Olesoxime (TRO19622): A Novel Mitochondrial-Targeted Neuroprotective Compound

Abstract
:1. Introduction
2. Discovery of Olesoxime
2.1. Chemistry

2.2. Molecular Pharmacology
3. Neuroprotection
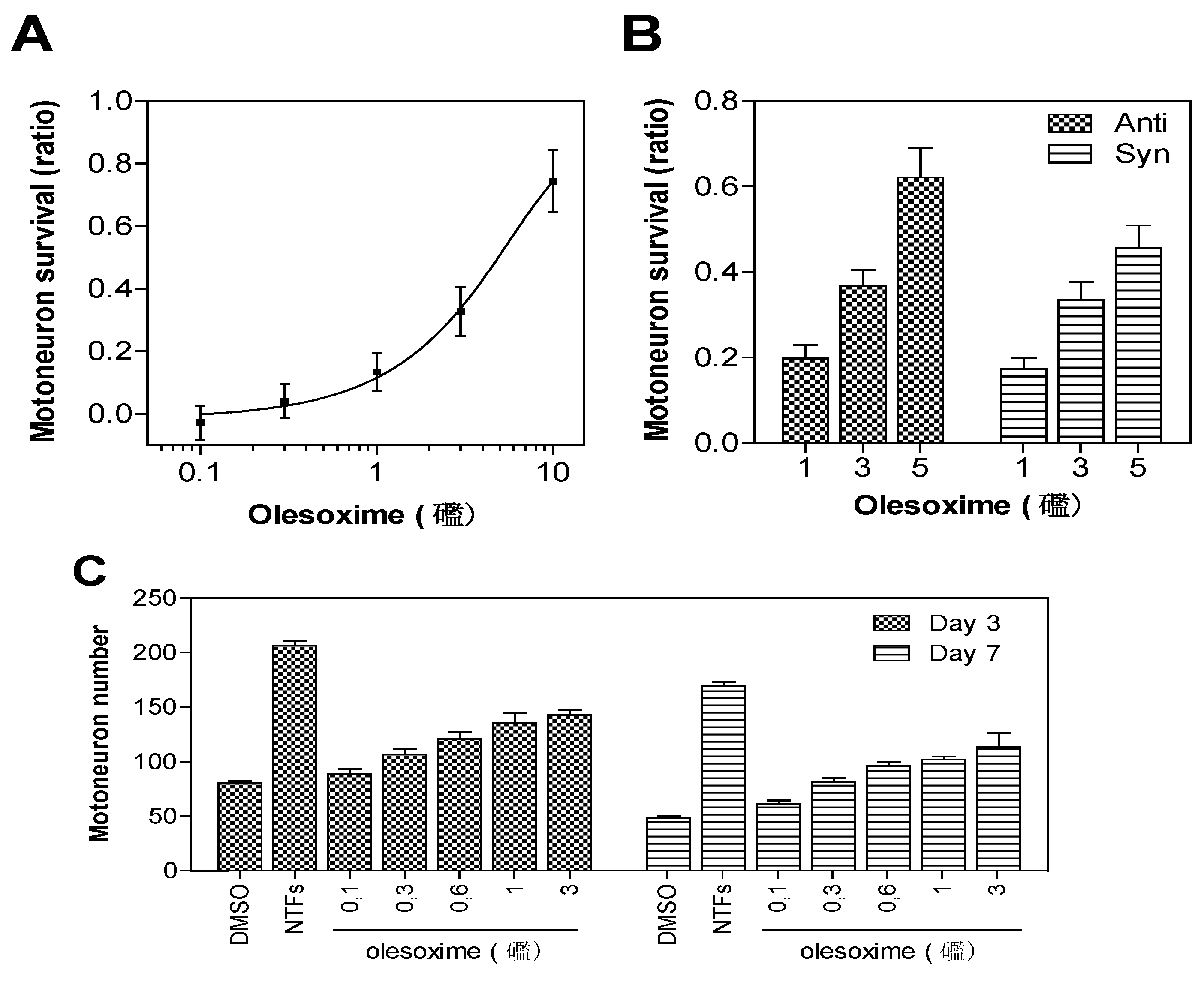
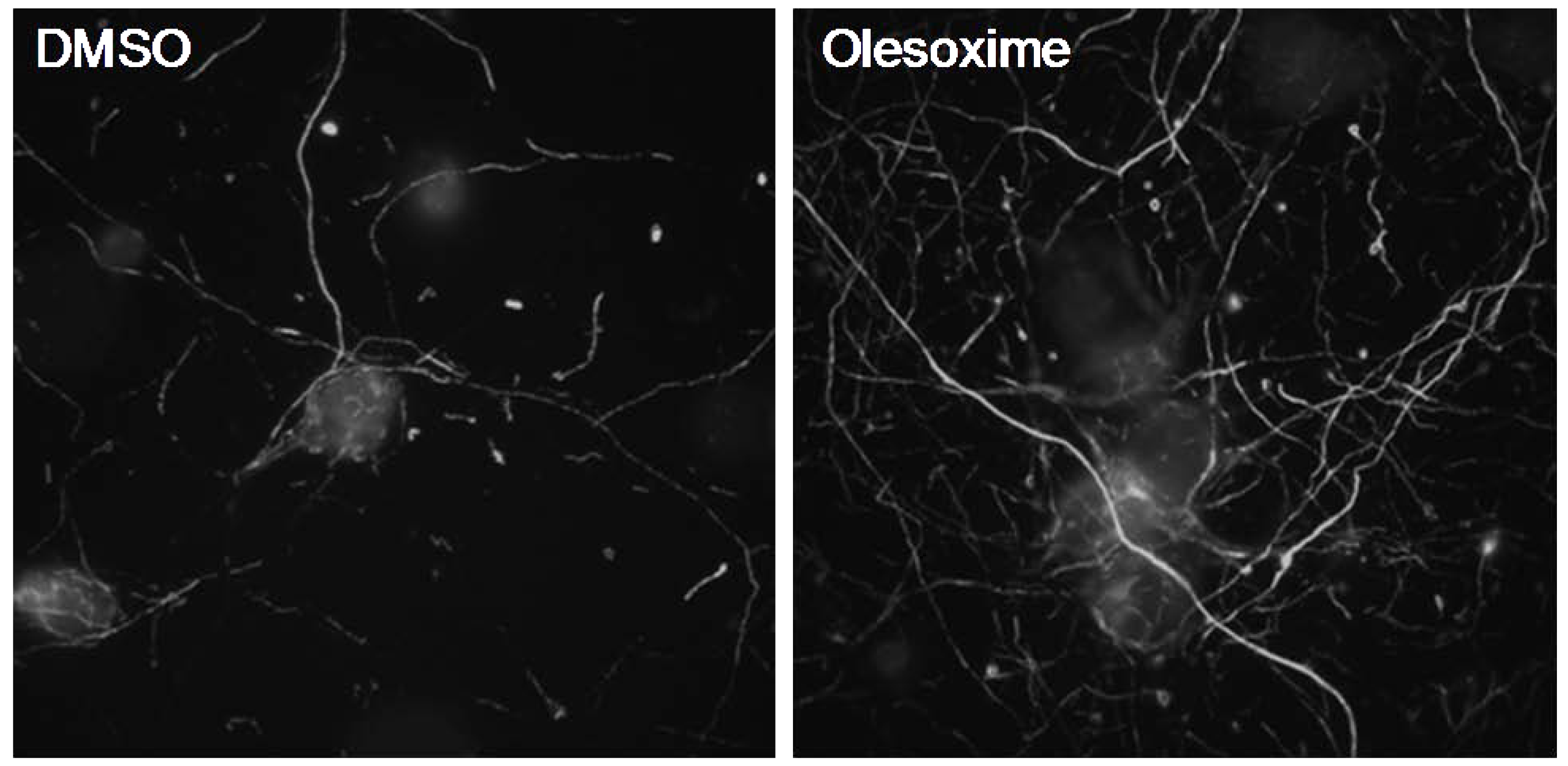
3.1. In Vitro Neuroprotection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell type | Stress condition | Outcome |
|---|---|---|
| Motor neuron | Trophic factor deprivation [2] | Neuronal survival and neurite outgrowth |
| Striatal neuron | Mutant Htt overexpression [7] | Neuronal survival |
| Cerebellar granule neuron | Low K+ [2] | Decreased cyt c release and neuronal survival |
| Cortical neuron | Camptothecin-induced toxicity (unpublished data) | Decreased cyt c release and neuronal survival |

3.2. In Vivo Neuroprotection—Nerve Lesion Models
3.2.1. Neonatal Motor Neuron Axotomy
3.2.2. Peripheral Nerve Trauma
3.3. In Vivo Neuroprotection—Chronic Motor Neuron Disease Mouse Models
3.3.1. SOD1G93A Transgenic Mouse Model of ALS
3.3.2. A Transgenic Mouse Model of SMA
3.4. In Vivo Neuroprotection—Painful Peripheral Neuropathies Models
3.4.1. Chemotherapy-Induced Peripheral Neuropathy (CIPN)
3.4.2. Diabetes-Induced Peripheral Neuropathy (DNP)
| Model | Route | Treatment (days) | MED (mg/kg) | Css,max (µg/mL) | |
|---|---|---|---|---|---|
| Axotomy model | PO | 5 | 30 | 20 | |
| Nerve crush model | SC | 42 | 3 | 0.6 | |
| SMA transgenic mice | SC | D21 to death | 30 | 12 | |
| ALS transgenic mice | SC | D60 to death | 3 | 0.6 | |
| Paclitaxel-induced NP in rat | Prevention of IEFNs degeneration | PO | 17 | < 3 | 0.16 |
| Treatment of mechano-allodynia & hyperalgesia | PO | 5 | 10 | 0.5 | |
| STZ diabetic rat | Improvement in motor nerve conduction | PO | 32 | 3 | 0.3 |
| Treatment of tactile allodynia | PO | 5 | 10 | 1.8 | |
4. Further Evidence that Olesoxime Targets Mitochondria
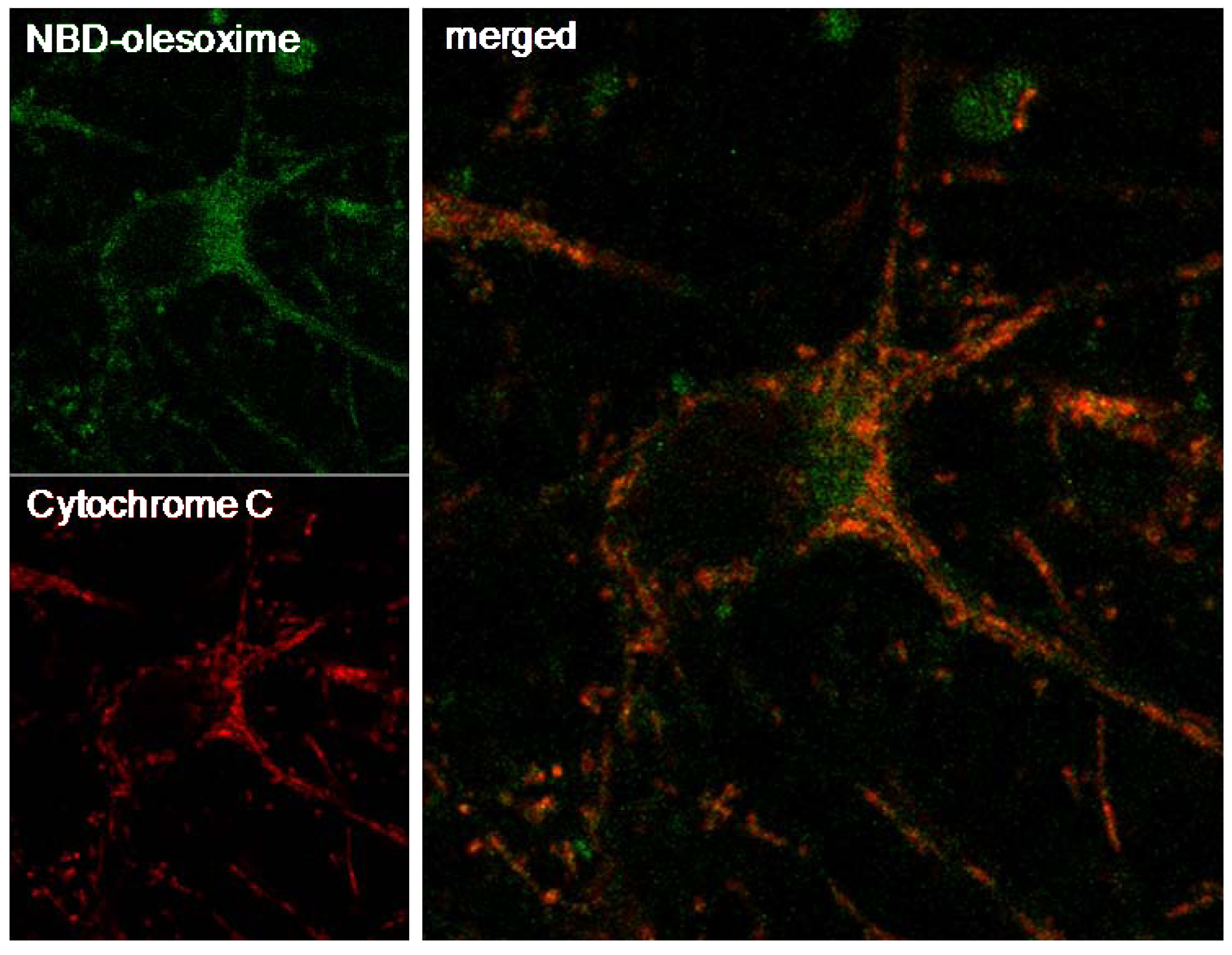
4.1. Olesoxime Concentrates at the Mitochondria

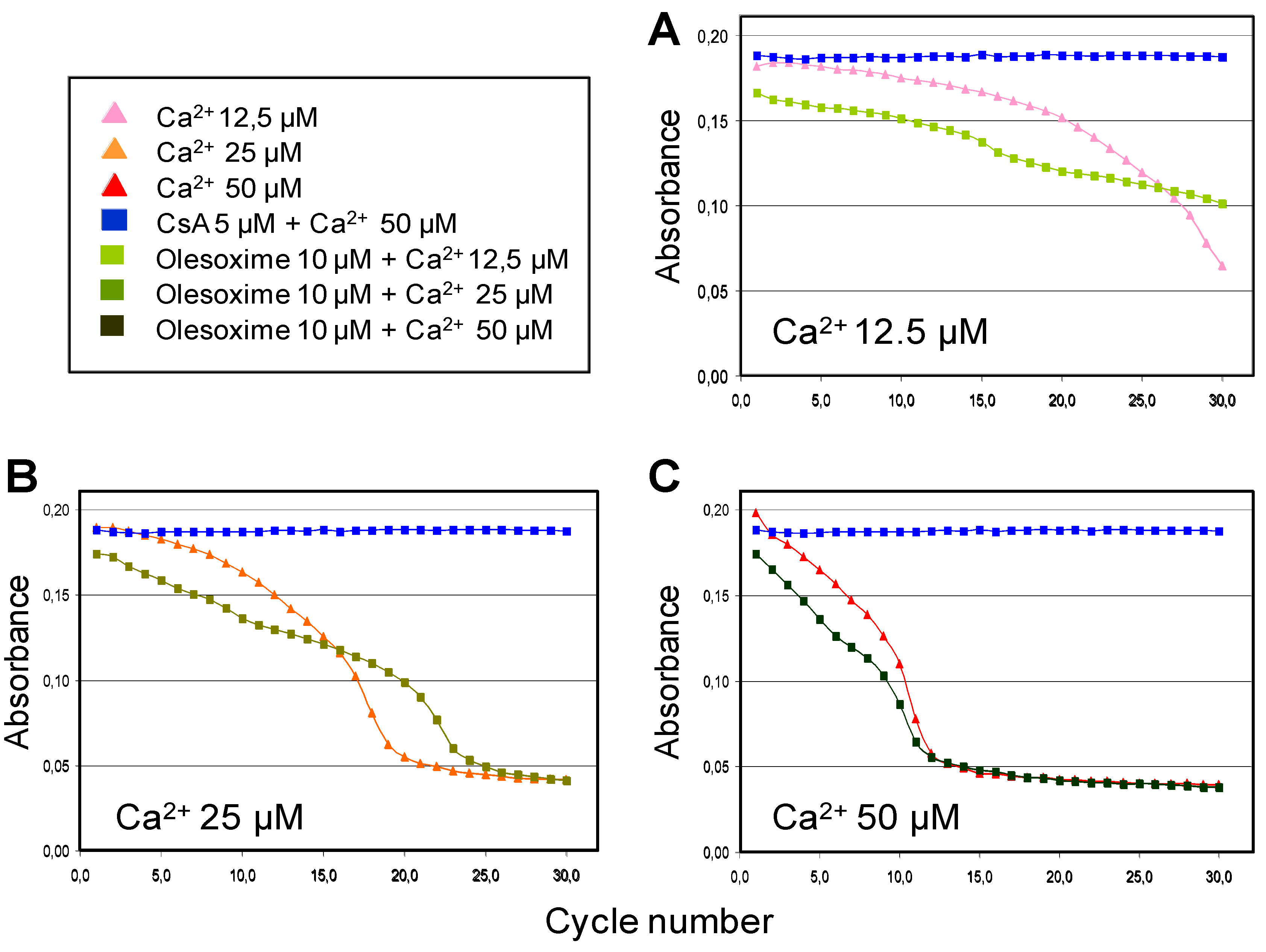
4.2. Olesoxime Does Not Increase Calcium Retention Capacity in Isolated Mitochondria

| μM | Control | Olesoxime | CsA | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 0.1 | 0.5 | 1 | 5 | 0.1 | 0.5 | 1 | 5 | ||
| mPTP induction (μM Ca2+) | 34.5 | 36.6 | 37.9 | 35.4 | 35.7 | 71.4 | 80.7 | 85.3 | 87.7 |
| SD | 1.72 | 0.80 | 2.28 | 1.04 | 0.57 | 1.12 | 3.48 | 1.25 | 2.71 |
| Norm. vs control | 1 | 2.1 | 3.4 | 0.8 | 1.2 | 36.9 | 46.2 | 50.8 | 53.1 |
4.3. Olesoxime Rescues Cardiomyocytes from Doxorubicin-Induced PTP Opening

4.4. Olesoxime Rescues Hela Cells from Arichidonic Acid-Induced PTP Opening

4.5. Olesoxime Inhibits Cytochrome C Release
5. Early CNS Safety and Brain Penetration

| Days of treatment | Dosemg/kg/d | Plasma levelsµg/mL | Brain levelsµg/g | Brain/plasma ratio |
|---|---|---|---|---|
| 7 | 0.3 | 0.06 ± 0.01 | BLQ | - |
| 3 | 0.55 ± 0.07 | 0.19 ± 0.02 | 0.35 | |
| 30 | 5.35 ± 1.19 | 1.23 ± 0.30 | 0.23 | |
| 42 | 0.3 | 0.06 ± 0.01 | BLQ | - |
| 3 | 0.47 ± 0.01 | 0.24 ± 0.04 | 0.51 | |
| 30 | 5.85 ± 1.06 | 2.66 ± 0.84 | 0.45 |
6. Conclusions
Acknowledgements
References
- Bordet, T.; Pruss, R.; Henderson, C.E. Screening for ALS drugs. In Amyotrophic Lateral Sclerosis; Mitsumoto, H., Przedborski, S., Gordon, P.H., Eds.; Taylor & Francis Group: New York, NY, USA, 2006; pp. 551–582. [Google Scholar]
- Bordet, T.; Buisson, B.; Michaud, M.; Drouot, C.; Galea, P.; Delaage, P.; Akentieva, N.P.; Evers, A.S.; Covey, D.F.; Ostuni, M.A.; Lacapere, J.J.; Massaad, C.; Schumacher, M.; Steidl, E.M.; Maux, D.; Delaage, M.; Henderson, C.E.; Pruss, R.M. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 2007, 322, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.E.; Bloch-Gallego, E.; Camu, W. Purified embryonic motoneurons. In Nerve Cell Culture: A practical approach; Cohen, J., Wilkin, G., Eds.; Oxford UP: London, UK, 1995; pp. 69–81. [Google Scholar]
- Greenlund, L.J.; Deckwerth, T.L.; Johnson, E.M. Superoxide dismutase delays neuronal apoptosis: A role for reactive oxygen species in programmed neuronal death. Neuron 1995, 14, 303–315. [Google Scholar]
- Estevez, A.G.; Spear, N.; Manuel, S.M.; Radi, R.; Henderson, C.E.; Barbeito, L.; Beckman, J.S. Nitric oxide and superoxide contribute to motor neuron apoptosis induced by trophic factor deprivation. J. Neurosci. 1998, 18, 923–931. [Google Scholar]
- Conde, C.; Caceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar]
- Valenza, M.; Rigamonti, D.; Goffredo, D.; Zuccato, C.; Fenu, S.; Jamot, L.; Strand, A.; Tarditi, A.; Woodman, B.; Racchi, M.; Mariotti, C.; Di Donato, S.; Corsini, A.; Bates, G.; Pruss, R.; Olson, J. M.; Sipione, S.; Tartari, M.; Cattaneo, E. Dysfunction of the cholesterol biosynthetic pathway in Huntington's disease. J. Neurosci. 2005, 25, 9932–9939. [Google Scholar]
- Sendtner, M.; Kreutzberg, G.W.; Thoenen, H. Ciliary neurotrophic factor prevents the degeneration of motor neurons after axotomy. Nature 1990, 345, 440–441. [Google Scholar]
- Rossiter, J.P.; Riopelle, R.J.; Bisby, M.A. Axotomy-induced apoptotic cell death of neonatal rat facial motoneurons: Time course analysis and relation to NADPH-diaphorase activity. Exp. Neurol. 1996, 138, 33–44. [Google Scholar]
- Vanderluit, J.L.; McPhail, L.T.; Fernandes, K.J.; McBride, C.B.; Huguenot, C.; Roy, S.; Robertson, G.S.; Nicholson, D.W.; Tetzlaff, W. Caspase-3 is activated following axotomy of neonatal facial motoneurons and caspase-3 gene deletion delays axotomy-induced cell death in rodents. Eur. J. Neurosci. 2000, 12, 3469–3480. [Google Scholar]
- Tong, J.X.; Rich, K.M. Diphenylpiperazines enhance regeneration after facial nerve injury. J. Neurocytol. 1997, 26, 339–347. [Google Scholar]
- Vanderluit, J.L.; McPhail, L.T.; Fernandes, K.J.; Kobayashi, N.R.; Tetzlaff, W. In vivo application of mitochondrial pore inhibitors blocks the induction of apoptosis in axotomized neonatal facial motoneurons. Cell Death Differ. 2003, 10, 969–976. [Google Scholar] [CrossRef] [PubMed]
- De Koning, P.; Brakkee, J.H.; Gispen, W.H. Methods for producing a reproducible crush in the sciatic and tibial nerve of the rat and rapid and precise testing of return of sensory function. Beneficial effects of melanocortins. J. Neurol. Sci. 1986, 74, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Loeffler, J.P. Neuromuscular junction destruction during amyotrophic lateral sclerosis: Insights from transgenic models. Curr. Opin. Pharmacol. 2009, 9, 341–346. [Google Scholar]
- Briese, M.; Esmaeili, B.; Sattelle, D.B. Is spinal muscular atrophy the result of defects in motor neuron processes? Bioessays 2005, 27, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Burghes, A.H.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [PubMed]
- Bordet, T.; Lesbordes, J.C.; Rouhani, S.; Castelnau-Ptakhine, L.; Schmalbruch, H.; Haase, G.; Kahn, A. Protective effects of cardiotrophin-1 adenoviral gene transfer on neuromuscular degeneration in transgenic ALS mice. Hum. Mol. Genet. 2001, 10, 1925–1933. [Google Scholar]
- Lesbordes, J.C.; Cifuentes-Diaz, C.; Miroglio, A.; Joshi, V.; Bordet, T.; Kahn, A.; Melki, J. Therapeutic benefits of cardiotrophin-1 gene transfer in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2003, 12, 1233–1239. [Google Scholar]
- Azzouz, M.; Hottinger, A.F.; Paterna, J.C.; Zurn, A.D.; Aebischer, P.; Büeler, H. Increased motoneuron survival and improved neuromuscular function in transgenic ALS mice after intraspinal injection of an adeno-associated virus encoding Bcl-2. Hum. Mol. Genet. 2000, 9, 803–811. [Google Scholar]
- Friedlander, R.M.; Brown, R.H.; Gagliardini, V.; Wang, J.; Yuan, J. Inhibition of ICE slows ALS in mice. Nature 1997, 388, 31. [Google Scholar]
- Li, M.; Ona, V.O.; Guégan, C.; Chen, M.; Jackson-Lewis, V.; Andrews, L.J.; Olszewski, A.J.; Stieg, P.E.; Lee, J.P.; Przedborski, S.; Friedlander, R.M. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science 2000, 288, 335–339. [Google Scholar]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; Chen, W; Zhai, P; Sufit, R.L.; Siddique, T. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [PubMed]
- Pearn, J. Incidence, prevalence and gene frequency studies of chronic childhood spinal muscular atrophy. J. Med. Genet. 1978, 15, 409–413. [Google Scholar]
- McAndrew, P.E.; Parsons, D.W.; Simard, L.R.; Rochette, C.; Ray, P.N.; Mendell, J.R.; Prior, T.W.; Burghes, A.H. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 1997, 60, 1411–1422. [Google Scholar]
- Scheffer, H.; Cobben, J.M.; Matthijs, G.; Wirth, B. Best practice guidelines for molecular analysis in spinal muscular atrophy. Eur. J. Hum. Genet. 2001, 9, 484–491. [Google Scholar]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; Le Paslier, D.; Frézal, J.; Cohen, D.; Weissenbach, J.; Munnich, A.; Melki, J. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar]
- Bussaglia, E.; Clermont, O.; Tizzano, E.; Lefebvre, S.; Burglen, L.; Cruaud, C.; Urtizberea, J. A.; Colomer, J.; Munnich, A.; Baiget, M.; Melki, J. A frame-shift deletion in the survival motor neuron gene in Spanish spinal muscular atrophy patients. Nat. Genet. 1995, 11, 335–337. [Google Scholar]
- Crawford, T.O.; Pardo, C.A. The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 1996, 3, 97–110. [Google Scholar]
- Schroth, M.K. Special considerations in the respiratory management of spinal muscular atrophy. Pediatrics 2009, 123, 245–249. [Google Scholar]
- Viollet, L.; Bertrandy, S.; Bueno Brunialti, A.L.; Lefebvre, S.; Burlet, P.; Clermont, O.; Cruaud, C.; Guenet, J.L.; Munnich, A.; Melki, J. cDNA isolation, expression, and chromosomal localization of the mouse survival motor neuron gene (Smn). Genomics 1997, 40, 185–188. [Google Scholar] [PubMed]
- DiDonato, C.J.; Chen, X.N.; Noya, D.; Korenberg, J.R.; Nadeau, J.H.; Simard, L.R. Cloning, characterization, and copy number of the murine survival motor neuron gene: Homolog of the spinal muscular atrophy-determining gene. Genome Res. 1997, 7, 339–352. [Google Scholar] [PubMed]
- Schrank, B.; Götz, R.; Gunnersen, J.M.; Ure, J.M.; Toyka, K.V.; Smith, A.G.; Sendtner, M. The inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. USA 1997, 94, 9920–9925. [Google Scholar]
- Hsieh-Li, H.M.; Chang, J.G.; Jong, Y.J.; Wu, M.H.; Wang, N.M.; Tsai, C.H.; Li, H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000, 24, 66–70. [Google Scholar]
- Frugier, T.; Tiziano, F.D.; Cifuentes-Diaz, C.; Miniou, P.; Roblot, N.; Dierich, A.; Le Meur, M.; Melki, J. Nuclear targeting defect of SMN lacking the C-terminus in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2000, 9, 849–858. [Google Scholar]
- Cifuentes-Diaz, C.; Nicole, S.; Velasco, M.E.; Borra-Cebrian, C.; Panozzo, C.; Frugier, T.; Millet, G.; Roblot, N.; Joshi, V.; Melki, J. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum. Mol. Genet. 2002, 11, 1439–1447. [Google Scholar]
- Authier, N.; Gillet, J.P.; Fialip, J.; Eschalier, A.; Coudore, F. Description of a short-term Taxol-induced nociceptive neuropathy in rats. Brain Res. 2000, 887, 239–249. [Google Scholar]
- Authier, N.; Gillet, J.P.; Fialip, J.; Eschalier, A.; Coudore, F. A new animal model of vincristine-induced nociceptive peripheral neuropathy. Neurotoxicology 2003, 24, 797–805. [Google Scholar]
- Thant, M.; Hawley, R.J.; Smith, M.T.; Cohen, M.H.; Minna, J.D.; Bunn, P.A.; Ihde, D.C.; West, W.; Matthews, M.J. Possible enhancement of vincristine neuropathy by VP-16. Cancer 1982, 49, 859–864. [Google Scholar]
- Polomano, R.C.; Mannes, A.J.; Clark, U.S.; Bennett, G.J. A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain 2001, 94, 293–304. [Google Scholar]
- Flatters, S.J.; Bennett, G.J. Ethosuximide reverses paclitaxel- and vincristine-induced painful peripheral neuropathy. Pain 2004, 109, 150–161. [Google Scholar]
- Flatters, S.J.; Bennett, G.J. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: Evidence for mitochondrial dysfunction. Pain 2006, 122, 245–257. [Google Scholar]
- Flatters, S.J.; Xiao, W.H.; Bennett, G.J. Acetyl-L-carnitine prevents and reduces paclitaxel-induced painful peripheral neuropathy. Neurosci Lett 2006, 397, 219–223. [Google Scholar]
- Siau, C.; Bennett, G.J. Dysregulation of cellular calcium homeostasis in chemotherapy-evoked painful peripheral neuropathy. Anesth. Analg. 2006, 102, 1485–1490. [Google Scholar]
- Xiao, W.; Bennett, G.J. Chemotherapy-evoked neuropathic pain: Abnormal spontaneous discharge in A-fiber and C-fiber primary afferent neurons and its suppression by acetyl-L-carnitineitine. Pain 2008, 135, 262–270. [Google Scholar]
- Xiao, W.H.; Zheng, F.Y.; Bennett, G.J.; Bordet, T.; Pruss, R.M. Olesoxime (cholest-4-en-3-one, oxime): Analgesic and neuroprotective effects in a rat model of painful peripheral neuropathy produced by the chemotherapeutic agent, paclitaxel. Pain 2009, 147, 202–209. [Google Scholar] [PubMed]
- Ahlgren, S.C.; Levine, J.D. Mechanical hyperalgesia in streptozotocin-diabetic rats. Neuroscience 1993, 52, 1049–1055. [Google Scholar]
- Courteix, C.; Eschalier, A.; Lavarenne, J. Streptozocin-induced diabetic rats: Behavioural evidence for a model of chronic pain. Pain 1993, 53, 81–88. [Google Scholar]
- Calcutt, N.A.; Li, L.; Yaksh, T.L.; Malmberg, A.B. Different effects of two aldose reductase inhibitors on nociception and prostaglandin E. Eur. J. Pharmacol. 1995, 285, 189–197. [Google Scholar]
- Calcutt, N.A.; Jorge, M.C.; Yaksh, T.L.; Chaplan, S.R. Tactile allodynia and formalin hyperalgesia in streptozotocin-diabetic rats: Effects of insulin, aldose reductase inhibition and lidocaine. Pain 1996, 68, 293–299. [Google Scholar]
- Malcangio, M.; Tomlinson, D.R. A pharmacologic analysis of mechanical hyperalgesia in streptozotocin/diabetic rats. Pain 1998, 76, 151–157. [Google Scholar]
- Field, M.J.; McCleary, S.; Hughes, J.; Singh, L. Gabapentin and pregabalin, but not morphine and amitriptyline, block both static and dynamic components of mechanical allodynia induced by streptozocin in the rat. Pain 1999, 80, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Cameron, N.E.; Cotter, M.A.; Low, P.A. Nerve blood flow in early experimental diabetes in rats: Relation to conduction deficits. Am. J. Physiol. 1991, 261, 1–8. [Google Scholar]
- Jakobsen, J. Axonal dwindling in early experimental diabetes. II. A study of isolated nerve fibres. Diabetologia 1976, 12, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Levine, J.D. Hyper-responsivity in a subset of C-fiber nociceptors in a model of painful diabetic neuropathy in the rat. Neuroscience 2001, 102, 185–192. [Google Scholar]
- Chen, X.; Levine, J.D. Altered temporal pattern of mechanically evoked C-fiber activity in a model of diabetic neuropathy in the rat. Neuroscience 2003, 121, 1007–1015. [Google Scholar]
- Orstavik, K.; Namer, B.; Schmidt, R.; Schmelz, M.; Hilliges, M.; Weidner, C.; Carr, R.W.; Handwerker, H.; Jorum, E.; Torebjork, H.E. Abnormal function of C-fibers in patients with diabetic neuropathy. J. Neurosci. 2006, 26, 11287–11294. [Google Scholar]
- Bordet, T.; Buisson, B.; Michaud, M.; Abitbol, J.L.; Marchand, F.; Grist, J.; Andriambeloson, E.; Malcangio, M.; Pruss, R.M. Specific antinociceptive activity of cholest-4-en-3-one, oxime (TRO19622) in experimental models of painful diabetic and chemotherapy-induced neuropathy. J. Pharmacol. Exp. Ther. 2008, 326, 623–632. [Google Scholar]
- Leung, A.W.; Halestrap, A.P. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 2008, 1777, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Rasola, A.; Bernardi, P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 2007, 12, 815–833. [Google Scholar]
- Waldmeier, P.C.; Zimmermann, K.; Qian, T.; Tintelnot-Blomley, M.; Lemasters, J.J. Cyclophilin D as a drug target. Curr. Med. Chem. 2003, 10, 1485–1506. [Google Scholar]
- Cardoso, S.; Santos, R.X.; Carvalho, C.; Correia, S.; Pereira, G.C.; Pereira, S.S.; Oliveira, P.J.; Santos, M.S.; Proenca, T.; Moreira, P.I. Doxorubicin increases the susceptibility of brain mitochondria to Ca(2+)-induced permeability transition and oxidative damage. Free Radic. Biol. Med. 2008, 45, 1395–1402. [Google Scholar]
- Yen, H.C.; Oberley, T.D.; Vichitbandha, S.; Ho, Y.S.; St Clair, D.K. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J. Clin. Invest 1996, 98, 1253–1260. [Google Scholar]
- Berthiaume, J.M.; Wallace, K.B. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol. Toxicol. 2007, 23, 15–25. [Google Scholar]
- d'Anglemont de Tassigny, A.; Berdeaux, A.; Souktani, R.; Henry, P.; Ghaleh, B. The volume-sensitive chloride channel inhibitors prevent both contractile dysfunction and apoptosis induced by doxorubicin through PI3kinase, Akt and Erk 1/2. Eur. J. Heart Fail 2008, 10, 39–46. [Google Scholar]
- Scorrano, L.; Penzo, D.; Petronilli, V.; Pagano, F.; Bernardi, P. Arachidonic acid causes cell death through the mitochondrial permeability transition. Implications for tumor necrosis factor-alpha aopototic signaling. J. Biol. Chem. 2001, 276, 12035–12040. [Google Scholar] [PubMed]
- Petronilli, V.; Penzo, D.; Scorrano, L.; Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem. 2001, 276, 12030–12034. [Google Scholar] [PubMed]
- Schousboe, A.; Meier, E.; Hertz, L.; Shaher, A.; DeVellis, J.; Vernadakis, A.; Haber, B. Preparation of cultures of mouse (rat) cerebellar granule cells. In Dissection and Tissue Culture Manual of the Nervous System; Shahar, A.D., De Vellis, J., Vernadakis, A., Haber, B., Eds.; A.R. Liss: New York, NY, USA, 1989; pp. 203–206. [Google Scholar]
- D'Mello, S.R.; Galli, C.; Ciotti, T.; Calissano, P. Induction of apoptosis in cerebellar granule neurons by low potassium: Inhibition of death by insulin-like growth factor I and cAMP. Proc. Natl. Acad. Sci. USA 1993, 90, 10989–10993. [Google Scholar]
- Steidl, E.M.; Neveu, E.; Bertrand, D.; Buisson, B. The adult rat hippocampal slice revisited with multi-electrode arrays. Brain Res. 2006, 1096, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Takasato, Y.; Rapoport, S.I.; Smith, Q.R. An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am. J. Physiol. 1984, 247, 484–493. [Google Scholar]
- Scott, S.; Kranz, J.E.; Cole, J.; Lincecum, J.M.; Thompson, K.; Kelly, N.; Bostrom, A.; Theodoss, J.; Al-Nakhala, B.M.; Vieira, F.G.; Ramasubbu, J.; Heywood, J.A. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler 2008, 9, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Kroemer, G. Mitochondria in cell death: Novel targets for neuroprotection and cardioprotection. Trends Mol. Med. 2003, 9, 196–205. [Google Scholar]
- Szeto, H.H. Mitochondria-targeted peptide antioxidants: Novel neuroprotective agents. Aaps. J. 2006, 8, e521–e531. [Google Scholar]
- Swerdlow, R.H. Mitochondrial medicine and the neurodegenerative mitochondriopathies. Pharmaceuticals 2009, 2, 150–167. [Google Scholar]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar]
- Kong, J.; Xu, Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 1998, 18, 3241–3250. [Google Scholar]
- Acsadi, G.; Lee, I.; Li, X.; Khaidakov, M.; Pecinova, A.; Parker, G.C.; Huttemann, M. Mitochondrial dysfunction in a neural cell model of spinal muscular atrophy. J. Neurosci. Res. 2009, 87, 2748–2756. [Google Scholar]
- Afifi, A.K.; Aleu, F.P.; Goodgold, J.; MacKay, B. Ultrastructure of atrophic muscle in amyotrophic lateral sclerosis. Neurology 1966, 16, 475–481. [Google Scholar]
- Atsumi, T. The ultrastructure of intramuscular nerves in amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 1981, 55, 193–198. [Google Scholar] [CrossRef]
- Hirano, A.; Nakano, I.; Kurland, L.T.; Mulder, D.W.; Holley, P.W.; Saccomanno, G. Fine structural study of neurofibrillary changes in a family with amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 1984, 43, 471–480. [Google Scholar]
- Sasaki, S.; Iwata, M. Ultrastructural study of synapses in the anterior horn neurons of patients with amyotrophic lateral sclerosis. Neurosci. Lett 1996, 204, 53–56. [Google Scholar]
- Echaniz-Laguna, A.; Zoll, J.; Ponsot, E.; N'Guessan, B.; Tranchant, C.; Loeffler, J.P.; Lampert, E. Muscular mitochondrial function in amyotrophic lateral sclerosis is progressively altered as the disease develops: A temporal study in man. Exp. Neurol. 2006, 198, 25–30. [Google Scholar]
- Vila, L.; Barrett, E.F.; Barrett, J.N. Stimulation-induced mitochondrial [Ca2+] elevations in mouse motor terminals: Comparison of wild-type with SOD1-G93A. J. Physiol. 2003, 549, 719–728. [Google Scholar]
- David, G.; Barrett, E.F. Quantal release from motor nerve terminals of mice expressing the G93A mutation of human superoxide dismutase 1 (SOD1G93A). In Neuroscience Meeting, New Orleans, LA, USA, 2003.
- Lewinski, F.V.; Keller, B.U. Ca(2+), mitochondria and selective motoneuron vulnerability: Implications for ALS. Trends Neurosci. 2005, 494–500. [Google Scholar]
- Mattiazzi, M.; D'Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [PubMed]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; Brannstrom, T.; Gredal, O.; Wong, P.C.; Williams, D.S.; Cleveland, D.W. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar]
- Kirkinezos, I.G.; Hernandez, D.; Bradley, W.G.; Moraes, C.T. An ALS mouse model with a permeable blood-brain barrier benefits from systemic cyclosporine A treatment. J. Neurochem. 2004, 88, 821–826. [Google Scholar]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar]
- Abitbol, J.L.; Cuvier, V.; Bordet, T.; Drouot, C.; Berna, P.; Pruss, R.M. Safety and pharmacokinetics of repeated doses of TRO19622, a drug candidate for the treatment of amyotrophic lateral sclerosis and spinal muscular atrophy. In ALS/MND International Symposium, Yokohama, Japan, 30 November-2 December 2006.
- Estournet, B.; Chabrol, B.; Cuisset, J.M.; Cances, C.; Strub-Wourgaft, N.; Cuvier, V.; Bassissi, F.; Bordet, T.; Pruss, R.; Abitbol, J.L. Safety and pharmacokinetics (PK) of TRO19622 in Spinal Muscular Atrophy (SMA) children and adults. In 61st Annual Meeting American Academy of Neurology, Seattle, WA, USA, 25 April-2 May 2009.
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bordet, T.; Berna, P.; Abitbol, J.-L.; Pruss, R.M. Olesoxime (TRO19622): A Novel Mitochondrial-Targeted Neuroprotective Compound. Pharmaceuticals 2010, 3, 345-368. https://doi.org/10.3390/ph3020345
Bordet T, Berna P, Abitbol J-L, Pruss RM. Olesoxime (TRO19622): A Novel Mitochondrial-Targeted Neuroprotective Compound. Pharmaceuticals. 2010; 3(2):345-368. https://doi.org/10.3390/ph3020345
Chicago/Turabian StyleBordet, Thierry, Patrick Berna, Jean-Louis Abitbol, and Rebecca M. Pruss. 2010. "Olesoxime (TRO19622): A Novel Mitochondrial-Targeted Neuroprotective Compound" Pharmaceuticals 3, no. 2: 345-368. https://doi.org/10.3390/ph3020345
APA StyleBordet, T., Berna, P., Abitbol, J. -L., & Pruss, R. M. (2010). Olesoxime (TRO19622): A Novel Mitochondrial-Targeted Neuroprotective Compound. Pharmaceuticals, 3(2), 345-368. https://doi.org/10.3390/ph3020345