Glioblastoma Multiforme Therapy and Mechanisms of Resistance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Glioma Chemotherapy: TMZ and Gliadel®



3. DNA Damage Repair
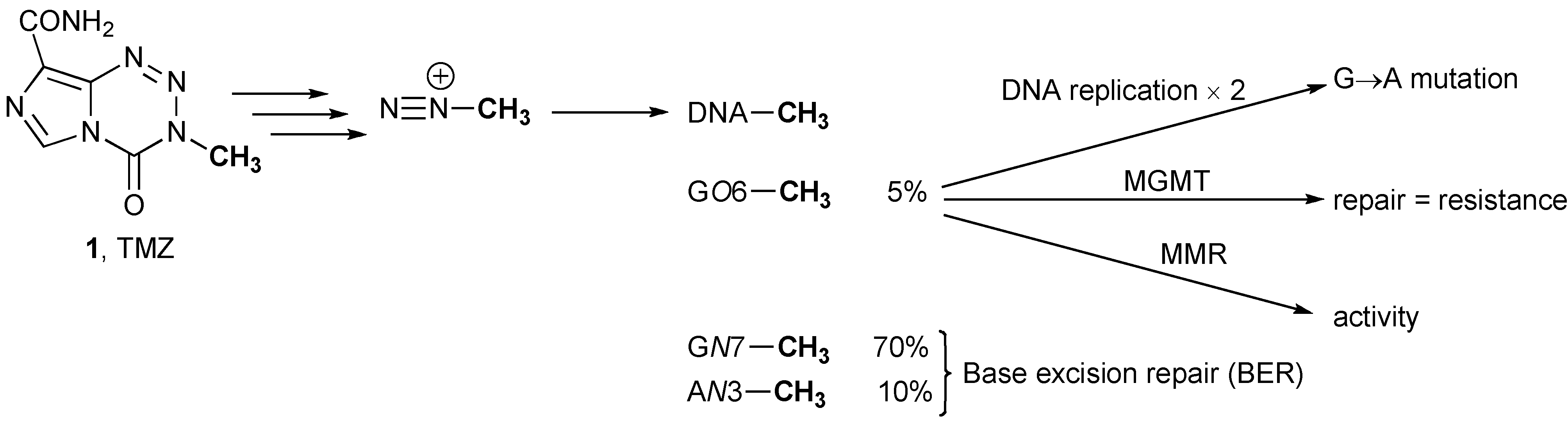
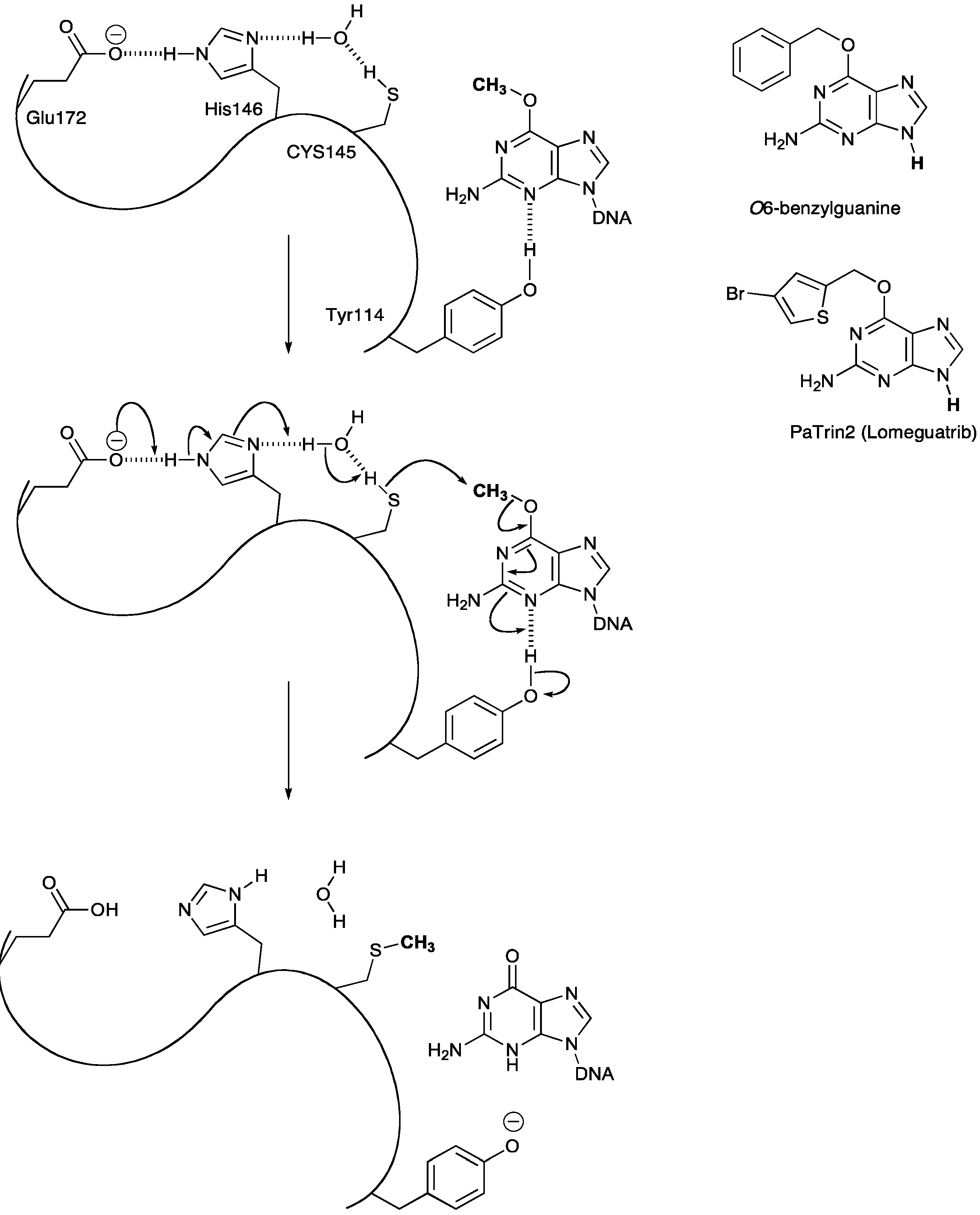
3.1. Methyl Guanine Methyl Transferase (MGMT)

3.2. MGMT Therapeutic Targets
3.3. Mismatch Repair (MMR)
4. Targeting a Complex Vasculature: GBM Cancer Stem Cell, GBM Endothelial Cells, and Angiogenic Resistant Mechanisms
4.1. Rationale for Targeting GBM Vasculature
4.2. Identification of Plastic Neural Stem Cells in the Adult Brain
4.3. GBM Cancer Stem Cells Differentiate into Glioblastoma-Derived Endothelial Cells
4.4. GBM Cancer Stem Cells and Their Microenvironment Contribute to a Chemotherapeutic Resistant Tumor
4.5. Bevacizumab: A Story of Success and Failure
5. Autophagy
5.1. Therapy Induced Autophagy
5.2. Radiosensitivity and Autophagy
6. Emerging Approaches to Therapies
6.1. Development of Novel TMZ-like Drugs


6.2. Drugs Directed Against Isocitrate Dehydrogenase
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar]
- Stommel, J.M.; Kimmelman, A.C.; Ying, H.; Nabioullin, R.; Ponugoti, A.H.; Wiedemeyer, R.; Stegh, A.H.; Bradner, J.E.; Ligon, K.L.; Brennan, C.; et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007, 318, 287–290. [Google Scholar]
- Wikstrand, C.J.; Reist, C.J.; Archer, C.E.; Zalutsky, M.R.; Bigner, D.D. The class III variant of the epidermal growth factor receptor (EGFRvIII): Characterization and utilization as an immunotherapeutic target. J. Neurovirol. 1998, 4, 148–158. [Google Scholar]
- Chen, J.; McKay, R.M.; Parada, L.F. Malignant glioma: Lessons from genomics, mouse models, and stem cells. Cell 2012, 149, 36–47. [Google Scholar]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-Year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.B.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar]
- Hall, E.; Giaccia, A. Radiobiology for the Radiologist, 6th ed.; Lippincott, Williams & Wilkins: Philadelphia, PA, USA, 2006. [Google Scholar]
- Kesari, S. Understanding glioblastoma tumor biology: The potential to improve current diagnosis and treatments. Semin. Oncol. 2011, 38, S2–S10. [Google Scholar] [CrossRef]
- Vredenburgh, J.J.; Desjardins, A.; Herndon, J.E.; Dowell, J.M.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Welissa, W.; et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin. Cancer Res. 2007, 13, 1253. [Google Scholar] [CrossRef]
- Perry, J.; Chambers, A.; Spithoff, K.; Laperriere, N. Gliadel wafers in the treatment of malignant glioma: A systematic review. Curr. Oncol. 2007, 14, 189–194. [Google Scholar] [CrossRef]
- Panigrahi, M.; Das, P.K.; Parikh, P.M. Brain tumor and Gliadel wafer treatment. Indian J. Cancer 2011, 48, 11–17. [Google Scholar] [CrossRef]
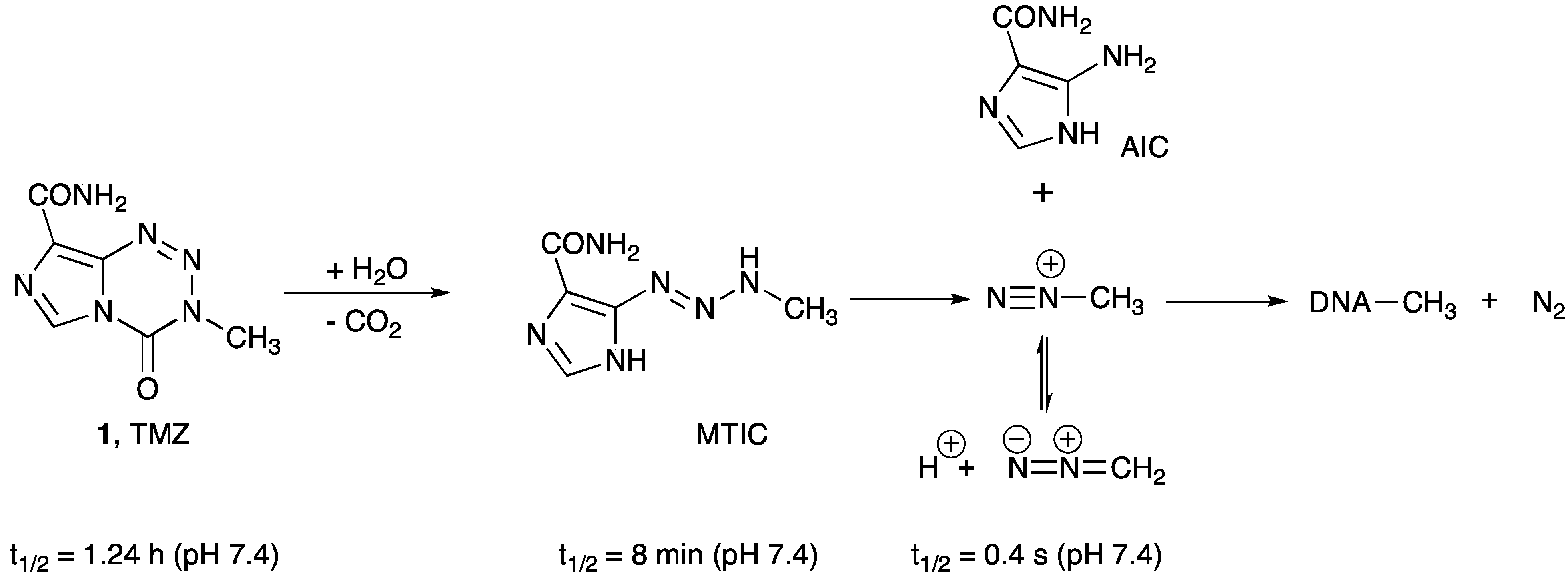
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar]
- Spiro, T.P.; Liu, L.; Majka, S.; Haaga, J.; Willson, J.K.; Gerson, S.L. Temozolomide: The effect of once- and twice-a-day dosing on tumor tissue levels of the DNA repair protein O(6)-alkylguanine-DNA-alkyltransferase. Clin. Cancer Res. 2001, 7, 2309–2317. [Google Scholar]
- Wheelhouse, R.T.; Stevens, M.F.G. Decomposition of the antitumour drug Temozolomide in deuterated phosphate buffer: Methyl group transfer is accompanied by deuterium exchange. J. Chem. Soc. Chem. Commun. 1993, 1993, 1177. [Google Scholar] [CrossRef]
- Denny, B.J.; Wheelhouse, R.T.; Stevens, M.F.; Tsang, L.L.; Slack, J.A. NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry 1994, 33, 9045–9051. [Google Scholar] [CrossRef]
- Pratt, W.B.; Ruddon, R.W.; Ensminger, W.D.; Maybaum, J. Anticancer Drugs, 2nd ed.; Oxford University Press: New York, NY, USA, 1994. [Google Scholar]
- Bleasdale, C.; Golding, B.T.; McGinnis, J.; Muller, S.; Watson, W.P. The mechanism of decomposition of N-methyl-N-nitrosourea in aqueous solution according to 13C- and 15N-NMR studies: Quantitative fragmentaion to cyanate. J. Chem. Soc. Chem. Commun. 1991, 1991, 1726–1728. [Google Scholar]
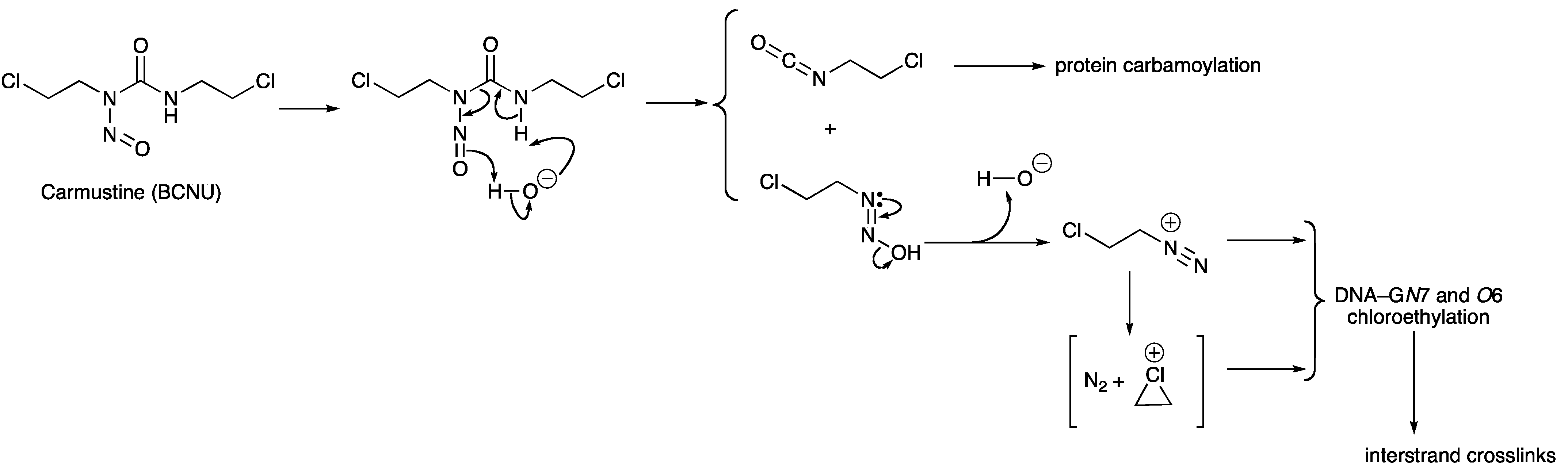
- Lown, J.W.; Chauhan, S.M. Mechanism of action of (2-haloethyl)nitrosoureas on DNA. Isolation and reactions of postulated 2-(alkylimino)-3-nitrosooxazolidine intermediates in the decomposition of 1,3-bis(2-chloroethyl)-, 1-(2-chloroethyl)-3-cyclohexyl-, and 1-(2-chloroethyl)-3-4'-trans-methylcyclohexyl)-1-nitrosourea. J. Med. Chem. 1981, 24, 270–279. [Google Scholar] [CrossRef]
- Lown, J.W.; Chauhan, S.M.S. Discrimination between alternative pathways of aqueous decomposition of anti-tumor (2-chloroethyl) nitrosoureas using specific O-18 labeling. J. Org. Chem. 1982, 47, 851–856. [Google Scholar] [CrossRef]
- Fung, L.K.; Ewend, M.G.; Sills, A.; Sipos, E.P.; Thompson, R.; Watts, M.; Colvin, O.M.; Brem, H.; Saltzman, W.M. Pharmacokinetics of interstitial delivery of carmustine, 4-hydroperoxycyclophosphamide, and paclitaxel from a biodegradable polymer implant in the monkey brain. Cancer Res. 1998, 58, 672. [Google Scholar]
- Grossman, S.A.; Reinhard, C.; Colvin, O.M.; Chasin, M.; Brundrett, R.; Tamargo, R.J.; Brem, H. The intracerebral distribution of BCNU delivered by surgically implanted biodegradable polymers. J. Neurosurg. 1992, 76, 640–647. [Google Scholar] [CrossRef]
- Kleinberg, L.R.; Weingart, J.; Burger, P.; Carson, K.; Grossman, S.A.; Li, K.; Olivi, A.; Wharam, M.D.; Brem, H. Clinical course and pathologic findings after Gliadel and radiotherapy for newly diagnosed malignant glioma: Implications for patient management. Cancer Invest. 2004, 22, 1–9. [Google Scholar]
- McGirt, M.J.; Than, K.D.; Weingart, J.D.; Chaichana, K.L.; Attenello, F.J.; Olivi, A.; Laterra, J.; Kleinberg, L.R.; Grossman, S.A.; Brem, H. Gliadel (BCNU) wafer plus concomitant temozolomide therapy after primary resection of glioblastoma multiforme. J. Neurosurg. 2009, 110, 583–588. [Google Scholar] [CrossRef]
- Park, C.K.; Kim, J.E.; Kim, J.Y.; Song, S.W.; Kim, J.W.; Choi, S.H.; Kim, T.M.; Lee, S.H.; Park, S.H. The Changes in MGMT Promoter Methylation Status in Initial and Recurrent Glioblastomas. Transl. Oncol. 2012, 5, 393–397. [Google Scholar]
- Pegg, A.E. Repair of O(6)-alkylguanine by alkyltransferases. Mutat Res. 2000, 462, 83–100. [Google Scholar] [CrossRef]
- Daniels, D.S.; Mol, C.D.; Arvai, A.S.; Kanugula, S.; Pegg, A.E.; Tainer, J.A. Active and alkylated human AGT structures: A novel zinc site, inhibitor and extrahelical base binding. EMBO J. 2000, 19, 1719–1730. [Google Scholar] [CrossRef]
- Hermisson, M.; Klumpp, A.; Wick, W.; Wischhusen, J.; Nagel, G.; Roos, W.; Kaina, B.; Weller, M. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J. Neurochem. 2006, 96, 766–776. [Google Scholar] [CrossRef]
- Sato, A.; Sunayama, J.; Matsuda, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Tachibana, K.; Tomiyama, A.; Kayama, T.; Kitanka, C. MEK-ERK signaling dictates DNA-repair gene MGMT expression and temozolomide resistance of stem-like glioblastoma cells via the MDM2-p53 axis. Stem Cells 2011, 29, 1942–1951. [Google Scholar] [CrossRef]
- Gerson, S.L. Clinical relevance of MGMT in the treatment of cancer. J. Clin. Oncol. 2002, 20, 2388–2399. [Google Scholar] [CrossRef]
- Van Nifterik, K.A.; van den Berg, J.; van der Meide, W.F.; Ameziane, N.; Wedekind, L.E.; Steenbergen, R.D.M.; Leenstra, S.; Lafleur, M.V.; Slotman, B.J.; Stalpers, L.J.A. Absence of the MGMT protein as well as methylation of the MGMT promoter predict the sensitivity for temozolomide. Br. J. Cancer 2010, 103, 29–35. [Google Scholar] [CrossRef]
- Villalva, C.; Cortes, U.; Wager, M.; Tourani, J.M.; Rivet, P.; Marquant, C.; Martin, S.; Turhan, A.G.; Karayan-Tapon, L. O6-Methylguanine-methyltransferase (MGMT) promoter methylation status in glioma stem-like cells is correlated to Temozolomide sensitivity under differentiation-promoting conditions. Int. J. Mol. Sci. 2012, 13, 6983–6994. [Google Scholar] [CrossRef]
- Kanzawa, T.; Bedwell, J.; Kondo, Y.; Kondo, S.; Germano, I.M. Inhibition of DNA repair for sensitizing resistant glioma cells to temozolomide. J. Neurosurg. 2003, 99, 1047–1052. [Google Scholar] [CrossRef]
- Turriziani, M.; Caporaso, P.; Bonmassar, L.; Buccisano, F.; Amadori, S.; Venditti, A.; Cantonetti, M.; D’Atri, S.; Bonmassar, E. O6-(4-bromothenyl)guanine (PaTrin-2), a novel inhibitor of O6-alkylguanine DNA alkyl-transferase, increases the inhibitory activity of temozolomide against human acute leukaemia cells in vitro. Pharmacol. Res. 2006, 53, 317–323. [Google Scholar]
- Clemons, M.; Kelly, J.; Watson, A.J.; Howell, A.; McElhinney, R.S.; McMurry, T.B.; Margison, G.P. O6-(4-bromothenyl)guanine reverses temozolomide resistance in human breast tumour MCF-7 cells and xenografts. Br. J. Cancer 2005, 93, 1152–1156. [Google Scholar] [CrossRef]
- Barvaux, V.A.; Ranson, M.; Brown, R.; McElhinney, R.S.; McMurry, T.B.; Margison, G.P. Dual repair modulation reverses Temozolomide resistance in vitro. Mol. Cancer. Ther. 2004, 3, 123–127. [Google Scholar]
- Hegi, M.E.; Diserens, A.C.; Godard, S.; Dietrich, P.Y.; Regli, L.; Ostermann, S.; Otten, P.; Melle, G.V.; de Tribolet, N.; Stupp, R. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin. Cancer Res. 2004, 10, 1871–1874. [Google Scholar] [CrossRef]
- Lalezari, S.; Chou, A.P.; Tran, A.; Solis, O.E.; Khanlou, N.; Chen, W.; Li, S.; Carrillo, J.A.; Chowdhury, R.; Selfridge, J.; et al. Combined analysis of O6-methylguanine-DNA methyltransferase protein expression and promoter methylation provides optimized prognostication of glioblastoma outcome. Neuro-oncology 2013, 15, 370–381. [Google Scholar] [CrossRef]
- Kreth, S.; Heyn, J.; Grau, S.; Kretzschmar, H.A.; Egensperger, R.; Kreth, F.W. Identification of valid endogenous control genes for determining gene expression in human glioma. Neuro-oncology 2010, 12, 570–579. [Google Scholar] [CrossRef]
- Quinn, J.A.; Desjardins, A.; Weingart, J.; Brem, H.; Dolan, M.E.; Delaney, S.M.; Vredenburgh, J.; Rich, J.; Friedman, A.H.; Reardon, D.A.; et al. Phase I trial of temozolomide plus O6-benzylguanine for patients with recurrent or progressive malignant glioma. J. Clin. Oncol. 2005, 23, 7178–7187. [Google Scholar] [CrossRef]
- Reese, J.S.; Qin, X.; Ballas, C.B.; Sekiguchi, M.; Gerson, S.L. MGMT expression in murine bone marrow is a major determinant of animal survival after alkylating agent exposure. J. Hematother. Stem. Cell Res. 2001, 10, 115–123. [Google Scholar] [CrossRef]
- Srinivasan, A.; Gold, B. Small-molecule inhibitors of DNA damage-repair pathways: An approach to overcome tumor resistance to alkylating anticancer drugs. Future Med. Chem. 2012, 4, 1093–1111. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Gerson, S.L.; Denis, L.; Geyer, C.; Hammond, L.A.; Patnaik, A.; Goetz, A.D.; Schwartz, G.; Edwards, T.; Reyderman, L.; et al. Marked inactivation of O6-alkylguanine-DNA alkyltransferase activity with protracted temozolomide schedules. Br. J. Cancer 2003, 88, 1004–1111. [Google Scholar] [CrossRef]
- Norden, A.D.; Lesser, G.J.; Drappatz, J.; Ligon, K.L.; Hammond, S.N.; Lee, E.Q.; Readon, E.R.; Fadul, C.E.; Plotkin, S.R.; Batchelor, T.T.; et al. Phase 2 study of dose-intense temozolomide in recurrent glioblastoma. Neuro-oncology 2013, 15, 930–935. [Google Scholar] [CrossRef]
- Kato, T.; Natsume, A.; Toda, H.; Iwamizu, H.; Sugita, T.; Hachisu, R.; Watanabe, R.; Yuki, K.; Motomura, K.; Bankiewicz, K.; et al. Efficient delivery of liposome-mediated MGMT-siRNA reinforces the cytotoxity of temozolomide in GBM-initiating cells. Gene Ther. 2010, 17, 1363–1371. [Google Scholar] [CrossRef]
- Viel, T.; Monfared, P.; Schelhaas, S.; Fricke, I.B.; Kuhlmann, M.T.; Fraefel, C.; Jacobs, A.H. Optimizing glioblastoma temozolomide chemotherapy employing lentiviral-based anti-MGMT shRNA technology. Mol. Ther. 2013, 21, 570–579. [Google Scholar] [CrossRef]
- Vlachostergios, P.J.; Hatzidaki, E.; Stathakis, N.E.; Koukoulis, G.K.; Papandreou, C.N. Bortezomib downregulates MGMT expression in T98G glioblastoma cells. Cell Mol. Neurobiol. 2013, 33, 313–318. [Google Scholar] [CrossRef]
- Gong, X.; Schwartz, P.H.; Linskey, M.E.; Bota, D.A. Neural stem/progenitors and glioma stem-like cells have differential sensitivity to chemotherapy. Neurology 2011, 76, 1126–1134. [Google Scholar] [CrossRef]
- Dy, G.K.; Thomas, J.P.; Wilding, G.; Bruzek, L.; Mandrekar, S.; Erlichman, C.; Alberti, D.; Binger, K.; Pitot, H.C.; Alberts, S.R.; et al. A phase I and pharmacologic trial of two schedules of the proteasome inhibitor, PS-341 (bortezomib, velcade), in patients with advanced cancer. Clin. Cancer Res. 2005, 11, 3410–3416. [Google Scholar]
- Phuphanich, S.; Supko, J.G.; Carson, K.A.; Grossman, S.A.; Burt Nabors, L.; Mikkelsen, T.; Lesser, G.; Rosenfeld, S.A.; Desideri, S.; Olson, J.J.; et al. Phase 1 clinical trial of bortezomib in adults with recurrent malignant glioma. J. Neurooncol. 2010, 100, 95–103. [Google Scholar] [CrossRef]
- Ghosal, G.; Chen, J. DNA damage tolerance: A double-edged sword guarding the genome. Transl. Cancer Res. 2013, 2, 107–129. [Google Scholar]
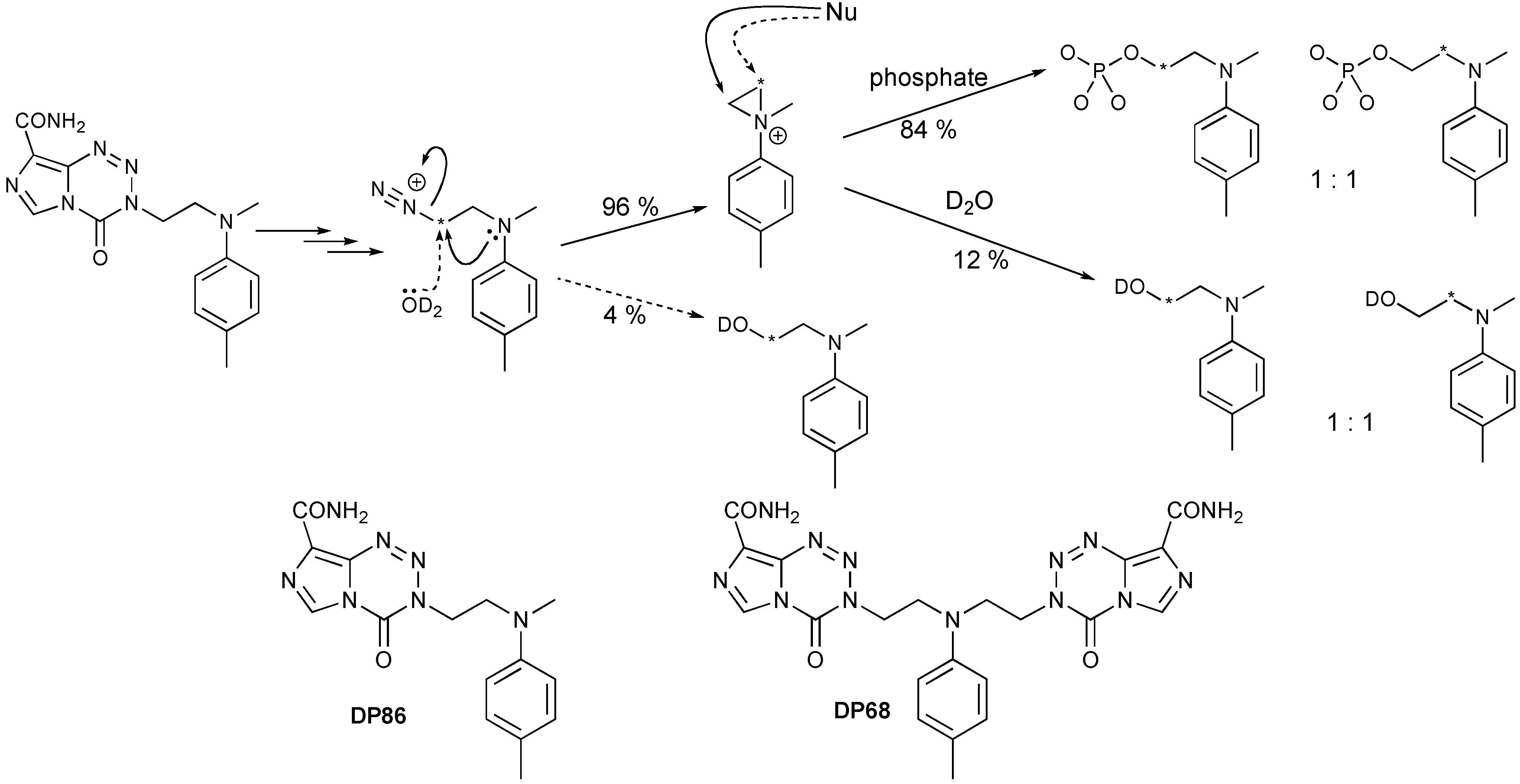
- Tuma, A.C.M.; Ramirez, Y.; Pletsas, D.; Wheelhouse, R.T.; Phillips, R.M.; Ross, A.H.; Knudson, K.; Sarkaria, J.N. Cytotoxicity of A Novel Bi-Functional Temozolomide Analog, DP68, is Independent of MGMT Status in Glioblastoma Models. In Proceedings of the 104th Annual Meeting of the American Association for Cancer Research, Washington, DC, USA, 6–10 April 2013.
- Martinez, R.; Schackert, H.K.; Appelt, H.; Plaschke, J.; Baretton, G.; Schackert, G. Low-level microsatellite instability phenotype in sporadic glioblastoma multiforme. J. Cancer Res. Clin. Oncol. 2005, 131, 87–93. [Google Scholar] [CrossRef]
- Maxwell, J.A.; Johnson, S.P.; McLendon, R.E.; Lister, D.W.; Horne, K.S.; Rasheed, A.; Quinn, J.A.; Ali-Osman, F.; Friedman, A.H.; Modrich, P.L.; et al. Mismatch repair deficiency does not mediate clinical resistance to temozolomide in malignant glioma. Clin Cancer Res. 2008, 14, 4859–4868. [Google Scholar] [CrossRef]
- Eckert, A.; Kloor, M.; Giersch, A.; Ahmadi, R.; Herold-Mende, C.; Hampl, J.A.; Heppner, F.L.; Zoubaa, S.; Holinski-Feder, E.; Pietsch, T.; et al. Microsatellite instability in pediatric and adult high-grade gliomas. Brain Pathol. 2007, 17, 146–150. [Google Scholar] [CrossRef]
- Pei, C.; Chen, H.; Jia, X.; Yan, L.; Zou, Y.; Jiang, C.; Jin, H.; Kang, C.; Jiang, T.; Ren, H. A high frequency of MSH6 G268A polymorphism and survival association in glioblastoma. Int. J. Neurosci. 2013, 123, 114–120. [Google Scholar] [CrossRef]
- Rellecke, P.; Kuchelmeister, K.; Schachenmayr, W.; Schlegel, J. Mismatch repair protein hMSH2 in primary drug resistance in in vitro human malignant gliomas. J. Neurosurg. 2004, 101, 653–658. [Google Scholar] [CrossRef]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992, 255, 1707–1710. [Google Scholar]
- Okano, H.; Sawamoto, K. Neural stem cells: Involvement in adult neurogenesis and CNS repair. Philos. Trans. R. Soc. Lond. 2008, 363, 2111–2122. [Google Scholar] [CrossRef]
- Ramon y Cajal, S. Degeneration and Regeneration of the Nervous System; Oxford Unisversity Press: Oxford, UK, 1928. [Google Scholar]
- Wurmser, A.E.; Nakashima, K.; Summers, R.G.; Toni, N.; D’Amour, K.A.; Lie, D.C.; Gage, F.H. Cell fusion-independent differentiation of neural stem cells to the endothelial lineage. Nature 2004, 430, 350–356. [Google Scholar] [CrossRef]
- Shen, Q.; Wang, Y.; Kokovay, E.; Lin, G.; Chuang, S.M.; Goderie, S.K.; Roysam, B.; Temple, S. Adult SVZ stem cells lie in a vascular niche: A quantitative analysis of niche cell-cell interactions. Cell Stem Cell 2008, 3, 289–300. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Lerocca, L.M.; de Maria, R. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Figelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef]
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; Verma, I.M. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Nat. Acad. Sci. USA. 2011, 108, 4274–4280. [Google Scholar] [CrossRef]
- Dong, J.; Zhao, Y.; Huang, Q.; Fei, X.; Diao, Y.; Shen, Y.; Xiao, H.; Zhang, T.; Lan, Q.; Gu, X. Glioma stem/progenitor cells contribute to neovascularization via transdifferentiation. Stem Cell Rev. 2011, 7, 141–152. [Google Scholar] [CrossRef]
- Rodriguez, F.J.; Orr, B.A.; Ligon, K.L.; Eberhart, C.G. Neoplastic cells are a rare component in human glioblastoma microvasculature. Oncotarget 2012, 3, 98–106. [Google Scholar]
- Borovski, T.; Beke, P.; van Tellingen, O.; Rodermond, H.M.; Verhoeff, J.J.; Lascano, V.; Daalhuisen, J.B.; Medema, J.P.; Sprick, M.R. Therapy-resistant tumor microvascular endothelial cells contribute to treatment failure in glioblastoma multiforme. Oncogene 2013, 32, 1539–1548. [Google Scholar] [CrossRef]
- Francescone, R.; Scully, S.; Bentley, B.; Yan, W.; Taylor, S.L.; Oh, D.; Moral, L.; Shao, R. Glioblastoma-derived Tumor Cells Induce Vasculogenic Mimicry through Flk-1 Protein Activation. J. Biol. Chem. 2012, 287, 24821–24831. [Google Scholar]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; de Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Kreso, A.; O’Brien, C.A.; van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef]
- Medema, J.P. Cancer stem cells: The challenges ahead. Nat. Cell. Biol. 2013, 15, 338–344. [Google Scholar]
- Quintana, E.; Shackleton, M.; Foster, H.R.; Fullen, D.R.; Sabel, M.S.; Johnson, T.M.; Morrison, S.J. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that Is reversible and not hierarchically organized. Cancer Cell. 2010, 18, 510–523. [Google Scholar] [CrossRef]
- Ishizawa, K.; Rasheed, Z.A.; Karisch, R.; Wang, Q.; Kowalski, J.; Susky, E.; Pereira, K.; Karamboulas, C.; Moghal, N.; Rajeshkumar, N.V.; et al. Tumor-initiating cells are rare in many human tumors. Cell Stem Cell 2010, 7, 279–282. [Google Scholar] [CrossRef]
- Lathia, J.D.; Gallagher, J.; Myers, J.T.; Li, M.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; Huang, A.Y.; Rich, J.N. Direct in vivo evidence for tumor propagation by glioblastoma cancer stem cells. PLoS One. 2011, 6, e24807. [Google Scholar] [CrossRef]
- Deleyrolle, L.P.; Harding, A.; Cato, K.; Siebzehnrubl, F.A.; Rahman, M.; Azari, H.; Olson, S.; Gabrielli, B.; Osborne, G.; Vescovi, A.; et al. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain 2011, 134, 1331–1343. [Google Scholar] [CrossRef]
- Weinberg, R.A. The Biology of Cancer, 2nd ed.; Garland Science: New York, NY, USA, 2013. [Google Scholar]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef]
- Folkins, C.; Shaked, Y.; Man, S.; Tang, T.; Lee, C.R.; Zhu, Z.; Hoffman, R.M.; Kerbel, R.S. Glioma tumor stem-like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal-derived factor 1. Cancer Res. 2009, 69, 7243–7251. [Google Scholar]
- Borovski, T.; Verhoeff, J.J.; ten Cate, R.; Cameron, K.; de Vries, N.A.; van Tellingen, O.; Richel, D.J.; van Furth, W.R.; Medema, J.P.; Sprick, M.R. Tumor microvasculature supports proliferation and expansion of glioma-propagating cells. Int. J. Cancer. 2009, 125, 1222–1230. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Facchino, S.; Abdouh, M.; Chatoo, W.; Bernier, G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J. Neurosci. 2010, 30, 10096–10111. [Google Scholar] [CrossRef]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.-F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Harvey-Jumper, S.L.; Heth, J.A.; Murazko, K.M.; et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef]
- Eyler, C.E.; Foo, W.C.; LaFiura, K.M.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells. 2008, 26, 3027–3036. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Reardon, D.A.; Galanis, E.; De Groot, J.F.; Cloughesy, T.F.; Wefel, J.S.; Lamborn, K.R.; Lassman, A.B.; Gilbert, M.R.; Sampson, J.H.; Wick, W.; et al. Clinical trial end points for high-grade glioma: The evolving landscape. Neuro-oncology 2011, 13, 353–361. [Google Scholar] [CrossRef]
- Raizer, J.J.; Grimm, S.; Chamberlain, M.C.; Nicholas, M.K.; Chandler, J.P.; Muro, K.; Dubner, S.; Rademaker, A.W.; Renfrow, J.; Bredel, M. A phase 2 trial of single-agent bevacizumab given in an every-3-week schedule for patients with recurrent high-grade gliomas. Cancer 2010, 116, 5297–5305. [Google Scholar] [CrossRef]
- Pope, W.B.; Lai, A.; Nghiemphu, P.; Mischel, P.; Cloughesy, T.F. MRI in patients with high-grade gliomas treated with bevacizumab and chemotherapy. Neurology 2006, 66, 1258–1260. [Google Scholar] [CrossRef]
- Johansson, F.; Ekman, S.; Blomquist, E.; Henriksson, R.; Bergstrom, S.; Bergqvist, M. A review of dose-dense temozolomide alone and in combination with bevacizumab in patients with first relapse of glioblastoma. Anticancer Res. 2012, 32, 4001–4006. [Google Scholar]
- De Groot, J.F.; Fuller, G.; Kumar, A.J.; Piao, Y.; Eterovic, K.; Ji, Y.; Conrad, C.A. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro-oncology 2010, 12, 233–242. [Google Scholar] [CrossRef]
- Kumar, K.; Wigfield, S.; Gee, H.E.; Devlin, C.M.; Singleton, D.; Li, J.L.; Buffa, F.; Huffman, M.; Sinn, A.L.; Silver, J.; et al. Dichloroacetate reverses the hypoxic adaptation to bevacizumab and enhances its antitumor effects in mouse xenografts. J. Mol. Med. 2013, 91, 749–758. [Google Scholar] [CrossRef]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Nat. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra4. [Google Scholar]
- Hoey, T.; Yen, W.C.; Axelrod, F.; Basi, J.; Donigian, L.; Dylla, S.; Fitch-Bruhns, M.; Lazetic, S.; Park, I.K.; Sato, A.; et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell 2009, 5, 168–177. [Google Scholar] [CrossRef]
- Thomas, M.; Augustin, H.G. The role of the Angiopoietins in vascular morphogenesis. Angiogenesis 2009, 12, 125–137. [Google Scholar] [CrossRef]
- Noguera-Troise, I.; Daly, C.; Papadopoulos, N.J.; Coetzee, S.; Boland, P.; Gale, N.W.; Lin, H.C.; Yancopoulos, G.D.; Thurston, G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006, 444, 1032–1037. [Google Scholar] [CrossRef]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer. 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Maes, H.; Rubio, N.; Garg, A.D.; Agostinis, P. Autophagy: shaping the tumor microenvironment and therapeutic response. Trends Mol. Med. 2013, 19, 428–446. [Google Scholar] [CrossRef]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Isaka, Y. Chloroquine in cancer therapy: A double-edged sword of autophagy. Cancer Res. 2013, 73, 3–7. [Google Scholar] [CrossRef]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef]
- Fan, Q.W.; Weiss, W.A. Autophagy and Akt promote survival in glioma. Autophagy 2011, 7, 536–538. [Google Scholar] [CrossRef]
- Firat, E.; Weyerbrock, A.; Gaedicke, S.; Grosu, A.L.; Niedermann, G. Chloroquine or chloroquine-PI3K/Akt pathway inhibitor combinations strongly promote gamma-irradiation-induced cell death in primary stem-like glioma cells. PLoS One 2012, 7, e47357. [Google Scholar]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.H.; Christmann, M.; Kaina, B. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS One 2013, 8, e55665. [Google Scholar]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int. J. Cancer 2011, 129, 2720–2731. [Google Scholar] [CrossRef]
- Palumbo, S.; Pirtoli, L.; Tini, P.; Cevenini, G.; Calderaro, F.; Toscano, M.; Miracco, C.; Comincini, S. Different involvement of autophagy in human malignant glioma cell lines undergoing irradiation and temozolomide combined treatments. J. Cell. Biochem. 2012, 113, 2308–2318. [Google Scholar] [CrossRef]
- Wang, W.J.; Long, L.M.; Yang, N.; Zhang, Q.Q.; Ji, W.J.; Zhao, J.H.; Qin, Z.H.; Wang, Z.; Chen, G.; Liang, Z.Q. NVP-BEZ235, a novel dual PI3K/mTOR inhibitor, enhances the radiosensitivity of human glioma stem cells in vitro. Acta Pharmacol. Sin. 2013, 34, 681–690. [Google Scholar]
- Carmo, A.; Carvalheiro, H.; Crespo, I.; Nunes, I.; Lopes, M.C. Effect of temozolomide on the U-118 glioma cell line. Oncol Lett. 2011, 2, 1165–1170. [Google Scholar]
- Filippi-Chiela, E.; Thorne, M.; Silva, M.B.; Pelegrini, A.; Ledur, P.; Garicochea, B.; Zamin, L.L.; Lenz, G. Resveratrol abrogates the Temozolomide-induced G2 arrest leading to mitotic catastrophe and reinforces the Temozolomide-induced senescence in glioma cells. BMC Cancer 2013, 13, 147–160. [Google Scholar] [CrossRef]
- Eimer, S.; Belaud-Rotureau, M.A.; Airiau, K.; Jeanneteau, M.; Laharanne, E.; Veron, N.; Vital, A.; Loiseau, H.; Merlio, J.P.; Belloc, F. Autophagy inhibition cooperates with erlotinib to induce glioblastoma cell death. Cancer Biol. Ther. 2011, 11, 1017–1027. [Google Scholar] [CrossRef]
- Fan, Q.W.; Cheng, C.; Hackett, C.; Feldman, M.; Houseman, B.T.; Nicolaides, T.; Haas-Kogan, D.; James, C.D.; Oakes, S.A.; Debnath, J.; et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci. Signal. 2010, 3, ra81. [Google Scholar] [CrossRef]
- Liu, T.J.; Koul, D.; LaFortune, T.; Tiao, N.; Shen, R.J.; Maira, S.M.; Garcia-Echevrria, C.; Yung, W.K. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol. Cancer Ther. 2009, 8, 2204–2210. [Google Scholar] [CrossRef]
- Munshi, A. Chloroquine in glioblastoma--new horizons for an old drug. Cancer 2009, 115, 2380–2383. [Google Scholar]
- Sotelo, J.; Briceno, E.; Lopez-Gonzalez, M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Briceno, E.; Calderon, A.; Sotelo, J. Institutional experience with chloroquine as an adjuvant to the therapy for glioblastoma multiforme. Surg Neurol. 2007, 67, 388–391. [Google Scholar] [CrossRef]
- Ding, W.X.; Chen, X.; Yin, X.M. Tumor cells can evade dependence on autophagy through adaptation. Biochem. Biophys. Res. Commun. 2012, 425, 684–688. [Google Scholar] [CrossRef]
- Cerniglia, G.J.; Karar, J.; Tyagi, S.; Christofidou-Solomidou, M.; Rengan, R.; Koumenis, C.; Malty, A. Inhibition of autophagy as a strategy to augment radiosensitization by the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Mol. Pharmacol. 2012, 82, 1230–1140. [Google Scholar] [CrossRef]
- Kuger, S.; Graus, D.; Brendtke, R.; Gunther, N.; Katzer, A.; Lutyj, P.; Polat, B.; Chatterjee, M.; Sukhorukov, V.L.; Flentje, M.; Djuzenove, C.S. Radiosensitization of glioblastoma cell lines by the dual PI3K and mTOR inhibitor NVP-BEZ235 depends on drug-irradiation schedule. Transl. Oncol. 2013, 6, 169–179. [Google Scholar]
- Pletsas, D.; Wheelhouse, R.T.; Pletsa, V.; Nicolaou, A.; Jenkins, T.C.; Bibby, M.C.; Kyrtopoulos, S.A. Polar, functionalized guanine-O6 derivatives resistant to repair by O6-alkylguanine-DNA alkyltransferase: implications for the design of DNA-modifying drugs. Eur. J. Med. Chem. 2006, 41, 330–339. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.; Hummersone, M.; Madhusudan, S.; Laughton, C.A.; Bradshaw, T.D. Certain imidazotetrazines escape O6-methylguanine-DNA methyltransferase and mismatch repair. Oncology 2011, 80, 195–207. [Google Scholar] [CrossRef]
- Shuker, D.E.; Margison, G.P. Nitrosated glycine derivatives as a potential source of O6-methylguanine in DNA. Cancer Res. 1997, 57, 366–369. [Google Scholar]
- Harrison, K.L.; Fairhurst, N.; Challis, B.C.; Shuker, D.E. Synthesis, characterization, and immunochemical detection of O6-(carboxymethyl)-2'-deoxyguanosine: a DNA adduct formed by nitrosated glycine derivatives. Chem. Res. Toxicol. 1997, 10, 652–659. [Google Scholar] [CrossRef]
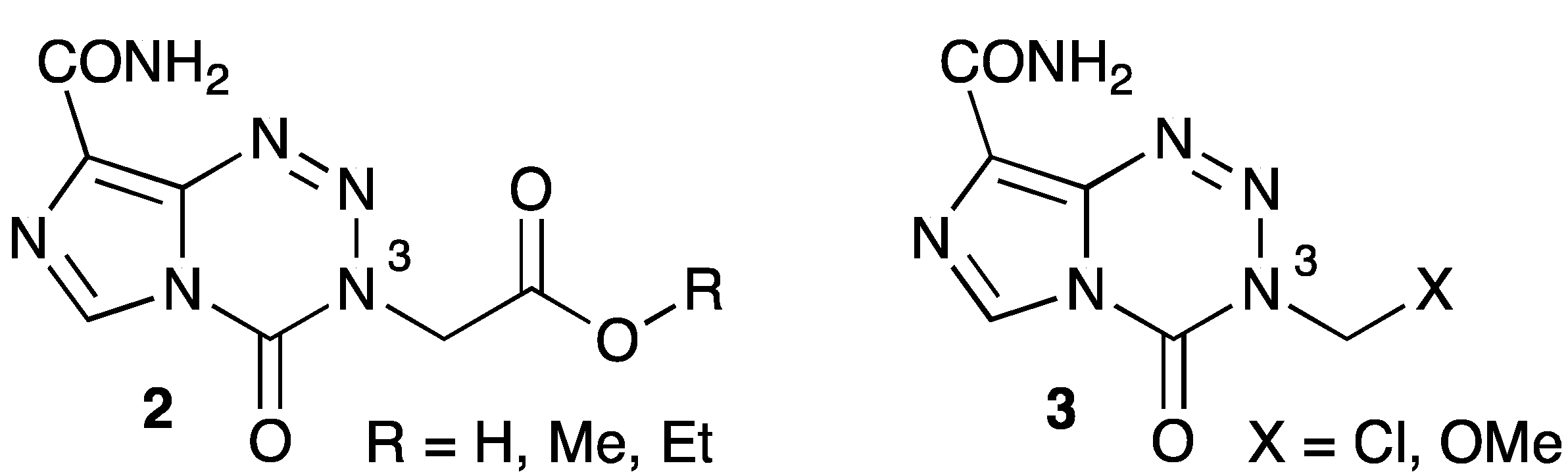
- Garelnabi, E.A.E.; Pletsas, D.; Li, L.; Kiakos, K.; Karodia, N.; Hartley, J.A.; Phillips, R.M.; Wheelhouse, R.T. Strategy for imidazotetrazine prodrugs with anticancer activity independent of MGMT and MMR. ACS Med. Chem. Lett. 2012, 3, 965–968. [Google Scholar] [CrossRef]
- Pletsas, D.; Garelnabi, E.A.; Li, L.; Phillips, R.M.; Wheelhouse, R.T. Synthesis and Quantitative Structure-Activity Relationship of Imidazotetrazine Prodrugs with Activity Independent of O6-Methylguanine-DNA-methyltransferase, DNA Mismatch Repair, and p53. J. Med. Chem. 2013, 56, 7120–7132. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Prensner, J.R.; Chinnaiyan, A.M. Metabolism unhinged: IDH mutations in cancer. Nat. Med. 2011, 17, 291–293. [Google Scholar] [CrossRef]
- Zhang, C.; Moore, L.M.; Li, X.; Yung, W.K.; Zhang, W. IDH1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro-oncology 2013, 15, 1114–1126. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Losman, J.A.; Kaelin, W.G., Jr. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef]
- Wang, F.; Travins, J.; DeLaBarre, B.; Penard-Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ramirez, Y.P.; Weatherbee, J.L.; Wheelhouse, R.T.; Ross, A.H. Glioblastoma Multiforme Therapy and Mechanisms of Resistance. Pharmaceuticals 2013, 6, 1475-1506. https://doi.org/10.3390/ph6121475
Ramirez YP, Weatherbee JL, Wheelhouse RT, Ross AH. Glioblastoma Multiforme Therapy and Mechanisms of Resistance. Pharmaceuticals. 2013; 6(12):1475-1506. https://doi.org/10.3390/ph6121475
Chicago/Turabian StyleRamirez, Yulian P., Jessica L. Weatherbee, Richard T. Wheelhouse, and Alonzo H. Ross. 2013. "Glioblastoma Multiforme Therapy and Mechanisms of Resistance" Pharmaceuticals 6, no. 12: 1475-1506. https://doi.org/10.3390/ph6121475
APA StyleRamirez, Y. P., Weatherbee, J. L., Wheelhouse, R. T., & Ross, A. H. (2013). Glioblastoma Multiforme Therapy and Mechanisms of Resistance. Pharmaceuticals, 6(12), 1475-1506. https://doi.org/10.3390/ph6121475