

AutoPepVax, a Novel Machine-Learning-Based Program for Vaccine Design: Application to a Pan-Cancer Vaccine Targeting EGFR Missense Mutations
 ,
, 
Abstract
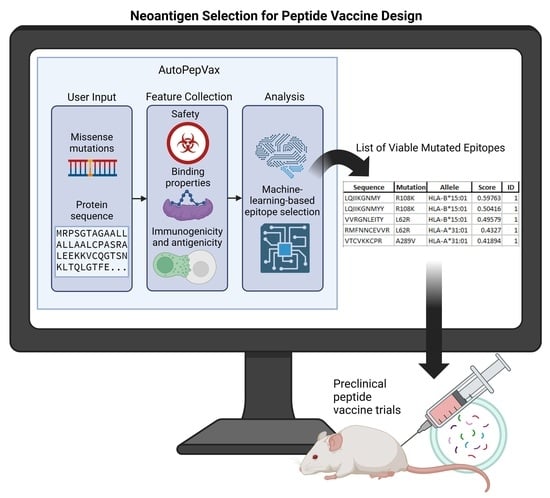
:
1. Introduction
2. Results
2.1. Workflow of Results
2.2. Validating AutoPepVax’s MHC-I-Restricted Epitope Assessment Model
2.3. AutoPepVax Operation
2.4. EGFR-Mutated Epitopes Identified by AutoPepVax
2.5. Pan-Cancer Vaccine Population Coverage
2.6. Population Coverage
2.7. TCR Models and Binding
3. Discussion
4. Limitations
5. Future Directions
6. Conclusions
7. Materials and Methods
7.1. AutoPepVax Data Collection: Developing Functions to Obtain Epitope Characteristics
7.2. AutoPepVax Selection and Ranking of EHLA-I Pairs with Machine-Learning-Based Models
7.3. AutoPepVax Filtration of EHLA-II Pairs
7.4. Applying AutoPepVax to Design of EGFR Peptide Vaccine
7.5. Determining Population Coverage of Composite Vaccines
7.6. Modeling of Peptide–MHC Complexes and TCR Interactions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, F.; Deng, L.; Jackson, K.R.; Talukder, A.H.; Katailiha, A.S.; Bradley, S.D.; Zou, Q.; Chen, C.; Huo, C.; Chiu, Y.; et al. Neoantigen vaccination induces clinical and immunologic responses in non-small cell lung cancer patients harboring EGFR mutations. J. Immunother. Cancer 2021, 9, e002531. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tang, H.; Li, L.; Wang, X.; Yu, Z.; Li, J. Peptide-based therapeutic cancer vaccine: Current trends in clinical application. Cell Prolif. 2021, 54, e13025. [Google Scholar] [CrossRef]
- Savsani, K.; Jabbour, G.; Dakshanamurthy, S. A New Epitope Selection Method: Application to Design a Multi-Valent Epitope Vaccine Targeting HRAS Oncogene in Squamous Cell Carcinoma. Vaccines 2021, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Savsani, K.; Dakshanamurthy, S. A Peptide Vaccine Design Targeting KIT Mutations in Acute Myeloid Leukemia. Pharmaceuticals 2023, 16, 932. [Google Scholar] [CrossRef] [PubMed]
- Suri, S.; Dakshanamurthy, S. IntegralVac: A Machine Learning-Based Comprehensive Multivalent Epitope Vaccine Design Method. Vaccines 2022, 10, 1678. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Gartner, J.J.; Parkhurst, M.R.; Gros, A.; Tran, E.; Jafferji, M.S.; Copeland, A.; Hanada, K.-I.; Zacharakis, N.; Lalani, A.; Krishna, S.; et al. A machine learning model for ranking candidate HLA class I neoantigens based on known neoepitopes from multiple human tumor types. Nat. Cancer 2021, 2, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; O’Rourke, D.M.; Tran, D.D.; Fink, K.L.; Nabors, L.B.; Li, G.; Bota, D.A.; Lukas, R.V.; et al. Rindopepimut with Bevacizumab for Patients with Relapsed EGFRvIII-Expressing Glioblastoma (ReACT): Results of a Double-Blind Randomized Phase II Trial. Clin. Cancer Res. 2020, 26, 1586–1594. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Celldex Therapeutics. A Phase II Study of Rindopepimut/GM-CSF in Patients With Relapsed EGFRvIII-Positive Glioblastoma. clinicaltrials.gov. 2020. Available online: https://clinicaltrials.gov/study/NCT01498328 (accessed on 4 August 2023).
- Celldex Therapeutics. An International, Randomized, Double-Blind, Controlled Study of Rindopepimut/GM-CSF With Adjuvant Temozolomide in Patients With Newly Diagnosed, Surgically Resected, EGFRvIII-Positive Glioblastoma. clinicaltrials.gov. 2018. Available online: https://clinicaltrials.gov/study/NCT01480479 (accessed on 4 August 2023).
- MD LZ. Phase I Study of Individualized Neoantigen Peptides in the Treatment of EGFR Mutant Non-Small Cell Lung Cancer. clinicaltrials.gov. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04397926 (accessed on 12 June 2023).
- Pan, J.; Xiong, D.; Zhang, Q.; Palen, K.; Shoemaker, R.H.; Johnson, B.; Sei, S.; Wang, Y.; You, M. Precision immunointerception of EGFR-driven tumorigenesis for lung cancer prevention. Front. Immunol. 2023, 14, 1036563. [Google Scholar] [CrossRef]
- Ebben, J.D.; Lubet, R.A.; Gad, E.; Disis, M.L.; You, M. Epidermal Growth Factor Receptor Derived Peptide Vaccination to Prevent Lung Adenocarcinoma Formation: An In Vivo Study in a Murine Model of EGFR Mutant Lung Cancer. Mol. Carcinog. 2015, 55, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Putzu, C.; Canova, S.; Paliogiannis, P.; Lobrano, R.; Sala, L.; Cortinovis, D.L.; Colonese, F. Duration of Immunotherapy in Non-Small Cell Lung Cancer Survivors: A Lifelong Commitment? Cancers 2023, 15, 689. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.; Reid, T.R.; Oronsky, A.; Sandhu, N.; Knox, S.J. A Review of Newly Diagnosed Glioblastoma. Front. Oncol. 2021, 10, 574012. [Google Scholar] [CrossRef] [PubMed]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [PubMed]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal Growth Factor Receptor in Glioma: Signal Transduction, Neuropathology, Imaging, and Radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Bonner, J.A.; Bredel, M. EGFR Mutations in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 3818. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Kim, J.; Choi, Y.J.; Kang, J.G. Clinical study of colorectal cancer operation: Survival analysis. Korean J. Clin. Oncol. 2020, 16, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Cho, D.; Kim, H.; Kim, S.; Cha, Y.; Greulich, H.; Bass, A.; Cho, H.; Cho, J. Colorectal adenocarcinoma-derived EGFR mutants are oncogenic and sensitive to EGFR-targeted monoclonal antibodies, cetuximab and panitumumab. Int. J. Cancer 2020, 146, 2194–2200. [Google Scholar] [CrossRef]
- Jia, W.; Zhang, T.; Huang, H.; Feng, H.; Wang, S.; Guo, Z.; Luo, Z.; Ji, X.; Cheng, X.; Zhao, R. Colorectal cancer vaccines: The current scenario and future prospects. Front. Immunol. 2022, 13, 942235. [Google Scholar] [CrossRef]
- Hung, K.; Hayashi, R.; Lafond-Walker, A.; Lowenstein, C.; Pardoll, D.; Levitsky, H. The Central Role of CD4+ T Cells in the Antitumor Immune Response. J. Exp. Med. 1998, 188, 2357–2368. [Google Scholar] [CrossRef]
- Giuntoli, R.L.; Lu, J.; Kobayashi, H.; Kennedy, R.; Celis, E. Direct Costimulation of Tumor-reactive CTL by Helper T Cells Potentiate Their Proliferation, Survival, and Effector Function. Clin Cancer Res. 2002, 8, 922–931. [Google Scholar] [PubMed]
- Bui, H.-H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006, 7, 153. [Google Scholar] [CrossRef]
- Gedeon, P.; Choi, B.; Sampson, J.; Bigner, D. Rindopepimut: Anti-EGFRvIII peptide vaccine, oncolytic. Drugs Futur. 2013, 38, 147–155. [Google Scholar] [CrossRef]
- MHCflurry 2.0: Improved Pan-Allele Prediction of MHC Class I-Presented Peptides by Incorporating Antigen Processing: Cell Systems. Available online: https://www.cell.com/cell-systems/fulltext/S2405-4712(20)30239-8?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS2405471220302398%3Fshowall%3Dtrue (accessed on 17 July 2023).
- Wang, M.; Kurgan, L.; Li, M. A comprehensive assessment and comparison of tools for HLA class I peptide-binding prediction. Briefings Bioinform. 2023, 24, bbad150. [Google Scholar] [CrossRef] [PubMed]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Pan-Specific Prediction of Peptide-MHC-I Complex Stability; A Correlate of T Cell Immunogenicity—PMC. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4976001/ (accessed on 17 July 2023).
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Springer Protocols Handbooks; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar] [CrossRef]
- EGFR—Epidermal Growth Factor Receptor—Homo sapiens (Human)|UniProtKB|UniProt. Available online: https://www.uniprot.org/uniprotkb/P00533/entry (accessed on 18 January 2024).
- Cosmic. EGFR Gene—COSMIC. Available online: https://cancer.sanger.ac.uk/cosmic/gene/analysis?ln=EGFR (accessed on 18 January 2024).
- Bank RPD. RCSB PDB: Homepage. Available online: https://www.rcsb.org/ (accessed on 16 January 2024).
- Zhou, P.; Jin, B.; Li, H.; Huang, S.-Y. HPEPDOCK: A web server for blind peptide–protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, 46, W443–W450. [Google Scholar] [CrossRef] [PubMed]
- PyMOL. pymol.org. Available online: https://pymol.org/2/ (accessed on 5 February 2024).
- Yin, R.; Ribeiro-Filho, H.V.; Lin, V.; Gowthaman, R.; Cheung, M.; Pierce, B.G. TCRmodel2: High-resolution modeling of T cell receptor recognition using deep learning. Nucleic Acids Res. 2023, 51, W569–W576. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| File | Description |
|---|---|
| CD4 Epitopes.csv | A list of all analyzed EHLA-II pairs and their pertinent characteristics. |
| CD4 Filtered Epitopes.csv | A filtered list of EHLA-II pairs that meet the exclusion criteria. |
| CD8 Epitopes.csv | A list of all analyzed EHLA-I pairs and their pertinent characteristics, including ID and score. |
| CD8 Filtered Epitopes.csv | A filtered list of EHLA-I pairs that meet the exclusion criteria. |
| Sequence.txt | A list of epitopes for internal use. |
| Cancer | Number of Mutations with Positive EHLA-I and EHLA-II Pairs | Total Mutations | Mutations with Overlapping Epitopes |
|---|---|---|---|
| Glioblastoma Multiforme | 1 | 8 | G598V |
| Colorectal Adenocarcinoma | 12 | 62 | R958H, G857R, L707S, E711V, P753L, S442R, G131R, L140V, E709K, R451C, S768G, T710A |
| Lung Adenocarcinoma | 5 | 11 | L861Q, E709K, L858R, G598V, S768I |
| Head and Neck Squamous Cell Carcinoma | 1 | 15 | E967A |
| Class | EHLA-I Optimized | EHLA-I Filtered | EHLA-II Optimized | EHLA-II Filtered |
|---|---|---|---|---|
| World Coverage a | 98.55% | 98.55% | 81.81% | 81.81% |
| Average Epitope Hit b | 2.3 | 30.88 | 1.11 | 38.74 |
| PC90 c | 1.51 | 11.2 | 0.55 | 19.24 |
| MHC Class I Alleles | HLA-A*01:01, HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*03:01, HLA-A*11:01, HLA-A*23:01, HLA-A*24:02, HLA-A*26:01, HLA-A*30:01, HLA-A*30:02, HLA-A*31:01, HLA-A*32:01, HLA-A*33:01, HLA-A*68:01, HLA-A*68:02, HLA-B*07:02, HLA-B*08:01, HLA-B*15:01, HLA-B*35:01, HLA-B*40:01, HLA-B*44:02, HLA-B*44:03, HLA-B*51:01, HLA-B*53:01, HLA-B*57:01, HLA-B*58:01 |
| MHC Class II Alleles | HLA-DRB1*01:01, HLA-DRB1*03:01, HLA-DRB1*04:01, HLA-DRB1*04:05, HLA-DRB1*07:01, HLA-DRB1*08:02, HLA-DRB1*09:01, HLA-DRB1*11:01, HLA-DRB1*12:01, HLA-DRB1*13:02, HLA-DRB1*15:01, HLA-DRB3*01:01, HLA-DRB3*02:02, HLA-DRB4*01:01, HLA-DRB5*01:01, HLA-DPA1*01:03/DPB1*02:01, HLA-DPA1*01:03/DPB1*04:01, HLA-DPA1*02:01/DPB1*01:01, HLA-DPA1*02:01/DPB1*05:01, HLA-DPA1*02:01/DPB1*14:01, HLA-DPA1*03:01/DPB1*04:02, HLA-DQA1*01:01/DQB1*05:01, HLA-DQA1*01:02/DQB1*06:02, HLA-DQA1*03:01/DQB1*03:02, HLA-DQA1*04:01/DQB1*04:02, HLA-DQA1*05:01/DQB1*02:01, HLA-DQA1*05:01/DQB1*03:01 |
| Parameter | Exclusion Criteria |
|---|---|
| Toxicity (EHLA-I and EHLA-II pairs) | Toxin |
| Half-life (EHLA-I and EHLA-II pairs) | >1 h |
| Instability Index (EHLA-I and EHLA-II pairs) | >40 |
| Allergenicity (EHLA-I and EHLA-II pairs) | Probable Allergen |
| IFNgamma (EHLA-II pairs) | Negative |
| Immunogenicity (EHLA-II pairs) | <50 |
| Antigenicity (EHLA-II pairs) | <0.4 |
| ID (EHLA-I pairs) | =0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bautista, E.; Jung, Y.H.; Jaramillo, M.; Ganesh, H.; Varma, A.; Savsani, K.; Dakshanamurthy, S. AutoPepVax, a Novel Machine-Learning-Based Program for Vaccine Design: Application to a Pan-Cancer Vaccine Targeting EGFR Missense Mutations. Pharmaceuticals 2024, 17, 419. https://doi.org/10.3390/ph17040419
Bautista E, Jung YH, Jaramillo M, Ganesh H, Varma A, Savsani K, Dakshanamurthy S. AutoPepVax, a Novel Machine-Learning-Based Program for Vaccine Design: Application to a Pan-Cancer Vaccine Targeting EGFR Missense Mutations. Pharmaceuticals. 2024; 17(4):419. https://doi.org/10.3390/ph17040419
Chicago/Turabian StyleBautista, Enrico, Young Hyun Jung, Manuela Jaramillo, Harrish Ganesh, Aryaan Varma, Kush Savsani, and Sivanesan Dakshanamurthy. 2024. "AutoPepVax, a Novel Machine-Learning-Based Program for Vaccine Design: Application to a Pan-Cancer Vaccine Targeting EGFR Missense Mutations" Pharmaceuticals 17, no. 4: 419. https://doi.org/10.3390/ph17040419
APA StyleBautista, E., Jung, Y. H., Jaramillo, M., Ganesh, H., Varma, A., Savsani, K., & Dakshanamurthy, S. (2024). AutoPepVax, a Novel Machine-Learning-Based Program for Vaccine Design: Application to a Pan-Cancer Vaccine Targeting EGFR Missense Mutations. Pharmaceuticals, 17(4), 419. https://doi.org/10.3390/ph17040419