenylyl cyclase III inhibitor (SQ22536), PLC inhibitor (U73122), SERCA inhibitor (thapsigargin), and CNG channel inhibitor (L-cis diltiazem). Thus, aldehyde 13-13’s Ca
2+ induction in VK2/E6E7 cells likely results from the activation of several Ca
2+ channels. A phosphokinase assay revealed the strong activation of AMPKα1 by aldehyde 13-13 in VK2/E6E7 cells, while immunoblotting illustrated the triggering of the CaMKK-β–AMPKα1–mTOR–autophagy signaling pathway due to OR2H2 activation in these cells. This pathway activation by aldehyde 13-13 was nullified in OR2H2 knockdown cells, emphasizing that OR2H2 is a functional receptor in VK2/E6E7 cells, with signaling pathway modulation by aldehyde 13-13 contingent upon OR2H2 activation.
OR2H2 activation stimulated autophagy. Metrics such as the LC3-II-to-LC3-I ratio, MDC staining, autophagic flux analysis, and transmission electron microscopy all depicted enhanced autophagy and the progression of autophagic turnover from the autophagosome to autolysosome in VK2/E6E7 cells. Autophagy stands at the crossroads of metabolic and proteostatic signaling, dictating key aging mechanisms and organism lifespan [
34]. A decline in autophagic activity is believed to lead to the buildup of damaged organelles and macromolecules with aging, whereas sustaining adequate autophagic activity aids in life extension [
35]. AMPK spurs autophagy either directly or indirectly through mTORC1 suppression or SIRT-1 activation. Indeed, AMPK oversees the aging process via an interconnected signaling network [
36].
AMPK is a central regulator of energy metabolism and homeostasis, influencing the aging process through interactions with several signaling networks [
37]. Enhanced AMPK activity potentially promotes healthspan and lifespan by mitigating cellular stress. This enhancement is achieved by activating signaling molecules such as FoxO/DAF-16, Nrf2/SKN-1, and SIRT1, stimulating the autophagic pathway via mTOR signaling, and restraining proinflammatory responses by inhibiting NF-κB signaling [
36]. As AMPK activity diminishes with age, its increased activity might extend the lifespan of simpler organisms.
Aldehyde 13-13 extended the lifespan of both C. elegans and budding yeast, potentially through the AMPK–autophagy pathway. However, neither C. elegans nor budding yeast have OR2H2 homologs. As such, pinpointing the target receptor and the signaling mechanisms of aldehyde 13-13 requires further exploration. We hypothesize that in organisms like C. elegans and yeast, aldehyde 13-13 might counteract aging through analogous (rather than homologous) mechanisms compared to the OR2H2-dependent pathway in human cells. However, the specific anti-aging action of aldehyde 13-13 on C. elegans and yeast remains elusive. In humans, OR2H2, being a receptor for aldehyde 13-13, might offer therapeutic avenues for mitigating the aging of human vaginal epithelial cells. This is evidenced by the fact that Ca2+ and cAMP signaling via aldehyde 13-13 was specifically observed in OR2H2-expressing VK2/E6E7 cells. This potential therapeutic approach could extend to other human tissues expressing OR2H2, a possibility we aim to probe further.
AMPK maintains cellular energy balance and combats senescence [
38,
39]. For instance, aging skeletal muscle stem cells (MuSCs) exhibit diminished autophagy, augmented apoptosis, and reduced phosphorylation of AMPK and its downstream target, p27
Kip1 [
15]. Activating AMPK in aged MuSCs in vitro curtailed apoptosis, boosted proliferation, and enhanced transplantation efficiency in vivo. Additionally, the AMPK/p27Kip1 pathway activation lowered senescence markers in older cells, partially relying on p27
Kip1 phosphorylation. Hence, the AMPK/p27
Kip1 pathway might modulate the autophagy/apoptosis equilibrium in aged MuSCs, serving as a prospective target for enhancing muscle regeneration in older adults. Our results indicate that the OR2H2–AMPK–autophagy pathway activation diminishes cellular senescence in VK2/E6E7 cells. To the best of our knowledge, this constitutes the pioneering assertion that ectopic olfactory receptors can impede aging and cellular senescence.
4. Materials and Methods
4.1. Antibodies and Reagents
Primary antibodies to β-actin (SC-47778, 1:1000), CaMKK-ß (SC-50341, 1:1000), and AMPKα1/2 (SC-25792, 1:1000) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Primary antibodies to p-CaMKKβ (12818S, 1:500), mTOR (2983S, 1:1000), S6 ribosomal protein (2217S, 1:1000), and p-S6 ribosomal protein (2211S, 1:1000) were purchased from Cell Signaling (Danvers, MA). Anti-p-AMPKα1/2 (11183, 1:1000) and anti-p16 (41296, 1:1000) antibodies were from Signalway (College Park, MD, USA). Anti-LC3B (NB100-2220, 1:500) and anti-p21 (NBP2-29463, 1:500) antibodies were purchased from Novus (St. Louis, MO, USA). Anti-p-mTOR (ab63552) and anti-Ki-67 (ab15580) antibodies were purchased from Abcam (Cambridge, UK). Anti-mouse (31430), anti-rabbit (31460), anti-goat (31402), and Alexa Fluor 488 (A11008) immunoglobin G secondary antibodies (1:5000) were purchased from Invitrogen (Carlsbad, CA, USA). The anti-OR2H2 primary antibody (1:1000) was purchased from Antikoerper-online.de (Aachen, Germany). L-Cis diltiazem was from Abcam (Cambridge, UK). SQ22536 and thapsigargin were purchased from Enzo Life Science (Farmingdale, NY, USA) and U73122 was from Sigma-Aldrich (St. Louis, MO, USA). Aldehyde 13-13 was obtained from Henkel (Düsseldorf, Germany). A769662, an AMPK agonist, was purchased from Cayman (Ann Arbor, MI, USA). Rapamycin was obtained from LC Laboratories (Woburn, MA, USA).
4.2. Cell Culture
VK2/E6E7 cells (ATCC, CRL-2616) were purchased from the American Type Culture Collection (ATCC, VA, USA), and cultured in keratinocyte-serum free medium (Gibco, New York, USA, 17005042), supplemented with 0.1% human recombinant epidermal growth factor, 0.3% bovine pituitary extract, and 0.4 mM calcium chloride as instructed in the manufacturer’s protocol. Culture medium was renewed every 2–3 days. Cells were grown in 37 °C with 5% CO2. All compounds except L-Cis diltiazem (Abcam) and aldehyde 13-13 (Henkel) were prediluted in DMSO. L-Cis diltiazem was prediluted in distilled water and aldehyde 13-13 in ethanol. All compounds were diluted 1:1000 in growth medium or Ringer’s solution and applied to VK2/E6E7 cells. All dilutions of aldehyde 13-13 were prepared fresh before use.
4.3. RNA Extraction and RT-PCR
Total cellular RNA was extracted with Total RNA extraction reagent RNAiso Plus (1 mL, Takara Bio, 9109, Japan) according to the manufacturer’s instructions. Complementary DNA was synthesized from 1 g RNA with ReverTra Ace® RT Master Mix kit (Toyobo, Osaka, Japan, FSQ-301) according to the manufacturer’s instructions. One g of cDNA was then used as a template for PCR reactions and specific primers were designed by the Primer-BLAST designer program (NCBI, Bethesda, MD, USA). RT-PCR was performed with EmeraldAmp® GT PCR Master Mix reagent according to the manufacturer’s instructions. The RT-PCR products were analyzed by agarose gel electrophoresis (3%) and image analysis was obtained by using a ChemiDoc touch imaging system with the Image Lab 5.2 software (Bio-Rad, Redmond, WA, USA). L32 was used to normalize the data.
4.4. Real Time qPCR
After a 24 h treatment in 6-well plates, total cellular RNA was extracted using the RNAiso Plus reagent (Takara Bio, Shiga, Japan), following the manufacturer’s guidelines. Complementary DNA (cDNA) was synthesized from 1 μg of RNA using the ReverTra Ace
® RT Master Mix kit (FSQ-301; Toyobo, Japan), again adhering to the manufacturer’s protocol. An amount of 1 μg of the resultant cDNA was utilized as a template for the polymerase chain reaction (PCR). Specific primers were crafted using the Primer-BLAST designer tool (NCBI, Bethesda, MD, USA). The cDNA was then subjected to PCR using the iQ5 i-Cycler system (Bio-Rad Laboratories, Hercules, CA, USA) and the Thunderbird SYBR
® qPCR Mix reagent (T00PS-201; Takara Bio). Amplification parameters for the cDNA included an initial 30 s denaturation at 95 °C, 40 cycles of 10 s denaturation at 95 °C, 20 s annealing at 59 °C, and 20 s extension at 68 °C. The fluorescent signal was quantified automatically post each PCR cycle. Gene expression was determined from the threshold cycle (Ct value), and L32 was employed for normalization. The oligonucleotide sequences can be found in
Supplementary Table S1.
4.5. OR2H2 Immunocytochemical Staining
Immunocytochemical staining was carried out using immunofluorescence microscopy. VK2/E6E7 cells were placed on coverslips inside a 24-well plate, containing 500 μL of cell culture medium, and incubated at 37 °C in a 5% CO
2 environment. Upon reaching 80% confluence, the medium was discarded, and the cells were rinsed with PBS. Post-fixation in ice-cold acetone, cells were treated with both primary (1:100, anti-OR2H2; Antikorperonline.de) and secondary antibodies (1:1000, goat anti-rabbit 546; Thermo Fisher, Carlsbad, CA, USA). Subsequently, cells were exposed to Alexa Fluor phalloidin 488 (1:200, Thermo Fisher) for 45 min at ambient temperature with gentle agitation. The cells were then sealed with Immu-Mount solution (Thermo Fisher) and inspected under an LSM 510 Meta Confocal Microscope (Carl Zeiss, Jena, Germany) employing a 40× oil immersion objective and the Leica Application Suite X Software. The specificity of the antibodies utilized was previously ascertained by rho-tagged transfection of Hana3A cells [
40].
4.6. Calcium Imaging and Calcium Inhibitor Treatments
Intracellular calcium concentration changes were monitored using the fluorescent dye Fura-2-acetoxylmethyl ester (Fura-2/AM; Invitrogen). VK2/E6E7 cells were cultured in 35 mm dishes and incubated in growth medium containing 3 μM Fura-2/AM for 30 min at 37 °C, protected from light. Next, growth medium was replaced with Ringer’s solution. Aldehyde 13-13 and calcium inhibitors were dissolved in Ringer’s solution (1:1000) and applied directly to the cells. In desensitization experiments, the cells were stimulated repetitively with aldehyde 13-13 (300 µM) for 2 min while control cells were treated with vehicle (ethanol in Ringer’s solution) repeatedly for 2 min. In experiments under calcium-free conditions, cells were stimulated with aldehyde 13-13 (500 µM) for 3 min, then washed for 3 min with Ringer’s solution. Cells were then treated with calcium-free Ringer’s solution for 7 min, and then aldehyde 13-13 was added for 3 min. A third application of aldehyde 13-13 was performed for 3 min. In experiments with calcium channel inhibitors, cells were stimulated with aldehyde 13-13 (500 µM) for 3 min. After 2 min washing with Ringer’s solution, the cells were stimulated with the calcium channel inhibitors. Subsequently, aldehyde 13-13 and inhibitors were co-applied for 3 min. A third application was also prepared under normal conditions after a 2 min wash-out step. Calcium channel inhibitors were incubated under the following conditions: SQ 22536 (10 µM) for 10 min; L-cis diltiazem (100 µM) for 3 min; U 73122 (5 µM) for 5 min; thapsigargin (5 µM) for 25 min. Intracellular calcium flux was observed using a DMI, 6000CS inverse microscope (Leica, Wetzlar, Germany), and calcium-dependent fluorescence changes were detected using a 10-fold fluorescence objective. Fluorescence emission intensities (340 and 380 nm) were detected by a DFX 360FX CCD camera (Leica). Regions of interest (ROIs) were analyzed using Advanced Fluorescence software LASX Office 1.4.5 27713 (LAS AF; Leica).
4.7. Phosphokinase Array
The human phosphokinase array kit by RnD systems was applied to investigate the phosphorylation of 43 different kinases under the influence of aldehyde 13-13 stimulation for an incubation time of 30 min. Before starting the array, according to the manufacturer’s instructions, protocol cells were cultivated in 75 cm2 culture flasks until a confluence of 90% was achieved. Medium was exchanged with fresh medium containing the diluted aldehyde 13-13 in a concentration of 500 µM. The control was treated with ethanol (<0.1%). After incubation, medium was aspirated, cells were covered with PBS and manually detached with a cell scraper. The detached cells were then subsequently taken up in the PBS and centrifuged for 10 min at 1000 rpm and 4 °C. The cell pellets thus obtained were then lysed in RIPA buffer and their protein concentration was determined using the PierceTM BCA protein assay kit. The total amount of protein was 250 µg for the treated and untreated samples. The evaluation of the membrane was performed by the chemiluminescence System Fusion SL 3500-WL. The intensity of the individual array dots was analyzed with the program Image J and a microarray plugin and normalized to the control.
4.8. OR2H2 Knockdown
For mRNA interference of OR2H2, VK2 cells (4 × 105) were seeded in six-well plates and transfected with a siRNA against OR2H2 (Cat No. sc-95450; Santa Cruz Biotechnology, Inc.) using Lipofectamine 2000 according to the manufacturer’s instructions. Briefly, cells were cultured with siRNA and Lipofectamine 2000 in Opti-MEM reduced-serum medium (Cat No. 32985070; Gibco, New York, NY, USA). After incubation for 5 h at 37 °C in a CO2 atmosphere, medium was removed, and cells were incubated for 19 h in serum-containing medium at 37 °C. To verify transfection, VK2 cells transfected with fluorescein-conjugated control siRNA (Cat No. SN1021; Bioneer, Dae-Jeon, Republic of Korea) were harvested. Intracellular fluorescence intensity was measured using a flow cytometer equipped with an FL1 detector (BD Biosciences, Franklin Lakes, NJ, USA).
4.9. cAMP Assay
Experiments were performed as previously reported (Wu et al., 2021). To measure intracellular cyclic adenosine monophosphate (cAMP) concentration changes, cells were cultivated in a 96-well plate until 90% confluence. Aldehyde 13-13 was diluted in induction buffer, consisting of Ringer’s solution, 500 μM isobutyl-1-methylxanthin, and 100 μM 4-imidazolidin-2-one (Sigma-Aldrich). After incubation for 30 min, intracellular cAMP concentrations were measured using cAMP Glo Assay Kit (Promega) and Fusion α Multi-well Plate Reader (Packard Bioscience, Wellesley, MA, USA). Data were normalized to the solvent control (ethanol). Treatment for 30 min with 10 μM forskolin (Sigma-Aldrich) served as the positive control.
4.10. Cell Viability Assay
The viability of siRNA-transfected VK2/E6E7 cells was assessed using a 2,5-diphenyl-2H-tetrazolium bromide (MTT) assay [
41]. Cells were seeded at a density of 1 × 10
4 cells per well in 96-well culture plates and were allowed to adhere overnight. The following day, the culture medium was discarded and replaced with a medium containing 10% MTT (Sigma Aldrich). Cells were then incubated for 3 h at 37 °C. Afterward, the MTT solution was carefully removed from each well, and dimethyl sulfoxide (DMSO; Bio Basic Inc., Markham, ON, Canada) was added to dissolve the formazan crystals. The absorbance of each sample was subsequently measured at 570 nm using a microplate spectrophotometer (Multiskan Go; Thermo Fisher Scientific).
4.11. Immunoblot Analysis
Total cellular proteins were extracted using radioimmunoprecipitation assay buffer supplemented with Halt protease and phosphatase inhibitor reagent (Thermo Fisher Scientific, Carlsbad, CA, USA). Total cellular proteins were centrifuged for 20 min at 12,000 rpm to obtain the soluble protein fraction. Protein concentration was measured using a protein assay dye reagent concentrate (Bio-Rad). Denatured proteins were run on a sodium dodecyl sulfate-polyacrylamide gel, and transferred to nitrocellulose membranes (GVS Filter Technology, Morecambe, UK). The membranes were blocked in TBS with 5% (w/v) non-fat dried milk for 1 h at room temperature. The primary antibodies were diluted in TBS-Tween 20 (0.1%) with 5% (w/v) non-fat dried milk. The secondary antibodies were diluted in TBS with 0.1% Tween-20. Immunoblot images were obtained using the ChemiDoc Touch Imaging System and analyzed with Image Lab 5.2 software (Bio-Rad). Protein levels were normalized with β-actin after stripping the identical blot using stripping buffer (Dyne Bio, CBS3180, Seong-Nam, Republic of Korea).
4.12. mRFP-GFP-LC3 Puncta Formation Assay
VK2/E6E7 cells (2 105) were seeded in a six-well confocal plate. After 24 h, cells were transfected with 1.5 ng of mRFP-GFP-LC3 plasmid using DharmaFECT transfection reagent (Horizon Discovery, Cambridge, UK) as per the manufacturer’s instructions and incubated for 48 h. Next, cells were treated with ethanol (control) or 500 µM aldehyde 13-13 for 30 min. Cells were washed once with PBS and fixed with 4% paraformaldehyde for 10 min. mRFP and GFP fluorescent puncta were observed under an LSM510 META confocal microscope and images were analyzed with LSM700 version 3.2 software (Carl Zeiss, Jena, Germany).
4.13. Monodansylcadaverine Staining Assay
VK2/E6E7 cells were seeded in a black or clear 96-well plate at 1 104 per well. After 24 h, cells were treated with ethanol, rapamycin (100 nM), or aldehyde 13-13 (500 µM) for 30 min and autophagy vacuoles were stained using a monodansylcadaverine (MDC) staining kit (Cayman, Ann Arbor, MI) as per the manufacturer’s protocol. Autophagy vacuoles were detected under a fluorescence microscope (Nikon Eclipse Ti) and images were analyzed using NIS-elements F software (Nikon Instruments, Inc., New York, NY, USA).
4.14. Autophagy Analysis by Transmission Electron Microscopy
VK2 cells (5 × 106) were cultured in 175T flasks for 24 h and treated with aldehyde 13-13 (500 µM) for 30 min. For TEM, cells were fixed with Karnovsky’s fixative and stored at 4 °C overnight. Cells were post-fixed with 1% osmium tetroxide and stained with 0.5% uranyl acetate overnight followed by dehydration in an ethanol gradient. The cells were infiltrated with Spurr’s resin overnight and visualized using a JEM1010 electron microscope at 120 kV (JEOL, Tokyo, Japan).
4.15. Senescence-Associated -Galactosidase Staining
VK2/E6E7 cells were seeded in 24-well plates at 5 × 104/well. Cells were grown for 24 h and treated with ethanol (control) or aldehyde 13-13 (15, 30, or 40 µM) for 1 h before H2O2 (300 µM) treatment. Next, the cells were incubated at 37 °C in 5% CO2 for 3 days to induce senescence.
4.16. Ki-67 Immunocytochemical Staining
VK2/E6E7 cells (3 104) were seeded on glass coverslips for 24 h. Cells were washed twice with PBS and fixed with 4% paraformaldehyde for 10 min. Fixed cells were incubated with 5% bovine serum albumin (BSA) in TBS-Tween 20 (0.1%) for 45 min and then with Ki-67 antibody (1:500, Abcam, UK) overnight at 4 °C. Next, cells were incubated with the Alexa Fluor 488-tagged anti-IgG secondary antibody (1:1000; Invitrogen) for 1 h in the dark. After washing, nuclei were stained with DAPI (Invitrogen) for 5 min. Coverslips were embedded in anti-fade mounting solution (Thermo Fisher Scientific) and slides were visualized using an LSM510 Meta confocal microscope and LSM700 v. 3.2 software (Carl Zeiss).
4.17. Annexin V and Propidium Iodide Staining
Cells were seeded in a six-well plate at 3 105/well and grown for 24 h. Proliferating cells were treated with vehicle and 40 µM aldehyde 13-13. Senescent cells were cotreated with 200 µM H2O2 plus vehicle and aldehyde 13-13. Next, cells were stained and incubated with 3 µL Annexin-V-FITC (BD Biosciences, CA, USA) and 10 µL propidium iodide (BD Biosciences, CA, USA) for 20 min and apoptosis analysis was performed using Accuri C6 Plus FACS (BD Biosciences).
4.18. Budding Yeast and Culture Conditions
Saccharomyces cerevisiae BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0, ura3Δ0) was streaked onto yeast extract peptone-dextrose (YPD) agar containing 2% Bacto agar, 1% Bacto yeast extract, 2% Bacto peptone, and 2% Difco dextrose (BD Biosciences). The plates were incubated at 30 °C to obtain isolated single colonies. A few picked colonies were inoculated into 10 mL of YPD medium (1% Bacto yeast extract, 2% Bacto peptone, and 2% Difco dextrose), and cultured overnight to make a seed culture. The seed culture was inoculated into 20 mL of YPD medium to an optical density at 600 nm of 0.2. All yeast cultures were incubated at 30 °C in an orbital shaker at 200 rpm.
4.19. Measurement of Chronological Lifespan by PI Staining and Flow Cytometry
Yeast cells were harvested by centrifugation, washed by resuspending in 1 mL of phosphate-buffered saline (PBS) and incubated for 20 min at 30 ℃ after adding 5 µL of PI solution (1 mg/mL, Sigma Aldrich). Stained cells were analyzed using a flow cytometer (FACS Verse; Becton Dickinson) with excitation at 488 nm and emission at 585 nm. Approximately 20,000 cells were analyzed per sample, and data were analyzed using Cell Quest software v3.3 (Becton Dickinson).
4.20. Colony-Forming Unit Assay of Budding Yeast
A colony-forming unit (CFU) assay was performed to determine yeast viability. Yeasts were harvested, serially diluted in PBS, counted using a hemocytometer, and 200 cells were spread onto YPD agar and incubated at 30 °C until colony formation.
4.21. Caenorhabditis elegans (C. elegans) Longevity Assay
C. elegans strains were cultured on fresh NGM plates for 2–3 generations without starvation, and lifespan analysis was conducted at 20 °C. Worms were synchronized by isolating eggs from gravid adults using hypochlorite and NaOH. When worms reached the L4 stage, 2′-deoxy-5-fluorouridine (FUdR, 0.6 mM) was added to prevent internal hatching. Aldehyde 13-13 (10 mM) was added to each well along with OP50 on the first day of the lifespan assay. Ethanol (0.1%) was vehicle in the control group. Wells containing > 15 worms were excluded from the analysis because of the risk of dietary restriction. Worm movement was used to determine whether the animals were alive or dead. Survival curves were analyzed by the log-rank (Mantel–Cox) method.
4.22. Statistical Analysis
Results are means ± standard error of the mean (SEM). To generate dose–response curves, SigmaPlot 10.0 was used, and curves were fitted using the Hill three-parameter equation. Significance was examined by two-tailed unpaired t-test. Data were subjected to one-way ANOVA followed by Tukey’s HSD test or Student’s t-test for multiple or two-group comparisons. Different letters indicate a significant difference at p < 0.05 (ANOVA).


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








