

Identification of mIDH1 R132C/S280F Inhibitors from Natural Products by Integrated Molecular Docking, Pharmacophore Modeling and Molecular Dynamics Simulations


Abstract
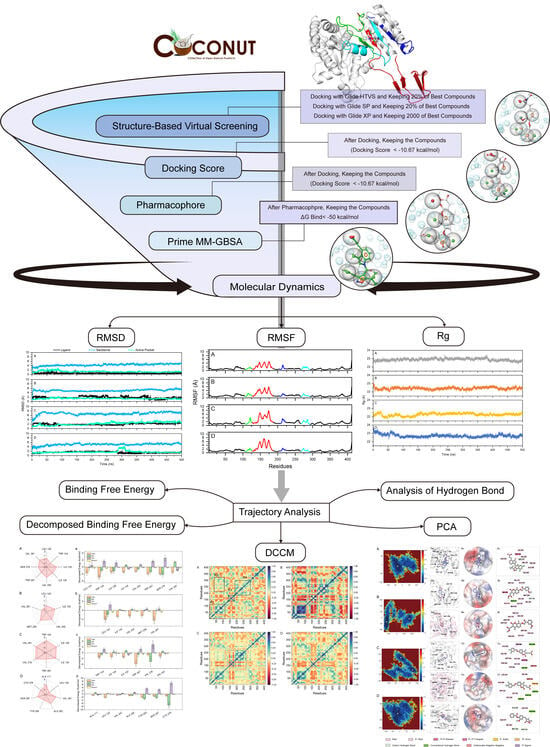
:
1. Introduction
2. Results and Discussion
2.1. Virtual Screening of Natural Compounds for mIDH1 Inhibitor
2.2. ADME Prediction
2.3. The Stability of the mIDH1 Inhibitor System
2.4. Dynamic Cross-Correlation Maps and Free Energy Landscapes
2.5. Analysis of Hydrogen Bond
2.6. Analysis of Binding Free Energy
3. Materials and Methods
3.1. Preparation of Receptor and Ligands
3.2. Structure-Based Virtual Screening
3.3. Pharmacophore-Based Virtual Screening
3.4. ADME Prediction and Prime MM-GBSA
3.5. Molecular Dynamics Simulations
3.6. Calculation of Binding Free Energy
3.7. The Computation of DCCM and PCA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cairns, R.A.; Mak, T.W. Oncogenic isocitrate dehydrogenase mutations: Mechanisms, models, and clinical opportunities. Cancer Discov. 2013, 3, 730–741. [Google Scholar] [CrossRef]
- Yang, H.; Ye, D.; Guan, K.L.; Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Abbas, S.; Lugthart, S.; Kavelaars, F.G.; Schelen, A.; Koenders, J.E.; Zeilemaker, A.; van Putten, W.J.; Rijneveld, A.W.; Löwenberg, B.; Valk, P.J. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: Prevalence and prognostic value. Blood 2010, 116, 2122–2126. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.C.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Chan, S.M.; Swords, R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 2017, 31, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. [Google Scholar] [CrossRef]
- Liu, S.; Cadoux-Hudson, T.; Schofield, C.J. Isocitrate dehydrogenase variants in cancer—Cellular consequences and therapeutic opportunities. Curr. Opin. Chem. Biol. 2020, 57, 122–134. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Schvartzman, J.M.; Reuter, V.P.; Koche, R.P.; Thompson, C.B. 2-hydroxyglutarate inhibits MyoD-mediated differentiation by preventing H3K9 demethylation. Proc. Natl. Acad. Sci. USA 2019, 116, 12851–12856. [Google Scholar] [CrossRef]
- Carbonneau, M.M.; Gagné, L.; Lalonde, M.E.; Germain, M.A.; Motorina, A.; Guiot, M.C.; Secco, B.; Vincent, E.E.; Tumber, A.; Hulea, L.; et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 2016, 7, 12700. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Finley, L.W.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, P.L.; Oeck, S.; Dow, J.; Economos, N.G.; Mirfakhraie, L.; Liu, Y.; Noronha, K.; Bao, X.; Li, J.; Shuch, B.M.; et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 2020, 582, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gong, Y. Isocitrate dehydrogenase inhibitors in acute myeloid leukemia. Biomark. Res. 2019, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Cleary, J.M.; Rouaisnel, B.; Daina, A.; Raghavan, S.; Roller, L.A.; Huffman, B.M.; Singh, H.; Wen, P.Y.; Bardeesy, N.; Zoete, V.; et al. Secondary IDH1 resistance mutations and oncogenic IDH2 mutations cause acquired resistance to ivosidenib in cholangiocarcinoma. NPJ Precis. Oncol. 2022, 6, 61. [Google Scholar] [CrossRef]
- Oltvai, Z.N.; Harley, S.E.; Koes, D.; Michel, S.; Warlick, E.D.; Nelson, A.C.; Yohe, S.; Mroz, P. Assessing acquired resistance to IDH1 inhibitor therapy by full-exon IDH1 sequencing and structural modeling. Cold Spring Harb. Mol. Case Stud. 2021, 7, a006007. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Shih, A.H.; Wang, B.; Nazir, A.; Rustenburg, A.S.; Albanese, S.K.; Patel, M.; Famulare, C.; Correa, F.M.; Takemoto, N.; et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018, 559, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Wang, H.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Watts, J.M.; Pollyea, D.A.; et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020, 4, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Daiichi Sankyo Co., Ltd. A Study of DS-1001b in Patients with Chemotherapy- and Radiotherapy-Naive IDH1 Mutated WHO Grade II Glioma. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04458272 (accessed on 9 September 2023).
- Natsume, A.; Arakawa, Y.; Narita, Y.; Sugiyama, K.; Hata, N.; Muragaki, Y.; Shinojima, N.; Kumabe, T.; Saito, R.; Motomura, K.; et al. The first-in-human phase I study of a brain-penetrant mutant IDH1 inhibitor DS-1001 in patients with recurrent or progressive IDH1-mutant gliomas. Neuro-Oncol. 2023, 25, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Machida, Y.; Nakagawa, M.; Matsunaga, H.; Yamaguchi, M.; Ogawara, Y.; Shima, Y.; Yamagata, K.; Katsumoto, T.; Hattori, A.; Itoh, M.; et al. A Potent Blood-Brain Barrier-Permeable Mutant IDH1 Inhibitor Suppresses the Growth of Glioblastoma with IDH1 Mutation in a Patient-Derived Orthotopic Xenograft Model. Mol. Cancer Ther. 2020, 19, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Reinbold, R.; Hvinden, I.C.; Rabe, P.; Herold, R.A.; Finch, A.; Wood, J.; Morgan, M.; Staudt, M.; Clifton, I.J.; Armstrong, F.A.; et al. Resistance to the isocitrate dehydrogenase 1 mutant inhibitor ivosidenib can be overcome by alternative dimer-interface binding inhibitors. Nat. Commun. 2022, 13, 4785. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, S.; Lai, H.; Jin, R.; Long, X.; Li, N.; Tang, Y.; Guo, H.; Yao, X.; Leung, E.L. Discovery of Novel IDH1 Inhibitor through Comparative Structure-Based Virtual Screening. Front. Pharmacol. 2020, 11, 579768. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, J.; Li, Y.; Huang, J.; Jiang, Z.; Wang, Y.; Liu, L.; Leung, E.L.H.; Yao, X. Identification of a Novel Protein Arginine Methyltransferase 5 Inhibitor in Non-small Cell Lung Cancer by Structure-Based Virtual Screening. Front. Pharmacol. 2018, 9, 173. [Google Scholar] [CrossRef]
- Ge, H.; Mao, L.; Zhao, J.; Wang, Y.; Shi, D.; Yang, X.; Wang, X.; Liu, H.; Yao, X. Discovery of novel IDO1 inhibitors via structure-based virtual screening and biological assays. J. Comput. Aided Mol. Des. 2021, 35, 679–694. [Google Scholar] [CrossRef]
- Tian, W.; Zhang, W.; Wang, Y.; Jin, R.; Wang, Y.; Guo, H.; Tang, Y.; Yao, X. Recent advances of IDH1 mutant inhibitor in cancer therapy. Front. Pharmacol. 2022, 13, 982424. [Google Scholar] [CrossRef]
- Zou, F.; Pusch, S.; Hua, J.; Ma, T.; Yang, L.; Zhu, Q.; Xu, Y.; Gu, Y.; von Deimling, A.; Zha, X. Identification of novel allosteric inhibitors of mutant isocitrate dehydrogenase 1 by cross docking-based virtual screening. Bioorganic Med. Chem. Lett. 2018, 28, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Thamim, M.; Agrahari, A.K.; Gupta, P.; Thirumoorthy, K. Rational Computational Approaches in Drug Discovery: Potential Inhibitors for Allosteric Regulation of Mutant Isocitrate Dehydrogenase-1 Enzyme in Cancers. Molecules 2023, 28, 2315. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Tang, R.; Deng, Y.; Yang, K.; Chen, L.; Li, H. Steroids from Ganoderma sinense as new natural inhibitors of cancer-associated mutant IDH1. Bioorganic Chem. 2018, 79, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Yang, F.; Qin, L.; Kuai, J.; Yang, L.; Zhang, L.; Sun, P.; Li, G.; Wang, X. Computational study on novel natural compound inhibitor targeting IDH1_R132H. Aging 2022, 14, 5478–5492. [Google Scholar] [CrossRef]
- Chan, W.J.; Adiwidjaja, J.; McLachlan, A.J.; Boddy, A.V.; Harnett, J.E. Interactions between natural products and cancer treatments: Underlying mechanisms and clinical importance. Cancer Chemother. Pharmacol. 2023, 91, 103–119. [Google Scholar] [CrossRef]
- Drasar, P.B.; Khripach, V.A. Growing Importance of Natural Products Research. Molecules 2019, 25, 6. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Sorokina, M.; Merseburger, P.; Rajan, K.; Yirik, M.A.; Steinbeck, C. COCONUT online: Collection of Open Natural Products database. J. Cheminformatics 2021, 13, 2. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, M.; Li, A.; Yao, X.; Chen, Y. Unraveling the allosteric inhibition mechanism of PARP-1 CAT and the D766/770A mutation effects via Gaussian accelerated molecular dynamics and Markov state model. Comput. Biol. Med. 2024, 168, 107682. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shao, X.; Leung, E.L.H.; Chen, Y.; Yao, X. Selectively targeting individual bromodomain: Drug discovery and molecular mechanisms. Pharmacol. Res. 2021, 172, 105804. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wang, Z.; Wang, L.; Wang, Q.; Tang, R.; Xiang, S.; Deng, Q.; Hou, T.; Sun, H. Deciphering the Shared and Specific Drug Resistance Mechanisms of Anaplastic Lymphoma Kinase via Binding Free Energy Computation. Research 2023, 6, 0170. [Google Scholar] [CrossRef] [PubMed]
- Prlic, A.; Kalro, T.; Bhattacharya, R.; Christie, C.; Burley, S.K.; Rose, P.W. Integrating genomic information with protein sequence and 3D atomic level structure at the RCSB protein data bank. Bioinformatics 2016, 32, 3833–3835. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC, New York, NY, 2015. Available online: https://www.schrodinger.com/training/maestro11/quick-start-guide/loading-and-preparing-a-protein-structure (accessed on 28 November 2022).
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKaprediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hy drophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Patel, H.M.; Ahmad, I.; Pawara, R.; Shaikh, M.; Surana, S. In silico search of triple mutant T790M/C797S allosteric inhibitors to conquer acquired resistance problem in non-small cell lung cancer (NSCLC): A combined approach of structure-based virtual screening and molecular dynamics simulation. J. Biomol. Struct. Dyn. 2021, 39, 1491–1505. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 2010, 29, 157–170. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, X.; Zhang, S.; Liang, S.; Zhang, Q.; Chen, J. Deciphering the binding mechanism of inhibitors of the SARS-CoV-2 main protease through multiple replica accelerated molecular dynamics simulations and free energy landscapes. Phys. Chem. Chem. Phys. PCCP 2022, 24, 22129–22143. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.S.; Liu, X.G.; Cui, Y.X.; Zhang, S.L.; Zhang, Q.G.; Chen, J.Z. Molecular mechanism concerning conformational changes of CDK2 mediated by binding of inhibitors using molecular dynamics simulations and principal component analysis. SAR QSAR Environ. Res. 2021, 32, 573–594. [Google Scholar] [CrossRef] [PubMed]
- Lua, R.C.; Lichtarge, O. PyETV: A PyMOL evolutionary trace viewer to analyze functional site predictions in protein complexes. Bioinformatics 2010, 26, 2981–2982. [Google Scholar] [CrossRef]
- Accelrys Software Inc., San Diego, CA, USA. Available online: https://accelrys-discovery-studio-visualizer.software.informer.com/3.0/ (accessed on 28 November 2022).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | MW | SP Docking Score (kcal/mol) | XP Docking Score (kcal/mol) | Phase Screen Score | Prime MM-GBSA (kcal/mol) |
|---|---|---|---|---|---|
| DS1001b | 535.79 | −10.67 | −14.09 | 2.61 | −84.90 |
| CNP0119040 | 410.42 | −8.60 | −10.75 | 1.53 | −65.57 |
| CNP0243438 | 407.25 | −8.84 | −11.83 | 1.54 | −60.72 |
| CNP0449118 | 357.36 | −9.61 | −11.34 | 1.51 | −59.10 |
| CNP0348579 | 352.39 | −8.72 | −11.65 | 1.50 | −57.48 |
| CNP0294912 | 366.41 | −9.89 | −10.72 | 1.60 | −54.00 |
| CNP0135500 | 416.81 | −8.68 | −10.68 | 1.62 | −53.26 |
| CNP0349353 | 399.31 | −8.79 | −11.24 | 1.60 | −52.91 |
| CNP0286492 | 390.35 | −9.57 | −11.27 | 1.51 | −52.79 |
| CNP0290966 | 437.45 | −10.03 | −11.64 | 1.50 | −51.28 |
| CNP0234840 | 406.43 | −6.72 | −11.21 | 1.63 | −51.13 |
| CNP0404801 | 449.46 | −9.09 | −11.66 | 1.66 | −50.77 |
| CNP0223368 | 492.52 | −9.49 | −11.08 | 1.56 | −50.47 |
| ID | a CNS | b DonorHB | c AccptHB | d QplogPo/w | e QPlogPC16 | f QPlogPoct | g QplogPw | h QPlogS | i CIQPlogS | j Qplog HERG | k QPPCaco | l QPlogBB | m QPPMDCK | n QPlogKp | # Metab | o Qplog Khsa | p Human Oral Absorption | q Percent Human Oral Absorption |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DS1001b | −1 | 1 | 6.50 | 5.82 | 15.14 | 22.340 | 10.80 | −7.55 | −8.73 | −3.76 | 79.38 | −0.91 | 394.98 | −3.25 | 1 | 0.82 | 1 | 69.09 |
| CNP0119040 | −2 | 0 | 8.00 | 3.09 | 12.75 | 18.87 | 10.13 | −4.69 | −5.04 | −5.88 | 424.82 | −1.40 | 196.10 | −2.81 | 6 | −0.05 | 3 | 92.10 |
| CNP0243438 | −2 | 1 | 5.25 | 4.63 | 13.06 | 19.12 | 9.41 | −6.21 | −6.36 | −4.19 | 84.18 | −1.12 | 222.44 | −2.95 | 5 | 0.35 | 1 | 88.54 |
| CNP0449118 | −2 | 1.25 | 7.00 | 2.66 | 12.63 | 19.00 | 13.97 | −3.98 | −3.77 | −3.23 | 32.97 | −1.73 | 28.88 | −3.00 | 3 | −0.39 | 2 | 69.67 |
| CNP0348579 | −2 | 1 | 5.25 | 4.03 | 12.14 | 17.86 | 9.54 | −5.48 | −5.24 | −4.29 | 82.57 | −1.44 | 42.46 | −2.87 | 6 | 0.29 | 3 | 84.88 |
| CNP0294912 | −1 | 0 | 5.25 | 4.33 | 12.01 | 16.94 | 7.69 | −6.05 | −5.38 | −6.05 | 1068.85 | −0.84 | 531.63 | −2.14 | 6 | 0.58 | 3 | 100.00 |
| CNP0135500 | 0 | 0 | 6.75 | 3.50 | 11.89 | 17.33 | 9.06 | −4.78 | −5.87 | −5.62 | 1056.24 | −0.63 | 1114.00 | −2.17 | 4 | 0.04 | 3 | 100.00 |
| CNP0349353 | −1 | 1 | 4.75 | 4.93 | 11.65 | 18.28 | 7.37 | −6.34 | −5.82 | −2.96 | 137.08 | −0.87 | 299.42 | −3.26 | 3 | 0.55 | 1 | 94.03 |
| CNP0286492 | −2 | 1 | 6.75 | 2.77 | 12.37 | 18.37 | 10.47 | −4.89 | −5.17 | −3.73 | 8.32 | −2.67 | 3.56 | −5.06 | 6 | 0.01 | 2 | 59.66 |
| CNP0290966 | 1 | 0 | 8.75 | 2.45 | 12.22 | 20.36 | 11.01 | −3.33 | −4.44 | −6.20 | 249.08 | −0.36 | 121.83 | −4.56 | 6 | −0.12 | 3 | 84.19 |
| CNP0234840 | −2 | 3 | 8.40 | 2.54 | 12.90 | 21.13 | 13.24 | −3.85 | −4.79 | −5.17 | 239.10 | −1.94 | 105.36 | −2.96 | 8 | −0.12 | 2 | 84.38 |
| CNP0404801 | −2 | 0 | 7.50 | 3.95 | 14.69 | 21.09 | 10.89 | −6.07 | −6.33 | −6.63 | 251.65 | −1.66 | 111.35 | −2.96 | 6 | 0.41 | 3 | 93.02 |
| CNP0223368 | −2 | 5 | 7.95 | 3.30 | 17.06 | 28.20 | 17.66 | −5.74 | −7.02 | −6.41 | 46.48 | −2.87 | 17.94 | −3.96 | 8 | 0.36 | 2 | 76.09 |
| Complex | Acceptor | Donor | Occupancy (%) | Distance (Å) | Angle (°) |
|---|---|---|---|---|---|
| mIDH1-DS-1001b | ligand@O1 | ILE_128@N-H | 30.30% | 3.09 | 143.57 |
| ligand@O2 | ALA_111@N-H | 2.93% | 3.06 | 153.10 | |
| ligand@O2 | ILE_128@N-H | 2.09% | 3.17 | 145.87 | |
| ligand@O2 | ARG_119@NH2-H | 1.91% | 3.12 | 131.69 | |
| mIDH1-CNP0119040 | ligand@O6 | LEU_120@N-H | 14.08% | 3.01 | 157.39 |
| ligand@O6 | SER_287@OG-H | 5.60% | 2.76 | 163.79 | |
| ligand@O1 | TRP_124@NE1-H | 5.33% | 2.94 | 152.86 | |
| ligand@O6 | SER_278@OG-H | 4.89% | 2.80 | 159.08 | |
| ligand@O1 | ARG_119@NH1-H | 1.42% | 2.89 | 152.98 | |
| mIDH1-CNP0243438 | ligand@O4 | TRP_267@NE1-H | 2.22% | 3.06 | 139.10 |
| ligand@O5 | TRP_267@NE1-H | 1.73% | 3.09 | 138.79 | |
| ligand@O4 | ASN_271@ND2-H | 1.47% | 3.11 | 153.96 | |
| ligand@O5 | TYR_135@OH-H | 1.07% | 2.93 | 151.19 | |
| ligand@O4 | SER_278@N-H | 1.07% | 3.19 | 130.08 | |
| ligand@O1 | TRP_267@NE1-H | 1.02% | 3.22 | 126.50 | |
| mIDH1-CNP0449118 | CYS_379@O | ligand@N1-H | 16.53% | 2.85 | 141.71 |
| ligand@O1 | SER_287@OG-H | 16.44% | 2.81 | 154.95 | |
| ligand@O3 | SER_287@OG-H | 5.82% | 3.23 | 136.59 | |
| ligand@O2 | SER_287@OG-H | 4.53% | 3.23 | 145.12 |
| Terms | DS-1001b | CNP0119040 | CNP0243438 | CNP0449118 |
|---|---|---|---|---|
| ΔEvdw | −49.51 ± 3.25 | −39.97 ± 3.76 | −43.70 ± 6.11 | −40.62 ± 2.18 |
| ΔEele | −57.11 ± 7.16 | −7.55 ± 3.15 | −74.58 ± 16.78 | −81.80 ± 8.72 |
| ΔGgas | −106.61 ± 8.10 | −47.52 ± 4.91 | −118.28 ± 20.38 | −122.42 ± 8.66 |
| ΔGGB | 76.29 ± 7.20 | 24.33 ± 2.97 | 92.22 ± 16.89 | 103.23 ± 8.21 |
| ΔGGBSUR | −6.63 ± 0.34 | −5.55 ± 0.44 | −5.26 ± 0.48 | −5.56 ± 0.19 |
| ΔGsol | 69.66 ± 7.12 | 18.78 ± 2.86 | 86.96 ± 16.62 | 97.67 ± 8.20 |
| ΔGbind | −36.95 ± 2.96 | −28.74 ± 3.93 | −31.32 ± 5.99 | −24.75 ± 2.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Bai, H.; Wang, Y.; Wang, X.; Jin, R.; Guo, H.; Lai, H.; Tang, Y.; Wang, Y. Identification of mIDH1 R132C/S280F Inhibitors from Natural Products by Integrated Molecular Docking, Pharmacophore Modeling and Molecular Dynamics Simulations. Pharmaceuticals 2024, 17, 336. https://doi.org/10.3390/ph17030336
Zhang W, Bai H, Wang Y, Wang X, Jin R, Guo H, Lai H, Tang Y, Wang Y. Identification of mIDH1 R132C/S280F Inhibitors from Natural Products by Integrated Molecular Docking, Pharmacophore Modeling and Molecular Dynamics Simulations. Pharmaceuticals. 2024; 17(3):336. https://doi.org/10.3390/ph17030336
Chicago/Turabian StyleZhang, Weitong, Hailong Bai, Yifan Wang, Xiaorui Wang, Ruyi Jin, Hui Guo, Huanling Lai, Yuping Tang, and Yuwei Wang. 2024. "Identification of mIDH1 R132C/S280F Inhibitors from Natural Products by Integrated Molecular Docking, Pharmacophore Modeling and Molecular Dynamics Simulations" Pharmaceuticals 17, no. 3: 336. https://doi.org/10.3390/ph17030336
APA StyleZhang, W., Bai, H., Wang, Y., Wang, X., Jin, R., Guo, H., Lai, H., Tang, Y., & Wang, Y. (2024). Identification of mIDH1 R132C/S280F Inhibitors from Natural Products by Integrated Molecular Docking, Pharmacophore Modeling and Molecular Dynamics Simulations. Pharmaceuticals, 17(3), 336. https://doi.org/10.3390/ph17030336