Metabolism and Pharmacokinetics of the Anti-Tuberculosis Drug Ethionamide in a Flavin-Containing Monooxygenase Null Mouse

Abstract
:1. Introduction

2. Results and Discussion
2.1. Linearity, Assay Precision, Accuracy, and Recovery Assessments

2.2. Stability
2.3. Modeling Fmo Genotype Differences in ETA Metabolic Profiles


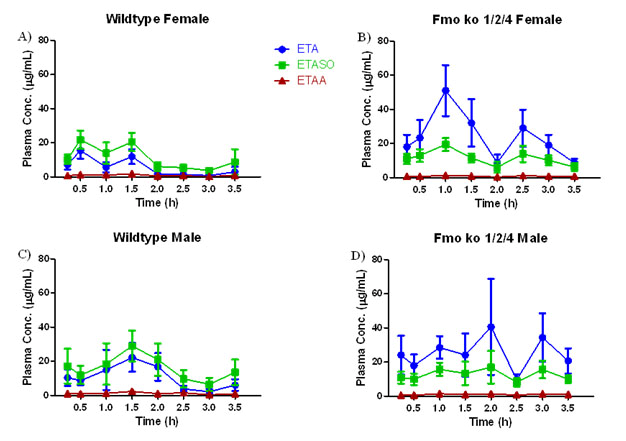
Kinetics of ETA Metabolism in Wild Type C57BL/6J and Fmo 1/2/4 Knockout Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacokinetic parameters | Value for mice a: | ||||
|---|---|---|---|---|---|
| C57BL6/J | Fmo 1/2/4 null | ||||
| ETA | ETASO | ETA | ETASO | ||
| Female | Cmax (µg/mL) | 15.1 ± 14.8 | 18.5± 20.6 | 31.0 ± 42.9 | 16.8 ± 7.9 |
| tmax (h) | 1.0 ± 0.5 | 1.0 ± 0.5 | 1.0 ± 0.0 | 1.0 ± 1.0 | |
| Male | Cmax (µg/mL) | 15.1 ± 34.4 | 18.5 ± 36.0 | 31.0 ± 93.6 | 16.8 ± 28.3 |
| tmax (h) | 1.0 ± 0.0 | 1.0 ± 0.0 | 1.0 ± 1.0 | 1.0 ± 1.0 | |
3. Experimental Section
3.1. Chemicals
3.2. Animals
3.3. In Vivo Single-Dose Ethionamide Study
3.4. Sample Extraction
3.5. Quantification of Ethionamide and Metabolites
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Reported Tuberculosis in the United States, 2010; U.S. Dept. Health aand Human Services: Atlanta, GA, USA, 2011; p. 149.
- Bastian, I.; Colebunders, R. Treatment and prevention of multidrug-resistant tuberculosis. Drugs 1999, 58, 633–661. [Google Scholar]
- Thee, S.; Seifart, H.I.; Rosekranz, B.; Hesseling, A.C.; Magdorf, K.; Donald, P.R.; Schaaf, H.S. Pharmacokinetics of ethionamide in children. Antimicrob. Agents Chemother. 2011, 55, 4594–4600. [Google Scholar]
- Schon, T.; Juréen, P.; Chryssanthour, E.; Giske, C.G.; Sturegård, E.; Kahlmeter, G.; Hoffner, S.; Angeby, K.A. Wild-type distributions of seven oral second-line drugs against Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 2011, 15, 502–509. [Google Scholar] [CrossRef]
- Qian, L.; de Montellano, P.R.O. Oxidative activation of thiacetazone by the Mycobacterium tuberculosis flavin monooxygenase EtaA and human FMO1 and FMO3. Chem. Res. Toxicol. 2006, 19, 443–449. [Google Scholar] [CrossRef]
- Dover, L.G.; Alahari, A.; Grataud, P.; Gomes, J.M.; Bhowruth, V.; Reynolds, R.C.; Besra, G.S.; Kremer, L. EthA, a common activator of thiocarbamide-containing drugs acting on different mycobacterial targets. Antimicrob. Agents Chemother. 2007, 51, 1055–1063. [Google Scholar] [CrossRef]
- Phillips, I.R.; Shephard, E.A. Flavin-containing monooxygenases: mutations, disease and drug response. Trends Pharmacol. Sci. 2008, 29, 294–301. [Google Scholar] [CrossRef] [Green Version]
- Furnes, B.; Feng, J.; Sommer, S.S.; Schlenk, D. Identification of novel variants of the flavin-containing monooxygenase gene family in African Americans. Drug Metab. Dispos. 2003, 31, 187–193. [Google Scholar]
- Cashman, J.R. Structural and catalytic properties of the mammalian flavin-containing monooxygenase. Chem. Res. Toxicol. 1995, 8, 165–181. [Google Scholar]
- Krueger, S.K.; Williams, D.E. Mammalian flavin-containing monooxygenases: Structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol. Ther. 2005, 106, 357–387. [Google Scholar] [CrossRef]
- Dolphin, C.T.; Beckett, D.J.; Janmohamed, A.; Cullingford, T.E.; Smith, R.L.; Shephard, E.A.; Phillips, I.R. The flavin-containing monooxygenase 2 gene (FMO2) of humans, but not of other primates, encodes a truncated, nonfunctional protein. J. Biol. Chem. 1998, 273, 30599–30607. [Google Scholar]
- Whestine, J.R.; Yueh, M.-F.; McCarver, D.G.; Williams, D.E.; Park, C.S.; Kang, J.H.; Cha, Y.N.; Dolphin, C.T.; Shephard, E.A.; Phillips, I.R.; Hines, R.N. Ethnic differences in human flavin-containing monooxygenase 2 (FMO2) polymorphisms: detection of expressed proteins in African Americans. Toxicol. Appl. Pharmacol. 2000, 168, 216–224. [Google Scholar] [CrossRef]
- Krueger, S.K.; Henderson, M.C.; Siddens, L.K.; VanDyke, J.E.; Benninghoff, A.D.; Karplus, P.A.; Furnes, B.; Schlenk, D.; Williams, D.E. Characterization of sulfoxygenation and structural implications of human flavin-containing monooxygenase isoform 2 (FMO2.1) variants S195L and N413K. Drug Metab. Dispos. 2009, 37, 1785–1791. [Google Scholar] [CrossRef]
- Krueger, S.K.; Siddens, L.K.; Martin, S.R.; Yu, Z.; Pereira, C.B.; Cabacungan, E.T.; Hines, R.N.; Ardlie, K.G.; Raucy, K.L.; Williams, D.E. Differences in FMO2*1 allelic frequency between Hispanics of Puerto Rican and Mexican descent. Drug Metab. Dispos. 2004, 32, 1337–1340. [Google Scholar] [CrossRef]
- Veeramah, K.R.; Thomas, M.G.; Weale, M.E.; Tarekegn, A.; Bekele, E.; Mendell, N.R.; Shephard, E.A.; Bradman, N.; Phillips, I.R. The potentially deleterious functional variant flavin-containing monooxygenase 2*1 is at high frequency throughout sub-Saharan Africa. Pharmacogenet. Genomics. 2008, 18, 877–886. [Google Scholar] [CrossRef]
- Henderson, M.C.; Siddens, L.K.; Morré, J.T.; Krueger, S.K.; Williams, D.E. Metabolism of the anti-tuberculosis drug ethionamide by mouse and human FMO1, FMO2 and FMO3 and mouse and human lung microsomes. Toxicol. Appl. Pharmacol. 2008, 233, 420–427. [Google Scholar]
- Henderson, M.C.; Krueger, S.K.; Stevens, J.F.; Williams, D.E. Human flavin-containing monooxygenase form 2 S-oxygenation: sulfenic acid formation from thioureas and oxidation of glutathione. Chem. Res. Toxicol. 2004, 17, 633–640. [Google Scholar] [CrossRef]
- Nishida, C.R.; de Montellano, P.R.O. Bioactivation of antituberculosis thioamide and thiourea prodrugs by bacterial and mammalian flavin monooxygenases. Chem. Biol. Interact. 2011, 192, 21–25. [Google Scholar] [CrossRef]
- Francois, A.A.; Nishida, C.R.; de Montellano, P.R.; Phillips, I.R.; Shephard, E.A. Human flavin-containing monooxygenase 2.1 catalyzes oxygenation of the antitubercular drugs thiacetazone and ethionamide. Drug Metab. Dispos. 2009, 37, 178–186. [Google Scholar] [CrossRef]
- Johnston, J.P.; Kane, P.O.; Kibby, M.R. The metabolism of ethionamide and its sulphoxide. J. Pharm. Pharmacol. 1967, 19, 1–9. [Google Scholar]
- Vilcheze, C.; Av-Gay, Y.; Attarian, R.; Chen, B.; Liu, W.; Alland, D.; Sacchettini, J.C.; Jacobs, W.R., Jr. Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol. Microbiol. 2008, 69, 1316–1329. [Google Scholar] [CrossRef]
- Hernandez, D.; Chandan, P.; Janohamed, A.; Phillips, I.R.; Shephard, E.A. Deletion of genes from the mouse genome using Cre/loxP technology. Methods Mol. Biol. 2006, 320, 307–319. [Google Scholar]
- Hernandez, D.; Janmohamed, A.; Chandan, P.; Omar, B.A.; Phillips, I.R.; Shephard, E.A. Deletion of the mouse Fmo1 gene results in enhanced pharmacological behavioural responses to imipramine. Pharmacogenet. Genomics 2009, 19, 289–299. [Google Scholar] [CrossRef]
- Bermudez, L.E.; Inderlied, C.B.; Kolonoski, P.; Petrofsky, M.; Young, L.S. Clarithromycin, dapsone, and a combination of both used to treat or prevent disseminated Mycobacterium avium infection in beige mice. Antimicrob. Agents Chemother. 1994, 38, 2717–2721. [Google Scholar] [CrossRef]
- Siddens, L.K.; Henderson, M.C.; VanDyke, J.E.; Williams, D.E.; Krueger, S.K. Characterization of mouse flavin-containing monooxygenase transcript levels in lung and liver, and activity of expressed isoforms. Biochem. Pharmacol. 2008, 75, 570–579. [Google Scholar]
- Falls, J.G.; Blake, B.L.; Cao, Y.; Levi, P.E.; Hodgson, E. Gender differences in hepatic expression of flavin-containing monooxygenase isoforms (FMO1, FMO3, and FMO5) in mice. J. Biochem. Toxicol. 1995, 10, 171–177. [Google Scholar] [CrossRef]
- Kato, R.; Yamazoe, Y. Sex-specific cytochrome P450 as a cause of sex- and species-related differences in drug toxicity. Toxicol. Lett. 1992, 64–65, 661–667. [Google Scholar] [CrossRef]
- Vannelli, T.A.; Dykman, A.; de Montellano, P.R.O. The antituberculosis drug ethionamide is activated by a flavoprotein monooxygenase. J. Biol. Chem. 2002, 277, 12824–12829. [Google Scholar]
- Hernandez, D.; Melidoni, A.N.; Phillips, I.R.; Shephard, E.A. Methods in Molecular Biology: Cytochrome P450 Protocols, 2nd; Phillips, I.R., Shephard, E.A., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2006; Volume 320, pp. 329–341. [Google Scholar]
- Hayhurst, G.P.; Lee, Y.H.; Lambert, G.; Ward, J.M.; Gonzalez, F.J. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell Biol. 2001, 21, 1393–1403. [Google Scholar] [CrossRef]
- GraphPad Prism, version 5; Data analysis and mapping software; In-Stat: La Jolla, CA, USA, 2011.
- Statistical Computing Program R, version 2.13.1; A free software environment for statistical computing and graphics; R Core Development Team; R Foundation for Statistical Computing: Vienna, Austria, 2011.
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Palmer, A.L.; Leykam, V.L.; Larkin, A.; Krueger, S.K.; Phillips, I.R.; Shephard, E.A.; Williams, D.E. Metabolism and Pharmacokinetics of the Anti-Tuberculosis Drug Ethionamide in a Flavin-Containing Monooxygenase Null Mouse. Pharmaceuticals 2012, 5, 1147-1159. https://doi.org/10.3390/ph5111147
Palmer AL, Leykam VL, Larkin A, Krueger SK, Phillips IR, Shephard EA, Williams DE. Metabolism and Pharmacokinetics of the Anti-Tuberculosis Drug Ethionamide in a Flavin-Containing Monooxygenase Null Mouse. Pharmaceuticals. 2012; 5(11):1147-1159. https://doi.org/10.3390/ph5111147
Chicago/Turabian StylePalmer, Amy L., Virginia L. Leykam, Andrew Larkin, Sharon K. Krueger, Ian R. Phillips, Elizabeth A. Shephard, and David E. Williams. 2012. "Metabolism and Pharmacokinetics of the Anti-Tuberculosis Drug Ethionamide in a Flavin-Containing Monooxygenase Null Mouse" Pharmaceuticals 5, no. 11: 1147-1159. https://doi.org/10.3390/ph5111147
APA StylePalmer, A. L., Leykam, V. L., Larkin, A., Krueger, S. K., Phillips, I. R., Shephard, E. A., & Williams, D. E. (2012). Metabolism and Pharmacokinetics of the Anti-Tuberculosis Drug Ethionamide in a Flavin-Containing Monooxygenase Null Mouse. Pharmaceuticals, 5(11), 1147-1159. https://doi.org/10.3390/ph5111147