
Peptides and Peptidomimetics for Antimicrobial Drug Design



Abstract
:1. Introduction

2. Antimicrobial Peptides Isolated from Mammals
2.1. Defensins
2.2. Cathelicidins


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Amino Acid Sequence a | Origin | Reference |
|---|---|---|---|
| α-defensin (HNP-2) | C1YC2RIPAC3IAGERRYGTC2IYQGRLWAFC3C1 | Human | [44] |
| β-defensin (BD2) | GIGDPVTC1LKSGAIC2HPVFC3PRRYKQIGTC2GLPGTKC1C3KKP | Human | [20] |
| LL-37 | LLGDFFRKSKEKIGKEFKIVQRIKDFLRNLVPRTES | Human | [45] |
| Protegrin | RGGRLC1YC2RRRFC2VC1VGR | Pig | [46,47] |
| Indolicidin | ILPWKWPWWPWRR-NH2 | Cattle | [34,35] |
| Magainin 2 | GIGKFLHSAKKFGKAFVGEIMNS | African clawed frog | [48] |
| Cecropine A | KWKLFKKIEKVGQNIRDGIIKAGPAVAVVGQATQIAK-NH2 | Hyalophora cecropia | [49] |
| Mellitin | GIGAVLKVLTTGLPALISWIKRKRQQ | Honey bee | [50] |
| Magainin II | GIGKFLHSAKKFGKAFVGEIMNS | African clawed frog | [10,11] |
| Polyphemusin | RRWC1FRVC2YRGFC2YRKC1R | Horseshoe crab | [5,51] |
| Gramicidin S | cyclo-(Val-Orn-Leu-D-Phe-Pro)2 | Bacillus brevis | [52] |
| Nisin A b | I-DHB-A1I-DHA-LA1-ABA2-PGA2K-ABA3-GALMGA3NMK-ABA4-A-ABA5-A4HA5SIHV-DHA-K | Lactococcus lactis | [53] |
3. Insect Antimicrobial Peptides
3.1. Cecropins
3.2. Melittin
4. Plant Antimicrobial Peptides
Thionins and Defensins

5. Antimicrobial Peptides Produced by Bacteria
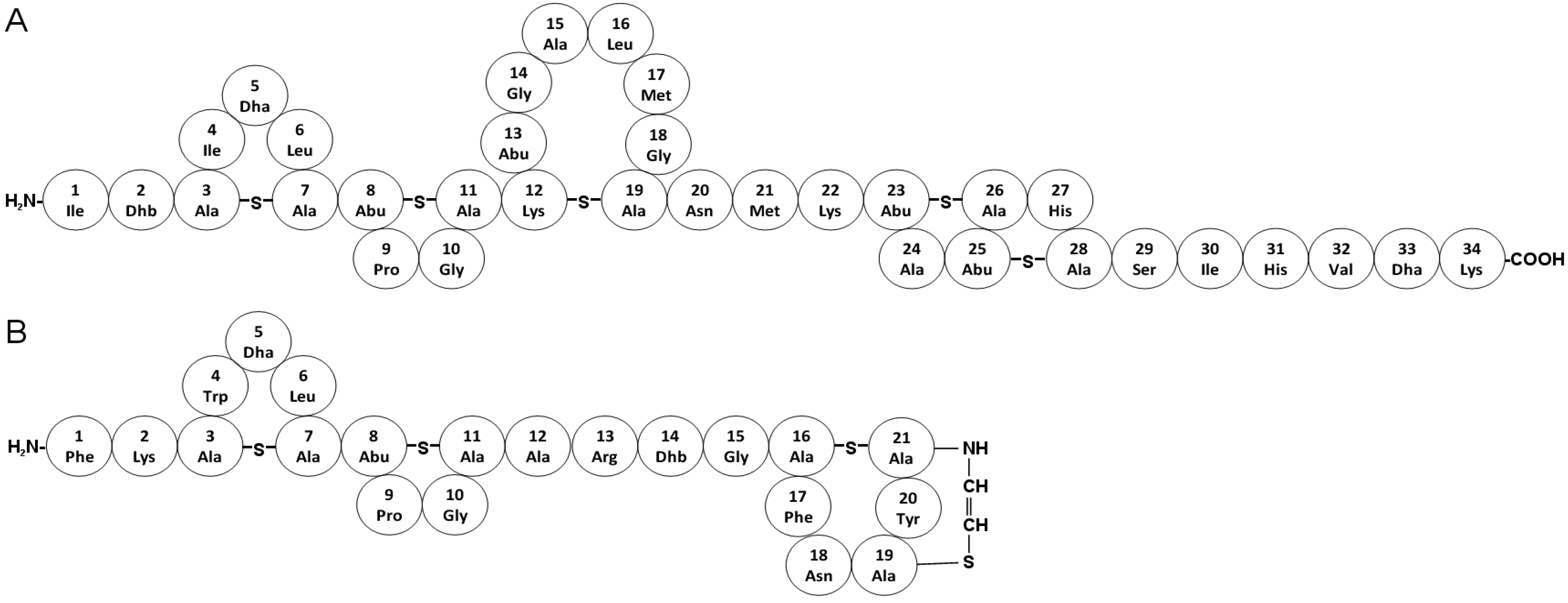
5.1. Nisin

5.2. Mutacin 1140
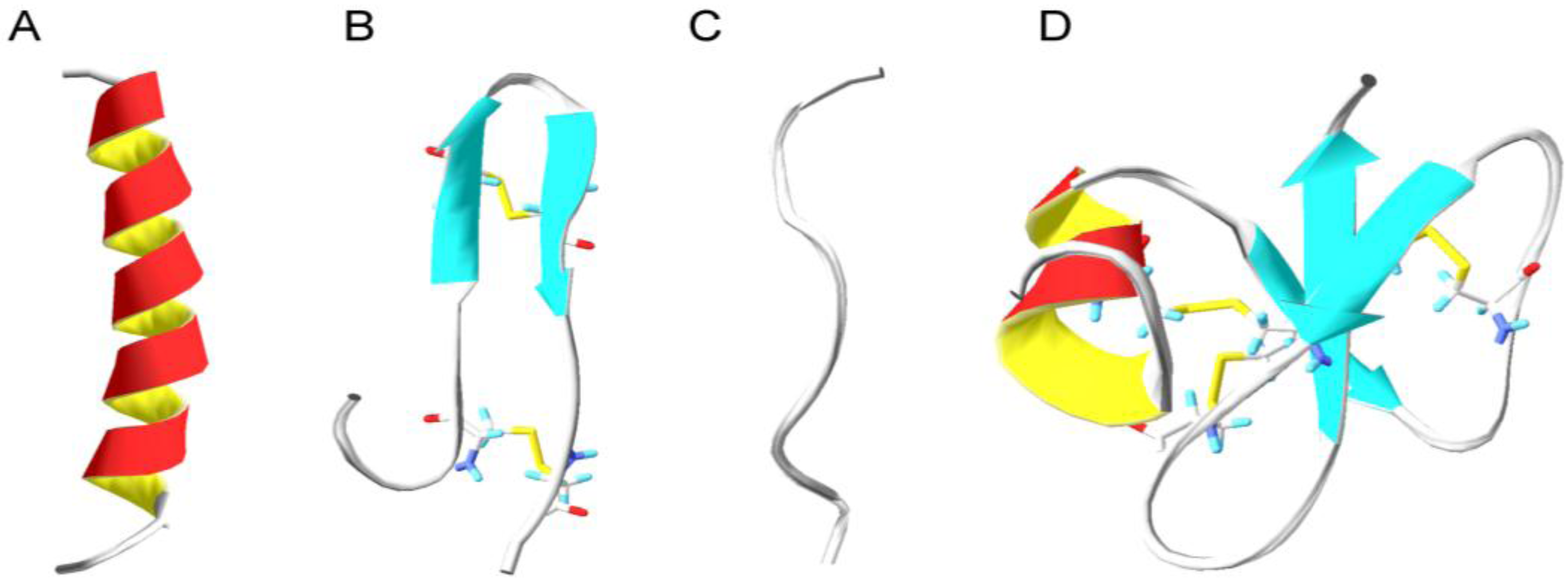
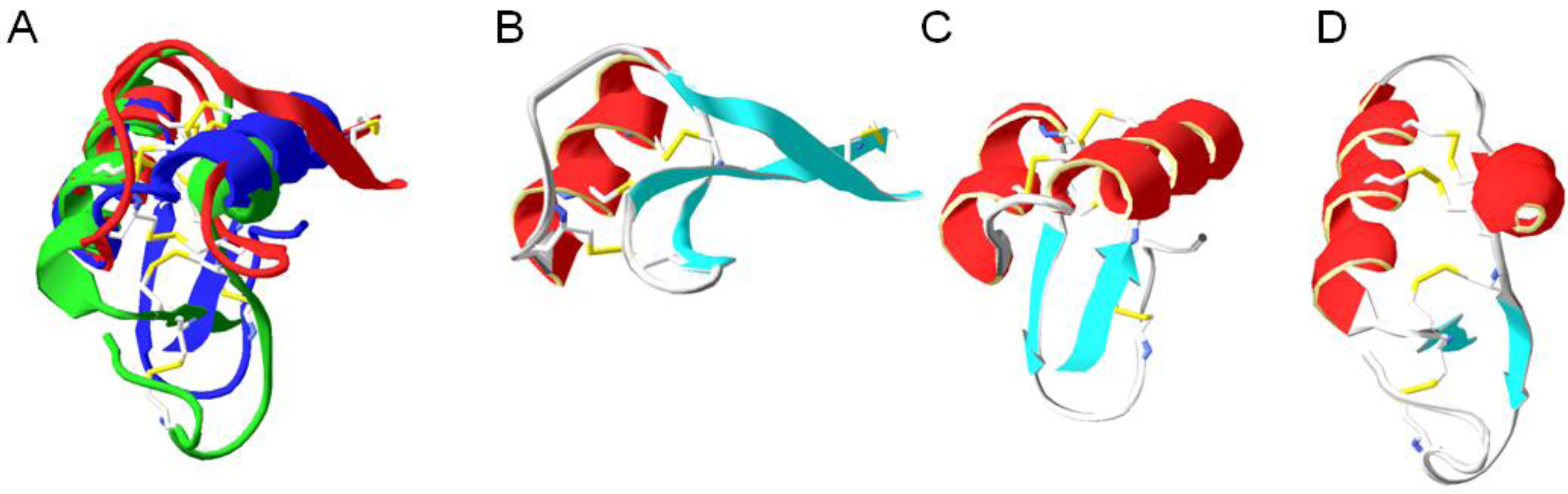
6. Structural Properties of Antimicrobial Peptides
6.1. Secondary Structure
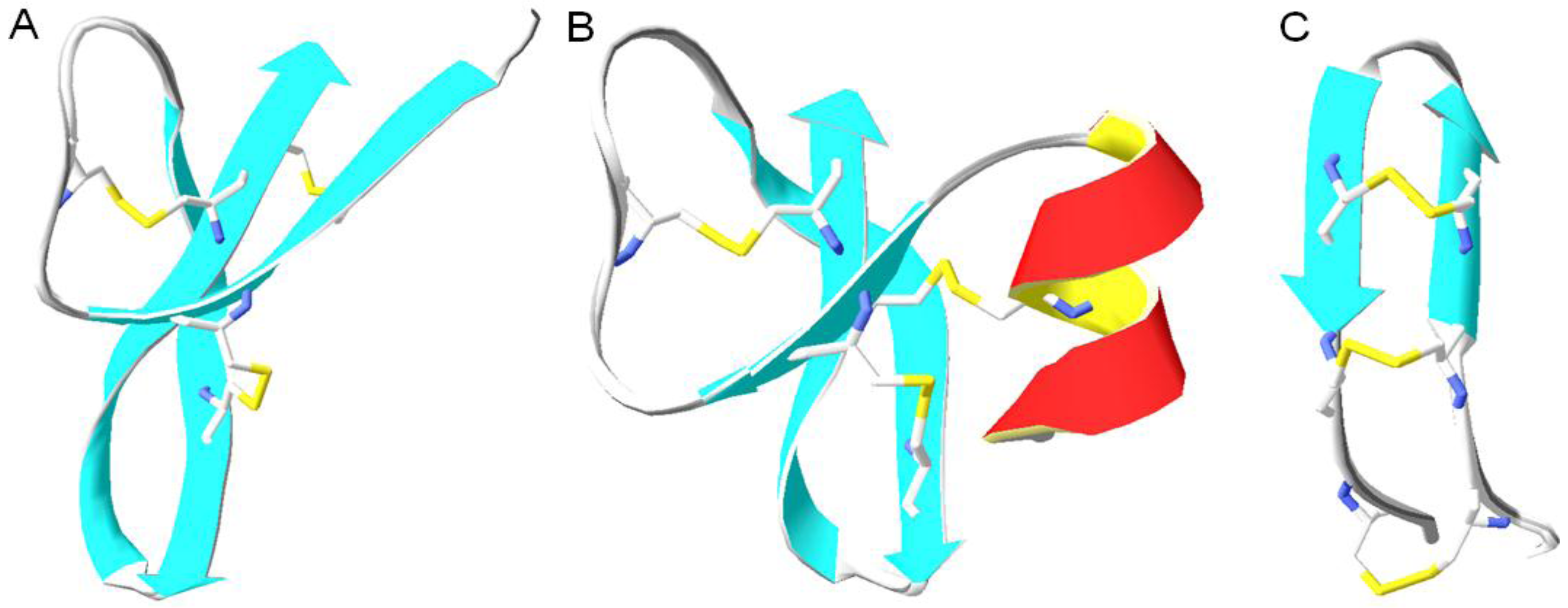
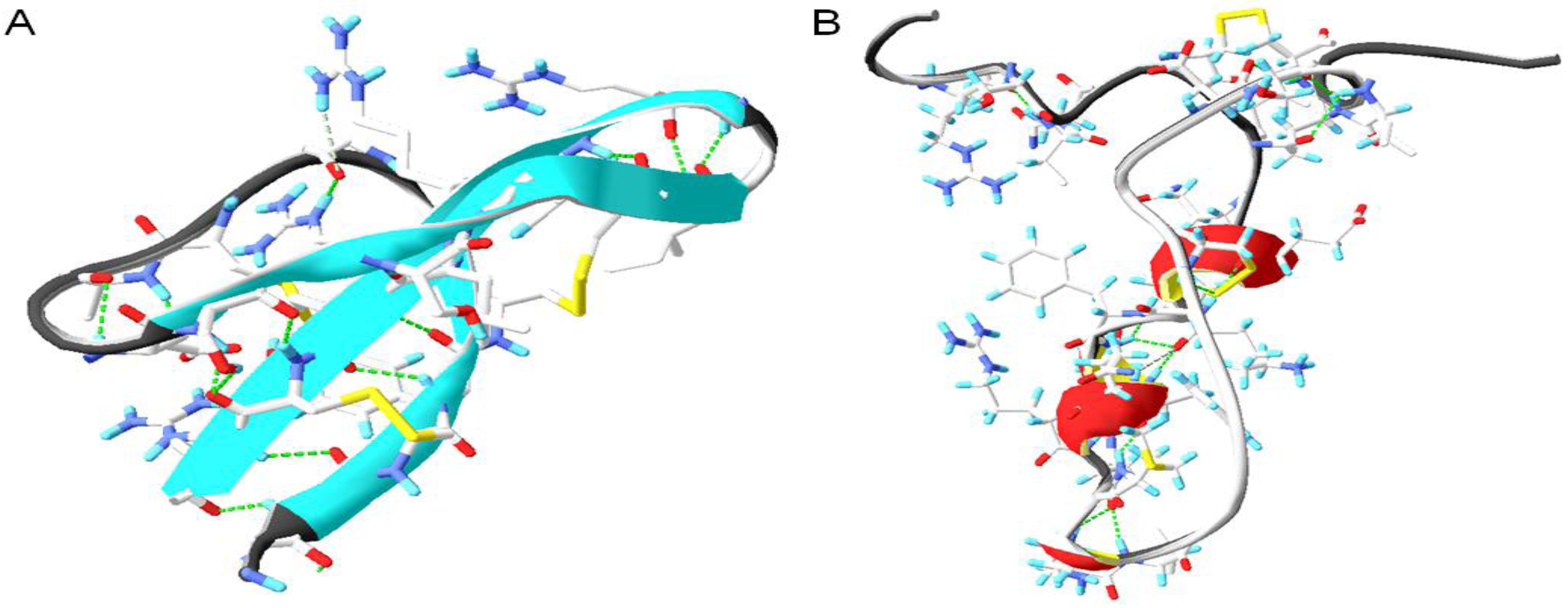
6.2. Conserved Salt Bridges

6.3. Cationicity
6.4. Hydrophobicity
6.5. Amphipathicity
6.6. Cyclic Antimicrobial Peptides

6.7. Length
7. Structural Properties of Specific Amino Acid Residues in Antimicrobial Peptide Sequences
7.1. Lysine (Lys, K) and Arginine (Arg, R)
7.2. Tryptophan (Trp, W)
7.3. Cysteine (Cys, C) and Disulfide Bonds
7.4. Proline (Pro, P)
8. Mechanism of Action of Antimicrobial Peptides
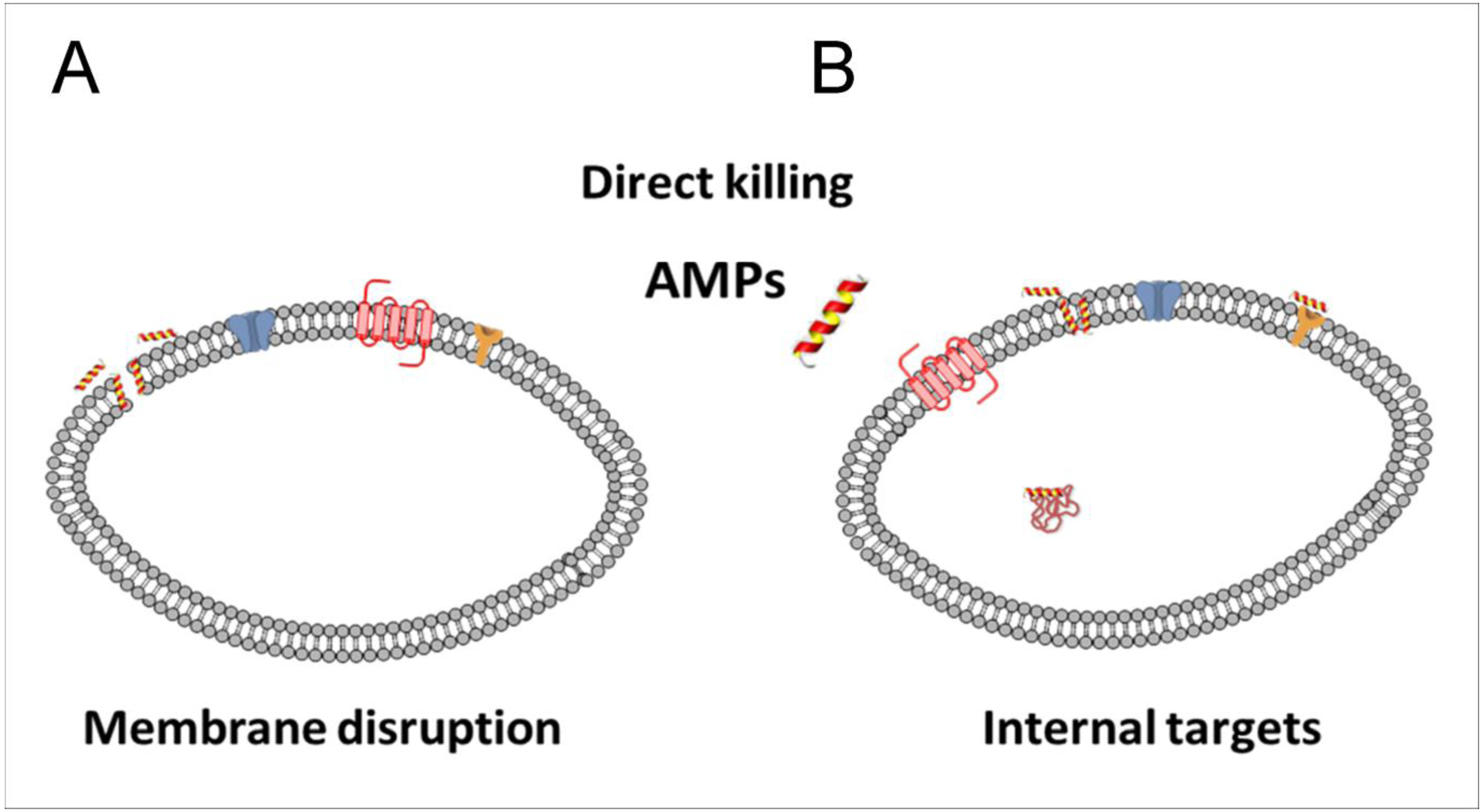
8.1. Direct Killing

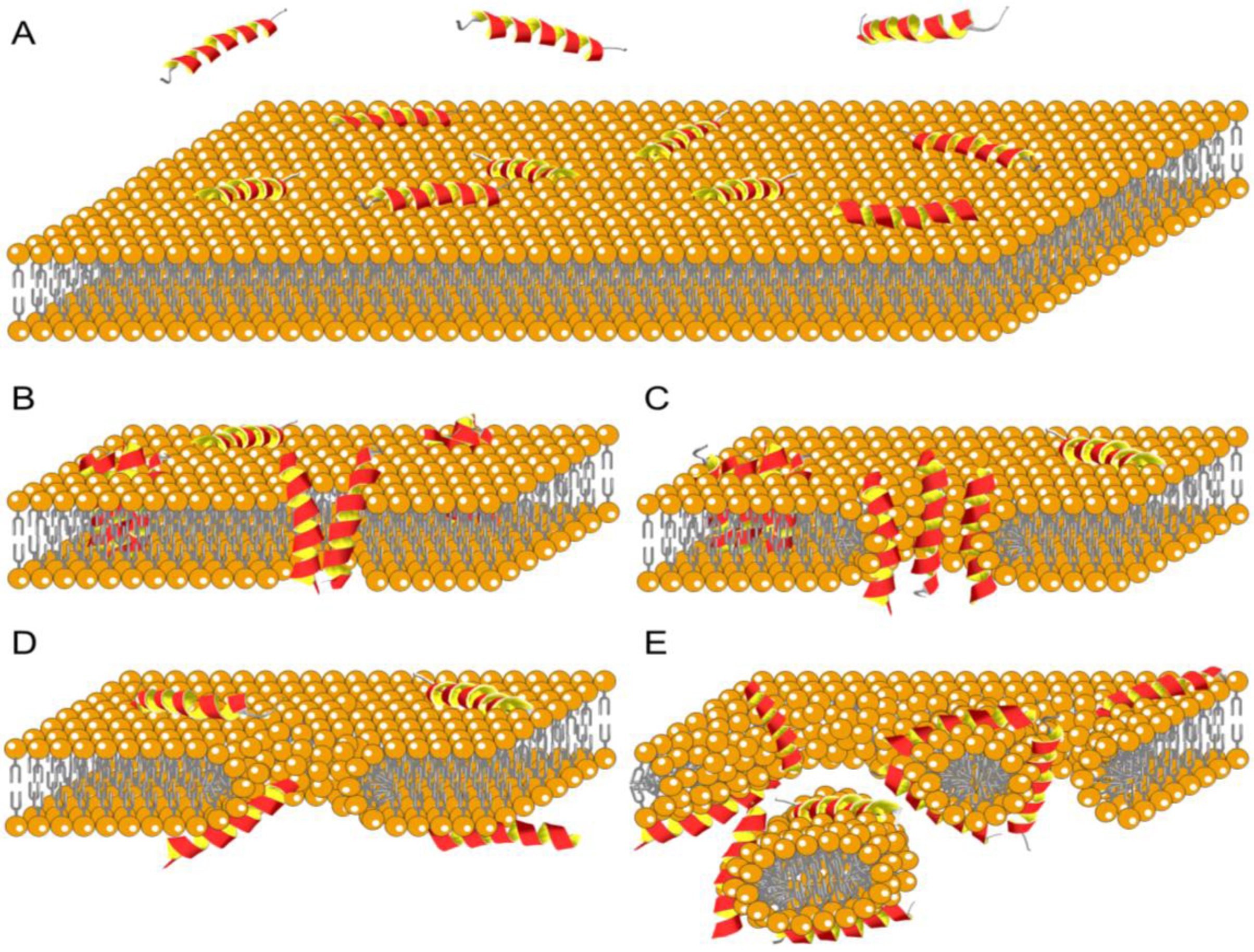
8.2. Membrane Interaction
8.3. Membrane Disruption by Antimicrobial Peptides
8.4. Other Mechanisms and Intracellular Antibacterial Targets

9. Mechanism of Bacterial Resistance towards Antimicrobial Peptides
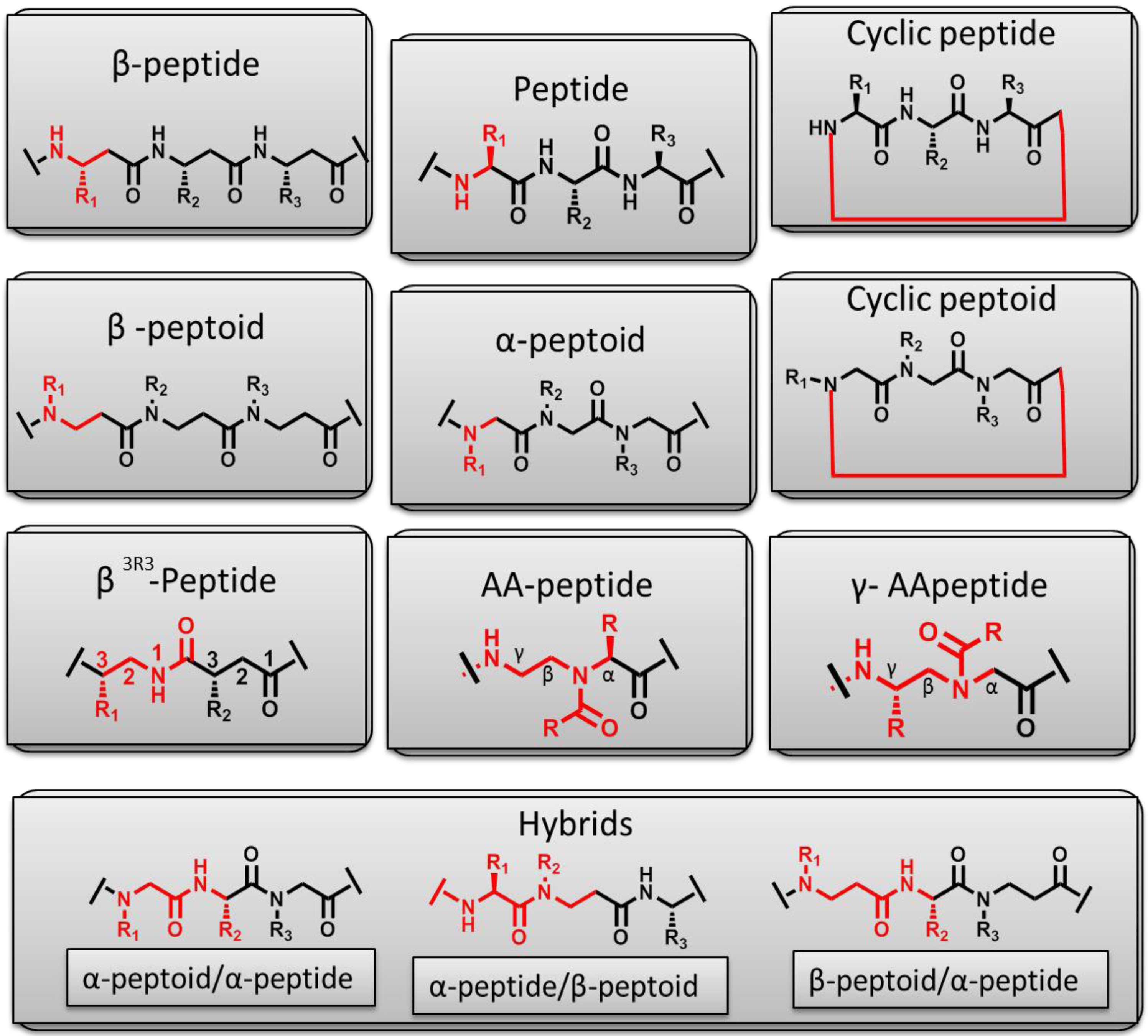
10. Peptidomimetics for Antimicrobial Research

10.1. Unnatural Amino Acid Sequences
10.2. Addition of Lipid Moieties
10.3. β-Peptidomimetics
10.4. Peptoids
10.5. Cyclic Peptoids
10.6. Hybrids
10.7. AApeptides
11. Towards the Design of Novel Antimicrobial Peptides
12. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Jenssen, H.; Hamill, P.; Hancock, R.E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Database-guided discovery of potent peptides to combat hiv-1 or superbugs. Pharmaceuticals (Basel) 2013, 6, 728–758. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Structures of human host defense cathelicidin ll-37 and its smallest antimicrobial peptide kr-12 in lipid micelles. J. Biol. Chem. 2008, 283, 32637–32643. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.P.; Rozek, A.; Hancock, R.E. Structure-activity relationships for the beta-hairpin cationic antimicrobial peptide polyphemusin i. Biochim. Biophys. Acta 2004, 1698, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Rozek, A.; Friedrich, C.L.; Hancock, R.E. Structure of the bovine antimicrobial peptide indolicidin bound to dodecylphosphocholine and sodium dodecyl sulfate micelles. Biochemistry 2000, 39, 15765–15774. [Google Scholar] [CrossRef] [PubMed]
- Sawai, M.V.; Jia, H.P.; Liu, L.; Aseyev, V.; Wiencek, J.M.; McCray, P.B., Jr.; Ganz, T.; Kearney, W.R.; Tack, B.F. The nmr structure of human beta-defensin-2 reveals a novel alpha-helical segment. Biochemistry 2001, 40, 3810–3816. [Google Scholar] [CrossRef] [PubMed]
- Andreu, D.; Rivas, L. Animal antimicrobial peptides: An overview. Biopolymers 1998, 47, 415–433. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. Apd2: The updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2009, 37, D933–D937. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.; Hultmark, D.; Engstrom, A.; Bennich, H.; Boman, H.G. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 1981, 292, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Pasupuleti, M.; Schmidtchen, A.; Malmsten, M. Antimicrobial peptides: Key components of the innate immune system. Crit. Rev. Biotechnol. 2012, 32, 143–171. [Google Scholar] [CrossRef] [PubMed]
- Boman, H.G. Peptide antibiotics and their role in innate immunity. Ann. Rev. immunol. 1995, 13, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.L.; Lehrer, R.I.; Rest, R.F. Human neutrophil antimicrobial activity. Rev. Infect. Dis. 1988, 10, S450–S456. [Google Scholar] [CrossRef] [PubMed]
- Selsted, M.E.; Ouellette, A.J. Mammalian defensins in the antimicrobial immune response. Nat. Immunol. 2005, 6, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Skerlavaj, B.; Romeo, D.; Gennaro, R. Rapid membrane permeabilization and inhibition of vital functions of gram-negative bacteria by bactenecins. Infect. Immun. 1990, 58, 3724–3730. [Google Scholar] [PubMed]
- Ganz, T.; Selsted, M.E.; Szklarek, D.; Harwig, S.S.; Daher, K.; Bainton, D.F.; Lehrer, R.I. Defensins. Natural peptide antibiotics of human neutrophils. J. Clin. Investig. 1985, 76, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Selsted, M.E.; Harwig, S.S.; Ganz, T.; Schilling, J.W.; Lehrer, R.I. Primary structures of three human neutrophil defensins. J. Clin. Investig. 1985, 76, 1436–1439. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nature Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Bowdish, D.M.; Davidson, D.J.; Hancock, R.E. Immunomodulatory properties of defensins and cathelicidins. Curr. Top. Microbiol. Immunol. 2006, 306, 27–66. [Google Scholar] [PubMed]
- Pazgier, M.; Hoover, D.M.; Yang, D.; Lu, W.; Lubkowski, J. Human beta-defensins. Cell. Mol. Life Sci. 2006, 63, 1294–1313. [Google Scholar] [CrossRef] [PubMed]
- Ouellette, A.J.; Selsted, M.E. Paneth cell defensins: Endogenous peptide components of intestinal host defense. FASEB J. 1996, 10, 1280–1289. [Google Scholar] [PubMed]
- Date, Y.; Nakazato, M.; Shiomi, K.; Toshimori, H.; Kangawa, K.; Matsuo, H.; Matsukura, S. Localization of human neutrophil peptide (hnp) and its messenger rna in neutrophil series. Ann. Hematol. 1994, 69, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Ericksen, B.; Wu, Z.; Lu, W.; Lehrer, R.I. Antibacterial activity and specificity of the six human {alpha}-defensins. Antimicrob. Agents Chemother. 2005, 49, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Porter, E.M.; van Dam, E.; Valore, E.V.; Ganz, T. Broad-spectrum antimicrobial activity of human intestinal defensin 5. Infect. Immun. 1997, 65, 2396–2401. [Google Scholar] [PubMed]
- Xu, Z.; Zhong, Z.; Huang, L.; Peng, L.; Wang, F.; Cen, P. High-level production of bioactive human beta-defensin-4 in escherichia coli by soluble fusion expression. Appl. Microbiol. Biotechnol. 2006, 72, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Nomura, I.; Goleva, E.; Howell, M.D.; Hamid, Q.A.; Ong, P.Y.; Hall, C.F.; Darst, M.A.; Gao, B.; Boguniewicz, M.; Travers, J.B.; et al. Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J. Immunol. 2003, 171, 3262–3269. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Ganz, T. Cathelicidins: A family of endogenous antimicrobial peptides. Curr. Opin. Hematol. 2002, 9, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Zaiou, M.; Gallo, R.L. Cathelicidins, essential gene-encoded mammalian antibiotics. J. Mol. Med. (Berl) 2002, 80, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Scocchi, M.; Pallavicini, A.; Salgaro, R.; Bociek, K.; Gennaro, R. The salmonid cathelicidins: A gene family with highly varied c-terminal antimicrobial domains. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2009, 152, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Putsep, K.; Carlsson, G.; Boman, H.G.; Andersson, M. Deficiency of antibacterial peptides in patients with morbus kostmann: An observation study. Lancet 2002, 360, 1144–1149. [Google Scholar] [CrossRef]
- Szyk, A.; Wu, Z.; Tucker, K.; Yang, D.; Lu, W.; Lubkowski, J. Crystal structures of human alpha-defensins hnp4, hd5, and hd6. Protein Sci. 2006, 15, 2749–2760. [Google Scholar] [CrossRef] [PubMed]
- Hoover, D.M.; Chertov, O.; Lubkowski, J. The structure of human beta-defensin-1: New insights into structural properties of beta-defensins. J. Biol. Chem. 2001, 276, 39021–39026. [Google Scholar] [CrossRef] [PubMed]
- Trabi, M.; Schirra, H.J.; Craik, D.J. Three-dimensional structure of rtd-1, a cyclic antimicrobial defensin from rhesus macaque leukocytes. Biochemistry 2001, 40, 4211–4221. [Google Scholar] [CrossRef] [PubMed]
- Falla, T.J.; Karunaratne, D.N.; Hancock, R.E.W. Mode of action of the antimicrobial peptide indolicidin. J. Biol. Chem. 1996, 271, 19298–19303. [Google Scholar] [CrossRef] [PubMed]
- Selsted, M.E.; Novotny, M.J.; Morris, W.L.; Tang, Y.Q.; Smith, W.; Cullor, J.S. Indolicidin, a novel bactericidal tridecapeptide amide from neutrophils. J. Biol. Chem. 1992, 267, 4292–4295. [Google Scholar] [PubMed]
- Hsu, C.H.; Chen, C.; Jou, M.L.; Lee, A.Y.; Lin, Y.C.; Yu, Y.P.; Huang, W.T.; Wu, S.H. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005, 33, 4053–4064. [Google Scholar] [CrossRef] [PubMed]
- Subbalakshmi, C.; Sitaram, N. Mechanism of antimicrobial action of indolicidin. FEMS Microbiol. Lett. 1998, 160, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Maier, E.; Benz, R.; Hancock, R.E. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of escherichia coli. Biochemistry 1999, 38, 7235–7242. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Kar, R.K.; Jana, J.; Saha, A.; Jana, B.; Krishnamoorthy, J.; Kumar, D.; Ghosh, S.; Chatterjee, S.; Bhunia, A. Indolicidin targets duplex DNA: Structural and mechanistic insight through a combination of spectroscopy and microscopy. ChemMedChem 2014, 9, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Ryge, T.S.; Doisy, X.; Ifrah, D.; Olsen, J.E.; Hansen, P.R. New indolicidin analogues with potent antibacterial activity. J. Pept. Res. 2004, 64, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, C.L.; Rozek, A.; Patrzykat, A.; Hancock, R.E. Structure and mechanism of action of an indolicidin peptide derivative with improved activity against gram-positive bacteria. J. Biol. Chem. 2001, 276, 24015–24022. [Google Scholar] [CrossRef] [PubMed]
- Falla, T.J.; Hancock, R.E. Improved activity of a synthetic indolicidin analog. Antimicrob. Agents Chemother. 1997, 41, 771–775. [Google Scholar] [PubMed]
- Rozek, A.; Powers, J.P.; Friedrich, C.L.; Hancock, R.E. Structure-based design of an indolicidin peptide analogue with increased protease stability. Biochemistry 2003, 42, 14130–14138. [Google Scholar] [CrossRef] [PubMed]
- Selsted, M.E.; Harwig, S.S. Determination of the disulfide array in the human defensin hnp-2. A covalently cyclized peptide. J. Biol. Chem. 1989, 264, 4003–4007. [Google Scholar] [PubMed]
- Tjabringa, G.S.; Rabe, K.F.; Hiemstra, P.S. The human cathelicidin ll-37: A multifunctional peptide involved in infection and inflammation in the lung. Pulm. Pharmacol. Ther. 2005, 18, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Liu, L.; Lehrer, R.I. Identification of a new member of the protegrin family by cdna cloning. FEBS Lett. 1994, 346, 285–288. [Google Scholar] [CrossRef]
- Cole, A.M.; Waring, A.J. The role of defensins in lung biology and therapy. Am. J. Respir. Med. 2002, 1, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Magainins, a class of antimicrobial peptides from xenopus skin: Isolation, characterization of two active forms, and partial cdna sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H. Secondary structure of the cecropins: Antibacterial peptides from the moth hyalophora cecropia. FEBS Lett. 1982, 137, 283–287. [Google Scholar] [CrossRef]
- Dempsey, C.E. The actions of melittin on membranes. Biochim. Biophys. Acta 1990, 1031, 143–161. [Google Scholar] [CrossRef]
- Miyata, T.; Tokunaga, F.; Yoneya, T.; Yoshikawa, K.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Antimicrobial peptides, isolated from horseshoe crab hemocytes, tachyplesin ii, and polyphemusins i and ii: Chemical structures and biological activity. J. Biochem. 1989, 106, 663–668. [Google Scholar] [PubMed]
- Gause, G.F.; Brazhnikova, M.G. Gramicin s - origin and mode of action. Lancet 1944, 2, 715–716. [Google Scholar]
- Gross, E.; Morell, J.L. The structure of nisin. J. Am. Chem. Soc. 1971, 93, 4634–4635. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, M. Cathelicidins, multifunctional peptides of the innate immunity. J. Leukoc. Biol. 2004, 75, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Gombart, A.F.; O'Kelly, J.; Saito, T.; Koeffler, H.P. Regulation of the camp gene by 1,25(oh)2d3 in various tissues. J. Steroid Biochem. Mol. Biol. 2007, 103, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Gombart, A.F.; Borregaard, N.; Koeffler, H.P. Human cathelicidin antimicrobial peptide (camp) gene is a direct target of the vitamin d receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin d3. FASEB J. 2005, 19, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C. Current understanding of the function of the nuclear vitamin d receptor in response to its natural and synthetic ligands. Recent Results Cancer Res. 2003, 164, 29–42. [Google Scholar] [PubMed]
- Guo, C.; Rosoha, E.; Lowry, M.B.; Borregaard, N.; Gombart, A.F. Curcumin induces human cathelicidin antimicrobial peptide gene expression through a vitamin d receptor-independent pathway. J. Nutr. Biochem. 2012, 24, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Barlow, P.G.; Li, Y.; Wilkinson, T.S.; Bowdish, D.M.; Lau, Y.E.; Cosseau, C.; Haslett, C.; Simpson, A.J.; Hancock, R.E.; Davidson, D.J. The human cationic host defense peptide ll-37 mediates contrasting effects on apoptotic pathways in different primary cells of the innate immune system. J. Leukoc. Biol. 2006, 80, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Hase, K.; Murakami, M.; Iimura, M.; Cole, S.P.; Horibe, Y.; Ohtake, T.; Obonyo, M.; Gallo, R.L.; Eckmann, L.; Kagnoff, M.F. Expression of ll-37 by human gastric epithelial cells as a potential host defense mechanism against helicobacter pylori. Gastroenterology 2003, 125, 1613–1625. [Google Scholar] [CrossRef] [PubMed]
- Travis, S.M.; Anderson, N.N.; Forsyth, W.R.; Espiritu, C.; Conway, B.D.; Greenberg, E.P.; McCray, P.B., Jr.; Lehrer, R.I.; Welsh, M.J.; Tack, B.F. Bactericidal activity of mammalian cathelicidin-derived peptides. Infect. Immun. 2000, 68, 2748–2755. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.G.; Davidson, D.J.; Gold, M.R.; Bowdish, D.; Hancock, R.E. The human antimicrobial peptide ll-37 is a multifunctional modulator of innate immune responses. J. Immunol. 2002, 169, 3883–3891. [Google Scholar] [CrossRef] [PubMed]
- Bowdish, D.M.; Davidson, D.J.; Lau, Y.E.; Lee, K.; Scott, M.G.; Hancock, R.E. Impact of ll-37 on anti-infective immunity. J. Leukoc. Biol. 2005, 77, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Barlow, P.G.; Beaumont, P.E.; Cosseau, C.; Mackellar, A.; Wilkinson, T.S.; Hancock, R.E.; Haslett, C.; Govan, J.R.; Simpson, A.J.; Davidson, D.J. The human cathelicidin ll-37 preferentially promotes apoptosis of infected airway epithelium. Am. J. Respir. Cell. Mol. Biol. 2010, 43, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Mookherjee, N.; Wee, K.; Bowdish, D.M.; Pistolic, J.; Li, Y.; Rehaume, L.; Hancock, R.E. Host defense peptide ll-37, in synergy with inflammatory mediator il-1beta, augments immune responses by multiple pathways. J. Immunol. 2007, 179, 7684–7691. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Niyonsaba, F.; Ushio, H.; Nagaoka, I.; Ikeda, S.; Okumura, K.; Ogawa, H. Cathelicidin ll-37 induces the generation of reactive oxygen species and release of human alpha-defensins from neutrophils. Br. J. Dermatol. 2007, 157, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Bals, R.; Weiner, D.J.; Moscioni, A.D.; Meegalla, R.L.; Wilson, J.M. Augmentation of innate host defense by expression of a cathelicidin antimicrobial peptide. Infect. Immun. 1999, 67, 6084–6089. [Google Scholar] [PubMed]
- Niyonsaba, F.; Iwabuchi, K.; Someya, A.; Hirata, M.; Matsuda, H.; Ogawa, H.; Nagaoka, I. A cathelicidin family of human antibacterial peptide ll-37 induces mast cell chemotaxis. Immunology 2002, 106, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A.; Pistolic, J.; Wyatt, A.; Tam, S.; Hancock, R.E. Human cathelicidin peptide ll-37 modulates the effects of ifn-gamma on apcs. J. Immunol. 2009, 183, 5788–5798. [Google Scholar] [CrossRef] [PubMed]
- Bulet, P.; Hetru, C.; Dimarcq, J.L.; Hoffmann, D. Antimicrobial peptides in insects; structure and function. Dev. Comp. Immunol. 1999, 23, 329–344. [Google Scholar] [CrossRef]
- Bulet, P.; Stocklin, R. Insect antimicrobial peptides: Structures, properties and gene regulation. Protein Pept. Lett. 2005, 12, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Vizioli, J.; Bulet, P.; Charlet, M.; Lowenberger, C.; Blass, C.; Muller, H.M.; Dimopoulos, G.; Hoffmann, J.; Kafatos, F.C.; Richman, A. Cloning and analysis of a cecropin gene from the malaria vector mosquito, anopheles gambiae. Insect Mol. Biol. 2000, 9, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, H.; Chattopadhyay, A. Melittin: A membrane-active peptide with diverse functions. Biosci. Rep. 2007, 27, 189–223. [Google Scholar] [CrossRef] [PubMed]
- Schroder, E.; Lubke, K.; Lehmann, M.; Beetz, I. Haemolytic activity and action on the surface tension of aqueous solutions of synthetic melittins and their derivatives. Experientia 1971, 27, 764–765. [Google Scholar] [CrossRef] [PubMed]
- Blondelle, S.E.; Houghten, R.A. Probing the relationships between the structure and hemolytic activity of melittin with a complete set of leucine substitution analogs. Peptide Res. 1991, 4, 12–18. [Google Scholar]
- Blondelle, S.E.; Houghten, R.A. Hemolytic and antimicrobial activities of the twenty-four individual omission analogues of melittin. Biochemistry 1991, 30, 4671–4678. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yu, R.Q.; Liu, Y.; Zhou, H.X.; Song, L.L.; Qiao, D.R. Design, recombinant expression, and antibacterial activity of the cecropins-melittin hybrid antimicrobial peptides. Curr. Microbiol. 2010, 61, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Li, W.; Zhang, L.; Zhang, Y.; Cao, B. Cecropin a-melittin mutant with improved proteolytic stability and enhanced antimicrobial activity against bacteria and fungi associated with gastroenteritis in vitro. Biochem. Biophys. Res. Commun. 2014, 451, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Marion, D.; Bakan, B.; Elmorjani, K. Plant lipid binding proteins: Properties and applications. Biotechnol. Adv. 2007, 25, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Stotz, H.U.; Thomson, J.G.; Wang, Y. Plant defensins: Defense, development and application. Plant Signal. Behav. 2009, 4, 1010–1012. [Google Scholar] [CrossRef] [PubMed]
- Beintema, J.J. Structural features of plant chitinases and chitin-binding proteins. FEBS Lett. 1994, 350, 159–163. [Google Scholar] [CrossRef]
- Burman, R.; Gunasekera, S.; Stromstedt, A.A.; Goransson, U. Chemistry and biology of cyclotides: Circular plant peptides outside the box. J. Nat. Prod. 2014, 77, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Stec, B. Plant thionins--the structural perspective. Cell. Mol. Life Sci. 2006, 63, 1370–1385. [Google Scholar] [CrossRef] [PubMed]
- Clifton, L.A.; Sanders, M.R.; Hughes, A.V.; Neylon, C.; Frazier, R.A.; Green, R.J. Lipid binding interactions of antimicrobial plant seed defence proteins: Puroindoline-a and beta-purothionin. Phys. Chem. Chem. Physics 2011, 13, 17153–17162. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, G.; Jayawardene, A.L. Isolation and characterization of viscotoxin 1-ps from viscum album l. Ssp. Austriacum (wiesb.) vollmann, growing on pinus silvestris. Acta Pharm. Suec. 1974, 11, 175–184. [Google Scholar] [PubMed]
- Giudici, M.; Pascual, R.; de la Canal, L.; Pfuller, K.; Pfuller, U.; Villalain, J. Interaction of viscotoxins a3 and b with membrane model systems: Implications to their mechanism of action. Biophys. J. 2003, 85, 971–981. [Google Scholar] [CrossRef]
- Stec, B.; Rao, U.; Teeter, M.M. Refinement of purothionins reveals solute particles important for lattice formation and toxicity. Part 2: Structure of beta-purothionin at 1.7 a resolution. Acta Crystallogr. D Biol. Crystallogr. 1995, 51, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Bruix, M.; Jimenez, M.A.; Santoro, J.; Gonzalez, C.; Colilla, F.J.; Mendez, E.; Rico, M. Solution structure of gamma 1-h and gamma 1-p thionins from barley and wheat endosperm determined by 1h-nmr: A structural motif common to toxic arthropod proteins. Biochemistry 1993, 32, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Milbradt, A.G.; Kerek, F.; Moroder, L.; Renner, C. Structural characterization of hellethionins from helleborus purpurascens. Biochemistry 2003, 42, 2404–2411. [Google Scholar] [CrossRef] [PubMed]
- Cotter, P.D.; Hill, C.; Ross, R.P. Bacteriocins: Developing innate immunity for food. Nat. Rev. Microbiol. 2005, 3, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Rogers, L.A. The inhibiting effect of streptococcus lactis on lactobacillus bulgaricus. J. Bacteriol. 1928, 16, 321–325. [Google Scholar] [PubMed]
- Wiedemann, I.; Breukink, E.; van Kraaij, C.; Kuipers, O.P.; Bierbaum, G.; de Kruijff, B.; Sahl, H.G. Specific binding of nisin to the peptidoglycan precursor lipid ii combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J. Biol. Chem. 2001, 276, 1772–1779. [Google Scholar] [CrossRef] [PubMed]
- Hasper, H.E.; de Kruijff, B.; Breukink, E. Assembly and stability of nisin-lipid ii pores. Biochemistry 2004, 43, 11567–11575. [Google Scholar] [CrossRef] [PubMed]
- Breukink, E.; Ganz, P.; de Kruijff, B.; Seelig, J. Binding of nisin z to bilayer vesicles as determined with isothermal titration calorimetry. Biochemistry 2000, 39, 10247–10254. [Google Scholar] [CrossRef] [PubMed]
- Hasper, H.E.; Kramer, N.E.; Smith, J.L.; Hillman, J.D.; Zachariah, C.; Kuipers, O.P.; de Kruijff, B.; Breukink, E. An alternative bactericidal mechanism of action for lantibiotic peptides that target lipid ii. Science 2006, 313, 1636–1637. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.C.; Leyland, M.; Clark, J.; Dodd, H.M.; Lian, L.Y.; Gasson, M.J.; Bycroft, B.W.; Roberts, G.C. Structure-activity relationships in the peptide antibiotic nisin: Antibacterial activity of fragments of nisin. FEBS Lett. 1996, 390, 129–132. [Google Scholar] [CrossRef]
- Strother, T.; Hamers, R.J.; Smith, L.M. Covalent attachment of oligodeoxyribonucleotides to amine-modified si (001) surfaces. Nucleic Acids Res. 2000, 28, 3535–3541. [Google Scholar] [CrossRef] [PubMed]
- Hillman, J.D.; Novak, J.; Sagura, E.; Gutierrez, J.A.; Brooks, T.A.; Crowley, P.J.; Hess, M.; Azizi, A.; Leung, K.; Cvitkovitch, D.; et al. Genetic and biochemical analysis of mutacin 1140, a lantibiotic from streptococcus mutans. Infect. Immun. 1998, 66, 2743–2749. [Google Scholar] [PubMed]
- Fox, J.L. Antimicrobial peptides stage a comeback. Nat. Biotechnol. 2013, 31, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Fan, H.; Huang, Y.; Peng, F.; Yuan, S.; Tong, Y. Recombinant lysostaphin protects mice from methicillin-resistant staphylococcus aureus pneumonia. BioMed Res. Int. 2014, 2014, 602185. [Google Scholar] [PubMed]
- Hill, C.P.; Yee, J.; Selsted, M.E.; Eisenberg, D. Crystal structure of defensin hnp-3, an amphiphilic dimer: Mechanisms of membrane permeabilization. Science 1991, 251, 1481–1485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Selsted, M.E.; Pardi, A. Nmr studies of defensin antimicrobial peptides. 1. Resonance assignment and secondary structure determination of rabbit np-2 and human hnp-1. Biochemistry 1992, 31, 11348–11356. [Google Scholar] [CrossRef] [PubMed]
- Pardi, A.; Zhang, X.L.; Selsted, M.E.; Skalicky, J.J.; Yip, P.F. Nmr studies of defensin antimicrobial peptides. 2. Three-dimensional structures of rabbit np-2 and human hnp-1. Biochemistry 1992, 31, 11357–11364. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, E.; Li, C.; Zeng, P.; Diepeveen-de Buin, M.; Lu, W.Y.; Breukink, E.; Lu, W. Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid ii. FEBS Lett. 2010, 584, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Shai, Y.; Segrest, J.P.; Anantharamaiah, G.M. Mechanisms for the modulation of membrane bilayer properties by amphipathic helical peptides. Biopolymers 1995, 37, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Jahnsen, R.D.; Frimodt-Moller, N.; Franzyk, H. Antimicrobial activity of peptidomimetics against multidrug-resistant escherichia coli : A comparative study of different backbones. J. Med. Chem. 2012, 55, 7253–7261. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Vogel, H.J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta 1999, 1462, 11–28. [Google Scholar] [CrossRef]
- Padmanabhan, S.; York, E.J.; Stewart, J.M.; Baldwin, R.L. Helix propensities of basic amino acids increase with the length of the side-chain. J. Mol. Biol. 1996, 257, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Scholtz, J.M. A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 1998, 75, 422–427. [Google Scholar] [CrossRef]
- Deslouches, B.; Phadke, S.M.; Lazarevic, V.; Cascio, M.; Islam, K.; Montelaro, R.C.; Mietzner, T.A. De novo generation of cationic antimicrobial peptides: Influence of length and tryptophan substitution on antimicrobial activity. Antimicrob. Agents Chemother. 2005, 49, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Javadpour, M.M.; Juban, M.M.; Lo, W.C.; Bishop, S.M.; Alberty, J.B.; Cowell, S.M.; Becker, C.L.; McLaughlin, M.L. De novo antimicrobial peptides with low mammalian cell toxicity. J. Med. Chem. 1996, 39, 3107–3113. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, M.; de Leeuw, E.; Pazgier, M.; Li, J.; Lubkowski, J.; Lu, W. The conserved salt bridge in human alpha-defensin 5 is required for its precursor processing and proteolytic stability. J. Biol. Chem. 2008, 283, 21509–21518. [Google Scholar] [CrossRef] [PubMed]
- Hunter, H.N.; Demcoe, A.R.; Jenssen, H.; Gutteberg, T.J.; Vogel, H.J. Human lactoferricin is partially folded in aqueous solution and is better stabilized in a membrane mimetic solvent. Antimicrob. Agents Chemother. 2005, 49, 3387–3395. [Google Scholar] [CrossRef] [PubMed]
- Wommack, A.J.; Robson, S.A.; Wanniarachchi, Y.A.; Wan, A.; Turner, C.J.; Wagner, G.; Nolan, E.M. Nmr solution structure and condition-dependent oligomerization of the antimicrobial peptide human defensin 5. Biochemistry 2012, 51, 9624–9637. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, E.; Rajabi, M.; Zou, G.; Pazgier, M.; Lu, W. Selective arginines are important for the antibacterial activity and host cell interaction of human alpha-defensin 5. FEBS Lett. 2009, 583, 2507–2512. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; de Leeuw, E.; Pazgier, M.; Yuan, W.R.; Zou, G.Z.; Wang, J.F.; Ericksen, B.; Lu, W.Y.; Lehrer, R.I.; Lu, W.Y. Through the looking glass, mechanistic insights from enantiomeric human defensins. J. Biol. Chem. 2009, 284, 29180–29192. [Google Scholar] [CrossRef] [PubMed]
- Pazgier, M.; Prahl, A.; Hoover, D.M.; Lubkowski, J. Studies of the biological properties of human beta-defensin 1. J. Biol. Chem. 2007, 282, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Harder, J.; Bartels, J.; Christophers, E.; Schroder, J.M. A peptide antibiotic from human skin. Nature 1997, 387, 861–861. [Google Scholar] [CrossRef] [PubMed]
- Schibli, D.J.; Hunter, H.N.; Aseyev, V.; Starner, T.D.; Wiencek, J.M.; McCray, P.B.; Tack, B.F.; Vogel, H.J. The solution structures of the human beta-defensins lead to a better understanding of the potent bactericidal activity of hbd3 against staphylococcus aureus. J. Biol. Chem. 2002, 277, 8279–8289. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Nikolenko, H.; Meyer, J.; Beyermann, M.; Bienert, M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001, 501, 146–150. [Google Scholar] [CrossRef]
- Hilpert, K.; Elliott, M.R.; Volkmer-Engert, R.; Henklein, P.; Donini, O.; Zhou, Q.; Winkler, D.F.H.; Hancock, R.E.W. Sequence requirements and an optimization strategy for short antimicrobial peptides. Chem. Biol. 2006, 13, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.G.; Martin, N.L.; Hancock, R.E.W. Interaction of macrophage cationic proteins with the outer-membrane of pseudomonas-aeruginosa. Infect. Immun. 1988, 56, 693–698. [Google Scholar] [PubMed]
- Piers, K.L.; Hancock, R.E.W. The interaction of a recombinant cecropin/melittin hybrid peptide with the outer-membrane of pseudomonas-aeruginosa. Mol. Microbiol. 1994, 12, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta 2009, 1788, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J. Biol. Chem. 2012, 287, 7738–7745. [Google Scholar] [CrossRef] [PubMed]
- Wieprecht, T.; Dathe, M.; Krause, E.; Beyermann, M.; Maloy, W.L.; MacDonald, D.L.; Bienert, M. Modulation of membrane activity of amphipathic, antibacterial peptides by slight modifications of the hydrophobic moment. FEBS Lett. 1997, 417, 135–140. [Google Scholar] [CrossRef]
- Gopal, R.; Seo, C.H.; Song, P.I.; Park, Y. Effect of repetitive lysine-tryptophan motifs on the bactericidal activity of antimicrobial peptides. Amino acids 2013, 44, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D. Three-dimensional structure of membrane and surface proteins. Ann. Rev. Biochem. 1984, 53, 595–623. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vidall, M.; Jayasinghe, S.; Ladokhin, A.S.; White, S.H. Folding amphipathic helices into membranes: Amphiphilicity trumps hydrophobicity. J. Mol. Biol. 2007, 370, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.; Vasil, M.L.; Hodges, R.S. Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar] [CrossRef] [PubMed]
- Thaker, H.D.; Cankaya, A.; Scott, R.W.; Tew, G.N. Role of amphiphilicity in the design of synthetic mimics of antimicrobial peptides with gram-negative activity. ACS Med. Chem. Lett. 2013, 4, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Wessolowski, A.; Bienert, M.; Dathe, M. Antimicrobial activity of arginine- and tryptophan-rich hexapeptides: The effects of aromatic clusters, d-amino acid substitution and cyclization. J. Pept. Res. 2004, 64, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Pathak, N.; Salas-Auvert, R.; Ruche, G.; Janna, M.H.; McCarthy, D.; Harrison, R.G. Comparison of the effects of hydrophobicity, amphiphilicity, and alpha-helicity on the activities of antimicrobial peptides. Proteins 1995, 22, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Andreu, D.; Ubach, J.; Boman, A.; Wahlin, B.; Wade, D.; Merrifield, R.B.; Boman, H.G. Shortened cecropin a-melittin hybrids. Significant size reduction retains potent antibiotic activity. FEBS Lett. 1992, 296, 190–194. [Google Scholar] [CrossRef]
- Kondejewski, L.H.; Jelokhani-Niaraki, M.; Farmer, S.W.; Lix, B.; Kay, C.M.; Sykes, B.D.; Hancock, R.E.W.; Hodges, R.S. Dissociation of antimicrobial and hemolytic activities in cyclic peptide diastereomers by systematic alterations in amphipathicity. J. Biol. Chem. 1999, 274, 13181–13192. [Google Scholar] [CrossRef] [PubMed]
- Kushibiki, T.; Kamiya, M.; Aizawa, T.; Kumaki, Y.; Kikukawa, T.; Mizuguchi, M.; Demura, M.; Kawabata, S.; Kawano, K. Interaction between tachyplesin i, an antimicrobial peptide derived from horseshoe crab, and lipopolysaccharide. Biochim. Biophys. Acta 2014, 1844, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, R.L.; Dieckmann, T.; Harwig, S.S.; Lehrer, R.I.; Eisenberg, D.; Feigon, J. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem. Biol. 1996, 3, 543–550. [Google Scholar] [CrossRef]
- Bagheri, M.; Keller, S.; Dathe, M. Interaction of w-substituted analogs of cyclo-rrrwfw with bacterial lipopolysaccharides: The role of the aromatic cluster in antimicrobial activity. Antimicrob. Agents Chemother. 2011, 55, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Scheinpflug, K.; Nikolenko, H.; Komarov, I.V.; Rautenbach, M.; Dathe, M. What goes around comes around-a comparative study of the influence of chemical modifications on the antimicrobial properties of small cyclic peptides. Pharmaceuticals (Basel) 2013, 6, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Kondejewski, L.H.; Farmer, S.W.; Wishart, D.S.; Hancock, R.E.; Hodges, R.S. Gramicidin s is active against both gram-positive and gram-negative bacteria. Int. J. Pept. Protein Res. 1996, 47, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Kondejewski, L.H.; Farmer, S.W.; Wishart, D.S.; Kay, C.M.; Hancock, R.E.; Hodges, R.S. Modulation of structure and antibacterial and hemolytic activity by ring size in cyclic gramicidin s analogs. J. Biol. Chem. 1996, 271, 25261–25268. [Google Scholar] [CrossRef] [PubMed]
- Appelt, C.; Wessolowski, A.; Soderhall, J.A.; Dathe, M.; Schmieder, P. Structure of the antimicrobial, cationic hexapeptide cyclo(rrwwrf) and its analogues in solution and bound to detergent micelles. Chembiochem 2005, 6, 1654–1662. [Google Scholar] [CrossRef] [PubMed]
- Trabi, M.; Craik, D.J. Circular proteins--no end in sight. Trends Biochem. Sci. 2002, 27, 132–138. [Google Scholar] [CrossRef]
- Bionda, N.; Stawikowski, M.; Stawikowska, R.; Cudic, M.; Lopez-Vallejo, F.; Treitl, D.; Medina-Franco, J.; Cudic, P. Effects of cyclic lipodepsipeptide structural modulation on stability, antibacterial activity, and human cell toxicity. ChemMedChem 2012, 7, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.G.; Brady, A.; Young, A.; Rasimick, B.; Chen, K.; Zhou, C.H.; Kallenbach, N.R. Length effects in antimicrobial peptides of the (rw)(n) series. Antimicrob. Agents Chemother. 2007, 51, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Niidome, T.; Matsuyama, N.; Kunihara, M.; Hatakeyama, T.; Aoyagi, H. Effect of chain length of cationic model peptides on antibacterial activity. Bull. Chem. Soc. Jpn. 2005, 78, 473–476. [Google Scholar] [CrossRef]
- Dong, N.; Ma, Q.; Shan, A.; Lv, Y.; Hu, W.; Gu, Y.; Li, Y. Strand length-dependent antimicrobial activity and membrane-active mechanism of arginine- and valine-rich beta-hairpin-like antimicrobial peptides. Antimicrob. Agents Chemother. 2012, 56, 2994–3003. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Mishra, B.; Epand, R.F.; Epand, R.M. High-quality 3d structures shine light on antibacterial, anti-biofilm and antiviral activities of human cathelicidin ll-37 and its fragments. Biochim. Biophys. Acta 2014, 1838, 2160–2172. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.M.; Schaus, K.A.; Vogel, H.J.; Juffer, A.H. Molecular dynamics study of peptide-bilayer adsorption. Biophys. J. 2001, 80, 579–596. [Google Scholar] [CrossRef]
- Aliste, M.P.; MacCallum, J.L.; Tieleman, D.P. Molecular dynamics simulations of pentapeptides at interfaces: Salt bridge and cation-pi interactions. Biochemistry 2003, 42, 8976–8987. [Google Scholar] [CrossRef] [PubMed]
- Zou, G.Z.; de Leeuw, E.; Li, C.; Pazgier, M.; Li, C.Q.; Zeng, P.Y.; Lu, W.Y.; Lubkowski, J.; Lu, W.Y. Toward understanding the cationicity of defensins. arg and lys versus their noncoded analogs. J. Biol. Chem. 2007, 282, 19653–19665. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Lee, M.K.; Kim, K.L.; Hahm, K.S. Structure-biological activity relationships of 11-residue highly basic peptide segment of bovine lactoferrin. Int. J. Pept. Protein Res. 1996, 48, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Gopal, R.; Park, S.C.; Ha, K.J.; Cho, S.J.; Kim, S.W.; Song, P.I.; Nah, J.W.; Park, Y.; Hahm, K.S. Effect of leucine and lysine substitution on the antimicrobial activity and evaluation of the mechanism of the hpa3nt3 analog peptide. J. Pept. Sci. 2009, 15, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.J.; Kim, D.T.; Steinman, L.; Fathman, C.G.; Rothbard, J.B. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 2000, 56, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Cordier, C.; Boutimah, F.; Bourdeloux, M.; Dupuy, F.; Met, E.; Alberti, P.; Loll, F.; Chassaing, G.; Burlina, F.; Saison-Behmoaras, T.E. Delivery of antisense peptide nucleic acids to cells by conjugation with small arginine-rich cell-penetrating peptide (r/w)9. PloS ONE 2014, 9, e104999. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Tagai, C.; Shiraishi, T.; Miyaji, K.; Iwamuro, S. Differential mode of antimicrobial actions of arginine-rich and lysine-rich histones against gram-positive staphylococcus aureus. Peptides 2013, 48, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Koyama, T.; Conlon, J.M.; Yamakura, F.; Iwamuro, S. Antimicrobial action of histone h2b in escherichia coli: Evidence for membrane translocation and DNA-binding of a histone h2b fragment after proteolytic cleavage by outer membrane proteinase t. Biochimie 2008, 90, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Tagai, C.; Morita, S.; Shiraishi, T.; Miyaji, K.; Iwamuro, S. Antimicrobial properties of arginine- and lysine-rich histones and involvement of bacterial outer membrane protease t in their differential mode of actions. Peptides 2011, 32, 2003–2009. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Saravanan, R.; Kwak, S.K.; Leong, S.S.J. Biomolecular engineering of a human beta defensin model for increased salt resistance. Chem. Eng. Sci. 2013, 95, 128–137. [Google Scholar] [CrossRef]
- Scudiero, O.; Galdiero, S.; Cantisani, M.; Di Noto, R.; Vitiello, M.; Galdiero, M.; Naclerio, G.; Cassiman, J.J.; Pedone, C.; Castaldo, G.; et al. Novel synthetic, salt-resistant analogs of human beta-defensins 1 and 3 endowed with enhanced antimicrobial activity. Antimicrob. Agents Chemother. 2010, 54, 2312–2322. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Young, A.W.; Hu, P.; Rice, A.J.; Zhou, C.; Zhang, Y.; Kallenbach, N.R. Tuning the membrane selectivity of antimicrobial peptides by using multivalent design. Chembiochem 2007, 8, 2063–2065. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Okumura, S.; Katayama, S.; Hirose, H.; Pujals, S.; Yamaguchi, H.; Arakawa, S.; Shimizu, S.; Futaki, S. Transformation of an antimicrobial peptide into a plasma membrane-permeable, mitochondria-targeted peptide via the substitution of lysine with arginine. Chem. Commun. (Camb) 2012, 48, 11097–11099. [Google Scholar] [CrossRef] [PubMed]
- Vogel, H.J.; Schibli, D.J.; Jing, W.; Lohmeier-Vogel, E.M.; Epand, R.F.; Epand, R.M. Towards a structure-function analysis of bovine lactoferricin and related tryptophan- and arginine-containing peptides. Biochem. Cell Biol. 2002, 80, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Khandelia, H.; Kaznessis, Y.N. Cation-pi interactions stabilize the structure of the antimicrobial peptide indolicidin near membranes: Molecular dynamics simulations. J. Phys. Chemi. B 2007, 111, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Wang, C.; Dong, W.; Zhu, W.; Shang, D. Antimicrobial properties and interaction of two trp-substituted cationic antimicrobial peptides with a lipid bilayer. J. Antibiot. 2014, 67, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Yau, W.M.; Wimley, W.C.; Gawrisch, K.; White, S.H. The preference of tryptophan for membrane interfaces. Biochemistry 1998, 37, 14713–14718. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.M.; Vogel, H.J.; Tieleman, D.P. Interactions of the designed antimicrobial peptide mb21 and truncated dermaseptin s3 with lipid bilayers: Molecular-dynamics simulations. Biochem. J. 2003, 370, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Wang, C.; Ma, L.; Sun, Y.; Shang, D. Investigation of the role of tryptophan residues in cationic antimicrobial peptides to determine the mechanism of antimicrobial action. J. Appl. Microbiol. 2013, 115, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, H.; Ayabe, T.; Maemoto, A.; Ishikawa, C.; Inaba, Y.; Sato, R.; Moriichi, K.; Okamoto, K.; Watari, J.; Kono, T.; et al. Denatured human alpha-defensin attenuates the bactericidal activity and the stability against enzymatic digestion. Biochem. Biophys. Res. Commun. 2007, 358, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Miles, K.; Clarke, D.J.; Lu, W.; Sibinska, Z.; Beaumont, P.E.; Davidson, D.J.; Barr, T.A.; Campopiano, D.J.; Gray, M. Dying and necrotic neutrophils are anti-inflammatory secondary to the release of alpha-defensins. J. Immunol. 2009, 183, 2122–2132. [Google Scholar] [CrossRef] [PubMed]
- Varkey, J.; Nagaraj, R. Antibacterial activity of human neutrophil defensin hnp-1 analogs without cysteines. Antimicrob. Agents Chemother. 2005, 49, 4561–4566. [Google Scholar] [CrossRef] [PubMed]
- Kluver, E.; Schulz-Maronde, S.; Scheid, S.; Meyer, B.; Forssmann, W.G.; Adermann, K. Structure-activity relation of human beta-defensin 3: Influence of disulfide bonds and cysteine substitution on antimicrobial activity and cytotoxicity. Biochemistry 2005, 44, 9804–9816. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Scott, M.G.; Yan, H.; Mayer, L.D.; Hancock, R.E. Interaction of polyphemusin i and structural analogs with bacterial membranes, lipopolysaccharide, and lipid monolayers. Biochemistry 2000, 39, 14504–14514. [Google Scholar] [CrossRef] [PubMed]
- Ostberg, N.; Kaznessis, Y. Protegrin structure-activity relationships: Using homology models of synthetic sequences to determine structural characteristics important for activity. Peptides 2005, 26, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Gottler, L.M.; de la Salud Bea, R.; Shelburne, C.E.; Ramamoorthy, A.; Marsh, E.N. Using fluorous amino acids to probe the effects of changing hydrophobicity on the physical and biological properties of the beta-hairpin antimicrobial peptide protegrin-1. Biochemistry 2008, 47, 9243–9250. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Falla, T.J.; Liu, H.; Hurst, M.A.; Fujii, C.A.; Mosca, D.A.; Embree, J.R.; Loury, D.J.; Radel, P.A.; Cheng Chang, C.; et al. Development of protegrins for the treatment and prevention of oral mucositis: Structure-activity relationships of synthetic protegrin analogues. Biopolymers 2000, 55, 88–98. [Google Scholar] [CrossRef]
- Mangoni, M.E.; Aumelas, A.; Charnet, P.; Roumestand, C.; Chiche, L.; Despaux, E.; Grassy, G.; Calas, B.; Chavanieu, A. Change in membrane permeability induced by protegrin 1: Implication of disulphide bridges for pore formation. FEBS Lett. 1996, 383, 93–98. [Google Scholar] [CrossRef]
- Harwig, S.S.; Waring, A.; Yang, H.J.; Cho, Y.; Tan, L.; Lehrer, R.I. Intramolecular disulfide bonds enhance the antimicrobial and lytic activities of protegrins at physiological sodium chloride concentrations. Eur. J. Biochem. 1996, 240, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, T.; He, Y.; Prieto, L. Membrane interactions and pore formation by the antimicrobial peptide protegrin. Biophys. J. 2013, 104, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.; Cady, S.D.; Tang, M.; Waring, A.J.; Lehrert, R.I.; Hong, M. Membrane-dependent oligomeric structure and pore formation of beta-hairpin antimicrobial peptide in lipid bilayers from solid-state nmr. Proc. Natl. Acad. Sci. USA 2006, 103, 16242–16247. [Google Scholar] [CrossRef] [PubMed]
- Aumelas, A.; Mangoni, M.; Roumestand, C.; Chiche, L.; Despaux, E.; Grassy, G.; Calas, B.; Chavanieu, A. Synthesis and solution structure of the antimicrobial peptide protegrin-1. Eur. J. Biochem. 1996, 237, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Mohanram, H.; Bhattacharjya, S. Cysteine deleted protegrin-1 (cdp-1): Anti-bacterial activity, outer-membrane disruption and selectivity. Biochim. Biophys. Acta 2014, 1840, 3006–3016. [Google Scholar] [CrossRef] [PubMed]
- Mosca, D.A.; Hurst, M.A.; So, W.; Viajar, B.S.; Fujii, C.A.; Falla, T.J. Ib-367, a protegrin peptide with in vitro and in vivo activities against the microflora associated with oral mucositis. Antimicrob. Agents Chemother. 2000, 44, 1803–1808. [Google Scholar] [CrossRef] [PubMed]
- Trotti, A.; Garden, A.; Warde, P.; Symonds, P.; Langer, C.; Redman, R.; Pajak, T.F.; Fleming, T.R.; Henke, M.; Bourhis, J.; et al. A multinational, randomized phase iii trial of iseganan hcl oral solution for reducing the severity of oral mucositis in patients receiving radiotherapy for head-and-neck malignancy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 674–681. [Google Scholar] [CrossRef]
- Scocchi, M.; Tossi, A.; Gennaro, R. Proline-rich antimicrobial peptides: Converging to a non-lytic mechanism of action. Cell. Mol. Life Sci. 2011, 68, 2317–2330. [Google Scholar] [CrossRef] [PubMed]
- Podda, E.; Benincasa, M.; Pacor, S.; Micali, F.; Mattiuzzo, M.; Gennaro, R.; Scocchi, M. Dual mode of action of bac7, a proline-rich antibacterial peptide. Biochim. Biophys. Acta 2006, 1760, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Boman, H.G.; Agerberth, B.; Boman, A. Mechanisms of action on escherichia coli of cecropin p1 and pr-39, two antibacterial peptides from pig intestine. Infect. Immun. 1993, 61, 2978–2984. [Google Scholar] [PubMed]
- Cabiaux, V.; Agerberth, B.; Johansson, J.; Homble, F.; Goormaghtigh, E.; Ruysschaert, J.M. Secondary structure and membrane interaction of pr-39, a pro+arg-rich antibacterial peptide. Eur. J. Biochem. 1994, 224, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, S.; Mita, K.; Ichinohe, K.; Hashimoto, S. Targeted engineering of the antibacterial peptide apidaecin, based on an in vivo monitoring assay system. Appl. Environ. Microbiol. 2009, 75, 1460–1464. [Google Scholar] [CrossRef] [PubMed]
- Casteels, P.; Ampe, C.; Jacobs, F.; Vaeck, M.; Tempst, P. Apidaecins: Antibacterial peptides from honeybees. EMBO J. 1989, 8, 2387–2391. [Google Scholar] [PubMed]
- Bulet, P.; Urge, L.; Ohresser, S.; Hetru, C.; Otvos, L., Jr. Enlarged scale chemical synthesis and range of activity of drosocin, an o-glycosylated antibacterial peptide of drosophila. Eur. J. Biochem. 1996, 238, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Kragol, G.; Hoffmann, R.; Chattergoon, M.A.; Lovas, S.; Cudic, M.; Bulet, P.; Condie, B.A.; Rosengren, K.J.; Montaner, L.J.; Otvos, L., Jr. Identification of crucial residues for the antibacterial activity of the proline-rich peptide, pyrrhocoricin. Eur. J. Biochem. 2002, 269, 4226–4237. [Google Scholar] [CrossRef] [PubMed]
- Cociancich, S.; Dupont, A.; Hegy, G.; Lanot, R.; Holder, F.; Hetru, C.; Hoffmann, J.A.; Bulet, P. Novel inducible antibacterial peptides from a hemipteran insect, the sap-sucking bug pyrrhocoris apterus. Biochem. J. 1994, 300(Pt 2), 567–575. [Google Scholar] [PubMed]
- Otvos, L., Jr.; Bokonyi, K.; Varga, I.; Otvos, B.I.; Hoffmann, R.; Ertl, H.C.; Wade, J.D.; McManus, A.M.; Craik, D.J.; Bulet, P. Insect peptides with improved protease-resistance protect mice against bacterial infection. Protein Sci. 2000, 9, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L.; Snyder, C.; Condie, B.; Bulet, P.; Wade, J.D. Chimeric antimicrobial peptides exhibit multiple modes of action. Int. J. Pept. Res. Ther. 2005, 11, 29–42. [Google Scholar] [CrossRef]
- Casteels, P.; Tempst, P. Apidaecin-type peptide antibiotics function through a non-poreforming mechanism involving stereospecificity. Biochem. Biophys. Res. Commun. 1994, 199, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Carneado, J.; Kogan, M.J.; Castel, S.; Giralt, E. Potential peptide carriers: Amphipathic proline-rich peptides derived from the n-terminal domain of gamma-zein. Angew. Chem. Int. Ed. Engl. 2004, 43, 1811–1814. [Google Scholar] [CrossRef] [PubMed]
- Cassone, M.; Vogiatzi, P.; La Montagna, R.; De Olivier Inacio, V.; Cudic, P.; Wade, J.D.; Otvos, L., Jr. Scope and limitations of the designer proline-rich antibacterial peptide dimer, a3-apo, alone or in synergy with conventional antibiotics. Peptides 2008, 29, 1878–1886. [Google Scholar] [CrossRef] [PubMed]
- Stensvag, K.; Haug, T.; Sperstad, S.V.; Rekdal, O.; Indrevoll, B.; Styrvold, O.B. Arasin 1, a proline-arginine-rich antimicrobial peptide isolated from the spider crab, hyas araneus. Dev. Comp. Immunol. 2008, 32, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, V.S.; Blencke, H.M.; Benincasa, M.; Haug, T.; Eksteen, J.J.; Styrvold, O.B.; Scocchi, M.; Stensvag, K. Structure-activity relationships of the antimicrobial peptide arasin 1 - and mode of action studies of the n-terminal, proline-rich region. PloS ONE 2013, 8, e53326. [Google Scholar] [CrossRef] [PubMed]
- Kragol, G.; Lovas, S.; Varadi, G.; Condie, B.A.; Hoffmann, R.; Otvos, L., Jr. The antibacterial peptide pyrrhocoricin inhibits the atpase actions of dnak and prevents chaperone-assisted protein folding. Biochemistry 2001, 40, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Crespo, L.; Sanclimens, G.; Montaner, B.; Perez-Tomas, R.; Royo, M.; Pons, M.; Albericio, F.; Giralt, E. Peptide dendrimers based on polyproline helices. J. Am. Chem. Soc. 2002, 124, 8876–8883. [Google Scholar] [CrossRef] [PubMed]
- Rozgonyi, F.; Szabo, D.; Kocsis, B.; Ostorhazi, E.; Abbadessa, G.; Cassone, M.; Wade, J.D.; Otvos, L., Jr. The antibacterial effect of a proline-rich antibacterial peptide a3-apo. Curr. Med. Chem. 2009, 16, 3996–4002. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Ye, G.; Cheng, X.; Yu, C.; Altosaar, I.; Hu, C. Characterization of an abaecin-like antimicrobial peptide identified from a pteromalus puparum cdna clone. J. Invertebr. Pathol. 2010, 105, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Dorn, A. Differential infectivity of two pseudomonas species and the immune response in the milkweed bug, oncopeltus fasciatus (insecta: Hemiptera). J. Invertebr. Pathol. 2001, 78, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, S.; Knappe, D.; Berthold, N.; von Buttlar, H.; Hoffmann, R.; Alber, G. Absence of in vitro innate immunomodulation by insect-derived short proline-rich antimicrobial peptides points to direct antibacterial action in vivo. J. Pept. Sci. 2012, 18, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Krizsan, A.; Volke, D.; Weinert, S.; Strater, N.; Knappe, D.; Hoffmann, R. Insect-derived proline-rich antimicrobial peptides kill bacteria by inhibiting bacterial protein translation at the 70 s ribosome. Angew. Chem. Int. Ed. Engl. 2014, 53, 12236–12239. [Google Scholar] [CrossRef] [PubMed]
- Bobone, S.; Roversi, D.; Giordano, L.; De Zotti, M.; Formaggio, F.; Toniolo, C.; Park, Y.; Stella, L. The lipid dependence of antimicrobial peptide activity is an unreliable experimental test for different pore models. Biochemistry 2012, 51, 10124–10126. [Google Scholar] [CrossRef] [PubMed]
- Roversi, D.; Luca, V.; Aureli, S.; Park, Y.; Mangoni, M.L.; Stella, L. How many antimicrobial peptide molecules kill a bacterium? The case of pmap-23. ACS Chem. Biol. 2014, 9, 2003–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.W. Action of antimicrobial peptides: Two-state model. Biochemistry 2000, 39, 8347–8352. [Google Scholar] [CrossRef] [PubMed]
- Andrushchenko, V.V.; Aarabi, M.H.; Nguyen, L.T.; Prenner, E.J.; Vogel, H.J. Thermodynamics of the interactions of tryptophan-rich cathelicidin antimicrobial peptides with model and natural membranes. Biochim. Biophys. Acta 2008, 1778, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Kagan, B.L.; Selsted, M.E.; Ganz, T.; Lehrer, R.I. Antimicrobial defensin peptides form voltage-dependent ion-permeable channels in planar lipid bilayer membranes. Proc. Natl. Acad. Sci. USA 1990, 87, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Papo, N.; Shai, Y. A molecular mechanism for lipopolysaccharide protection of gram-negative bacteria from antimicrobial peptides. J. Biol. Chem. 2005, 280, 10378–10387. [Google Scholar] [CrossRef] [PubMed]
- Andra, J.; Gutsmann, T.; Garidel, P.; Brandenburg, K. Mechanisms of endotoxin neutralization by synthetic cationic compounds. J. Endotoxin Res. 2006, 12, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Bobone, S.; Gerelli, Y.; De Zotti, M.; Bocchinfuso, G.; Farrotti, A.; Orioni, B.; Sebastiani, F.; Latter, E.; Penfold, J.; Senesi, R.; et al. Membrane thickness and the mechanism of action of the short peptaibol trichogin ga iv. Biochim. Biophys. Acta Biomembr. 2013, 1828, 1013–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourie, J.I.; Shorthouse, A.A. Properties of cytotoxic peptide-formed ion channels. Am. J. Physiol. Cell. Physiol. 2000, 278, C1063–C1087. [Google Scholar] [PubMed]
- Tarao, K.; Shimizu, A.; Ohkawa, S.; Harada, M.; Ito, Y.; Tamai, S.; Kuni, Y.; Nagaoka, T.; Hoshino, H. Increased uptake of bromodeoxyuridine by hepatocytes from early stage of primary biliary-cirrhosis. Gastroenterology 1991, 100, 725–730. [Google Scholar] [PubMed]
- Steiner, H.; Andreu, D.; Merrifield, R.B. Binding and action of cecropin and cecropin analogues: Antibacterial peptides from insects. Biochim. Biophys. Acta 1988, 939, 260–266. [Google Scholar] [CrossRef]
- Lichtenstein, A. Mechanism of mammalian cell lysis mediated by peptide defensins. Evidence for an initial alteration of the plasma membrane. J. Clin. Investig. 1991, 88, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Barton, A.; Daher, K.A.; Harwig, S.S.; Ganz, T.; Selsted, M.E. Interaction of human defensins with escherichia coli. Mechanism of bactericidal activity. J. Clin. Investig. 1989, 84, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Ehrenstein, G.; Lecar, H. Electrically gated ionic channels in lipid bilayers. Q. Rev. Biophys. 1977, 10, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.F.; Nagarajan, R.; Camesano, T.A. Antimicrobial peptide alamethicin insertion into lipid bilayer: A qcm-d exploration. Colloid. Surface. B 2014, 116, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Laver, D.R. The barrel-stave model as applied to alamethicin and its analogs reevaluated. Biophys. J. 1994, 66, 355–359. [Google Scholar] [CrossRef]
- De Zotti, M.; Biondi, B.; Peggion, C.; Formaggio, F.; Park, Y.; Hahm, K.S.; Toniolo, C. Trichogin ga iv: A versatile template for the synthesis of novel peptaibiotics. Org. Biomol. Chem. 2012, 10, 1285–1299. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Harroun, T.A.; Weiss, T.M.; Ding, L.; Huang, H.W. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 2001, 81, 1475–1485. [Google Scholar] [CrossRef]
- Sengupta, D.; Leontiadou, H.; Mark, A.E.; Marrink, S.J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta 2008, 1778, 2308–2317. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Lohner, K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B. Rationalizing the membrane interactions of cationic amphipathic antimicrobial peptides by their molecular shape. Curr. Opin. Colloid IN 2009, 14, 349–355. [Google Scholar] [CrossRef]
- Orioni, B.; Bocchinfuso, G.; Kim, J.Y.; Palleschi, A.; Grande, G.; Bobone, S.; Park, Y.; Kim, J.I.; Hahm, K.S.; Stella, L. Membrane perturbation by the antimicrobial peptide pmap-23: A fluorescence and molecular dynamics study. Biochim. Biophys. Acta Biomembr. 2009, 1788, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Pokorny, A.; Birkbeck, T.H.; Almeida, P.F.F. Mechanism and kinetics of delta-lysin interaction with phospholipid vesicles. Biochemistry 2002, 41, 11044–11056. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Sood, R.; Jutila, A.; Bose, S.; Fimland, G.; Nissen-Meyer, J.; Kinnunen, P.K.J. Interaction of the antimicrobial peptide pheromone plantaricin a with model membranes: Implications for a novel mechanism of action. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Epand, R.F. Bacterial membrane lipids in the action of antimicrobial agents. J. Pept. Sci. 2011, 17, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Pag, U.; Oedenkoven, M.; Sass, V.; Shai, Y.; Shamova, O.; Antcheva, N.; Tossi, A.; Sahl, H.G. Analysis of in vitro activities and modes of action of synthetic antimicrobial peptides derived from an alpha-helical 'sequence template'. J. Antimicrob. Chemother. 2008, 61, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Sass, V.; Schneider, T.; Wilmes, M.; Korner, C.; Tossi, A.; Novikova, N.; Shamova, O.; Sahl, H.G. Human beta-defensin 3 inhibits cell wall biosynthesis in staphylococci. Infect. Immun. 2010, 78, 2793–2800. [Google Scholar] [CrossRef] [PubMed]
- Sass, V.; Pag, U.; Tossi, A.; Bierbaum, G.; Sahl, H.G. Mode of action of human beta-defensin 3 against staphylococcus aureus and transcriptional analysis of responses to defensin challenge. Int. J. Med. Microbiol. 2008, 298, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Kai-Larsen, Y.; Frithiof, R.; Sorensen, O.E.; Kenne, E.; Scharffetter-Kochanek, K.; Eriksson, E.E.; Herwald, H.; Agerberth, B.; Lindbom, L. Neutrophil primary granule proteins hbp and hnp1–3 boost bacterial phagocytosis by human and murine macrophages. J. Clin. Investig. 2008, 118, 3491–3502. [Google Scholar] [CrossRef] [PubMed]
- Funderburg, N.; Lederman, M.M.; Feng, Z.; Drage, M.G.; Jacllowsky, J.; Harding, C.V.; Weinberg, A.; Sieg, S.F. Human beta-defensin-3 activates professional antigen-presenting cells via toll-like receptors 1 and 2. Proc. Natl. Acad. Sci. USA 2007, 104, 18631–18635. [Google Scholar] [CrossRef] [PubMed]
- Park, C.B.; Kim, M.S.; Kim, S.C. A novel antimicrobial peptide from bufo bufo gargarizans. Biochem. Biophys. Res. Commun. 1996, 218, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of action of the antimicrobial peptide buforin ii: Buforin ii kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 1998, 244, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Li, W.F.; Ma, G.X.; Zhou, X.X. Apidaecin-type peptides: Biodiversity, structure-function relationships and mode of action. Peptides 2006, 27, 2350–2359. [Google Scholar] [CrossRef] [PubMed]
- Gruenheid, S.; Le Moual, H. Resistance to antimicrobial peptides in gram-negative bacteria. FEMS Microbiol. Lett. 2012, 330, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Belas, R.; Manos, J.; Suvanasuthi, R. Proteus mirabilis zapa metalloprotease degrades a broad spectrum of substrates, including antimicrobial peptides. Infect. Immun. 2004, 72, 5159–5167. [Google Scholar] [CrossRef] [PubMed]
- Hritonenko, V.; Stathopoulos, C. Omptin proteins: An expanding family of outer membrane proteases in gram-negative enterobacteriaceae. Mol. Membr. Biol. 2007, 24, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Chromek, M.; Kai-Larsen, Y.; Luthje, P.; Wang, X.; Holm, A.A.; Hedlund, K.O.; Johansson, J.; Chapman, M.R.; Jacobson, S.H.; Romling, U.; et al. The antimicrobial peptide cathelicidin interferes with polymerization of curli fimbrie and thus inhibits the formation of uropathogenic e-coli biofilm. Pediatr. Nephrol. 2010, 25, 1849–1849. [Google Scholar]
- Ilg, K.; Endt, K.; Misselwitz, B.; Stecher, B.; Aebi, M.; Hardt, W.D. O-antigen-negative salmonella enterica serovar typhimurium is attenuated in intestinal colonization but elicits colitis in streptomycin-treated mice. Infect. Immun. 2009, 77, 2568–2575. [Google Scholar] [CrossRef] [PubMed]
- Foschiatti, M.; Cescutti, P.; Tossi, A.; Rizzo, R. Inhibition of cathelicidin activity by bacterial exopolysaccharides. Mol. Microbiol. 2009, 72, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Llobet, E.; Tomas, J.M.; Bengoechea, J.A. Capsule polysaccharide is a bacterial decoy for antimicrobial peptides. Microbiology. 2008, 154, 3877–3886. [Google Scholar] [CrossRef] [PubMed]
- Campos, M.A.; Vargas, M.A.; Regueiro, V.; Llompart, C.M.; Alberti, S.; Bengoechea, J.A. Capsule polysaccharide mediates bacterial resistance to antimicrobial peptides. Infect. Immun. 2004, 72, 7107–7114. [Google Scholar] [CrossRef] [PubMed]
- Gunn, J.S. The salmonella pmrab regulon: Lipopolysaccharide modifications, antimicrobial peptide resistance and more. Trends Microbiol. 2008, 16, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Ribeiro, A.A.; Guan, Z.Q.; Abraham, S.N.; Raetz, C.R.H. Attenuated virulence of a francisella mutant lacking the lipid a 4'-phosphatase. Proc. Natl. Acad. Sci. USA 2007, 104, 4136–4141. [Google Scholar] [CrossRef] [PubMed]
- Hein-Kristensen, L.; Franzyk, H.; Holch, A.; Gram, L. Adaptive evolution of escherichia coli to an alpha-peptide/beta-peptoid peptidomimetic induces stable resistance. PloS ONE 2013, 8, e73620. [Google Scholar] [CrossRef] [PubMed]
- Ernst, R.K.; Guina, T.; Miller, S.I. How intracellular bacteria survive: Surface modifications that promote resistance to host innate immune responses. J. Infect. Dis. 1999, 179, S326–S330. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lim, K.B.; Gunn, J.S.; Bainbridge, B.; Darveau, R.P.; Hackett, M.; Miller, S.I. Regulation of lipid a modifications by salmonella typhimurium virulence genes phop-phoq. Science 1997, 276, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lim, K.B.; Poduje, C.M.; Daniel, M.; Gunn, J.S.; Hackett, M.; Miller, S.I. Lipid a acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell 1998, 95, 189–198. [Google Scholar] [CrossRef]
- Nawrocki, K.L.; Crispell, E.K.; McBride, S.M. Antimicrobial peptide resistance mechanisms of gram-positive bacteria. Antibiotics (Basel) 2014, 3, 461–492. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Rolain, T.; Courtin, P.; Guillot, A.; Langella, P.; Hols, P.; Chapot-Chartier, M.P. Characterization of o-acetylation of n-acetylglucosamine: A novel structural variation of bacterial peptidoglycan. J. Biol. Chem. 2011, 286, 23950–23958. [Google Scholar]
- Saar-Dover, R.; Bitler, A.; Nezer, R.; Shmuel-Galia, L.; Firon, A.; Shimoni, E.; Trieu-Cuot, P. D-Alanylation of lipoteichoic acids confers resistance to cationic peptides in group b streptococcus by increasing the cell wall density. PLoS Pathog 2012, 8, e1002891. [Google Scholar]
- Shafer, W.M.; Qu, X.D.; Waring, A.J.; Lehrer, R.I. Modulation of neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a, member of the resistance/nodulation/division efflux pump family. Proc. Natl. Acad. Sci. USA 1998, 95, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- Parralopez, C.; Baer, M.T.; Groisman, E.A. Molecular-genetic analysis of a locus required for resistance to antimicrobial peptides in salmonella-typhimurium. EMBO J. 1993, 12, 4053–4062. [Google Scholar]
- Chakraborty, K.; Ghosh, S.; Koley, H.; Mukhopadhyay, A.K.; Ramamurthy, T.; Saha, D.R.; Mukhopadhyay, D.; Roychowdhury, S.; Hamabata, T.; Takeda, Y.; et al. Bacterial exotoxins downregulate cathelicidin (hcap-18/ll-37) and human beta-defensin 1 (hbd-1) expression in the intestinal epithelial cells. Cell. Microbiol. 2008, 10, 2520–2537. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Mikami, Y.; Fukushima, K.; Utsumi, T.; Yazawa, K. A new antibiotic, leucinostatin, derived from penicillium lilacinum. J. Antibiot. 1973, 26, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; de Boer, L.; Zaat, S.A.; Vogel, H.J. Investigating the cationic side chains of the antimicrobial peptide tritrpticin: Hydrogen bonding properties govern its membrane-disruptive activities. Biochim. Biophys. Acta 2011, 1808, 2297–2303. [Google Scholar] [CrossRef] [PubMed]
- Grauer, A.; Konig, B. Peptidomimetics - a versatile route to biologically active compounds. Eur. J. Org. Chem. 2009, 5099–5111. [Google Scholar] [CrossRef]
- Bessalle, R.; Kapitkovsky, A.; Gorea, A.; Shalit, I.; Fridkin, M. All-d-magainin: Chirality, antimicrobial activity and proteolytic resistance. FEBS Lett. 1990, 274, 151–155. [Google Scholar] [CrossRef]
- Giuliani, A.; Rinaldi, A.C. Beyond natural antimicrobial peptides: Multimeric peptides and other peptidomimetic approaches. Cell. Mol. Life Sci. 2011, 68, 2255–2266. [Google Scholar] [CrossRef] [PubMed]
- Shalev, D.E.; Rotem, S.; Fish, A.; Mor, A. Consequences of n-acylation on structure and membrane binding properties of dermaseptin derivative k4-s4-(1–13). J. Biol. Chem. 2006, 281, 9432–9438. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, A.; Pirri, G.; Bozzi, A.; Di Giulio, A.; Aschi, M.; Rinaldi, A.C. Antimicrobial peptides: Natural templates for synthetic membrane-active compounds. Cell. Mol. Life Sci. 2008, 65, 2450–2460. [Google Scholar] [CrossRef] [PubMed]
- Bionda, N.; Pastar, I.; Davis, S.C.; Cudic, P. In vitro and in vivo activities of novel cyclic lipopeptides against staphylococcal biofilms. Protein pept. Lett. 2014, 21, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Jacob, B.; Gunasekaran, P.; Murugan, R.N.; Ryu, E.K.; Lee, G.H.; Hyun, J.K.; Cheong, C.; Kim, N.H.; Shin, S.Y.; et al. Poly-lysine peptidomimetics having potent antimicrobial activity without hemolytic activity. Amino Acids 2014, 46, 2259–2269. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.; Alst, T.; Havelkova, M.; Strom, M.B. Antimicrobial activity of small beta-peptidomimetics based on the pharmacophore model of short cationic antimicrobial peptides. J. Med. Chem. 2010, 53, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Mosca, S.; Keller, J.; Azzouz, N.; Wagner, S.; Titz, A.; Seeberger, P.H.; Brezesinski, G.; Hartmann, L. Amphiphilic cationic beta(3r3)-peptides: Membrane active peptidomimetics and their potential as antimicrobial agents. Biomacromolecules 2014, 15, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.W.; Sanborn, T.J.; Zuckermann, R.N.; Barron, A.E. Peptoid oligomers with alpha-chiral, aromatic side chains: Effects of chain length on secondary structure. J. Am. Chem. Soc. 2001, 123, 2958–2963. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.W.; Sanborn, T.J.; Huang, K.; Zuckermann, R.N.; Barron, A.E. Peptoid oligomers with alpha-chiral, aromatic side chains: Sequence requirements for the formation of stable peptoid helices. J. Am. Chem. Soc. 2001, 123, 6778–6784. [Google Scholar] [CrossRef] [PubMed]
- Sanborn, T.J.; Wu, C.W.; Zuckermann, R.N.; Barron, A.E. Extreme stability of helices formed by water-soluble poly-n-substituted glycines (polypeptoids) with alpha-chiral side chains. Biopolymers 2002, 63, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.C.; Yu, P.; Kwon, Y.U.; Kodadek, T. High-throughput evaluation of relative cell permeability between peptoids and peptides. Bioorg. Med. Chem. 2008, 16, 5853–5861. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.K.; Nan, Y.H.; Lee, E.K.; Shin, S.Y. A novel trp-rich model antimicrobial peptoid with increased protease stability. Bull. Korean Chem. Soc. 2010, 31, 2509–2513. [Google Scholar] [CrossRef]
- Chongsiriwatana, N.P.; Patch, J.A.; Czyzewski, A.M.; Dohm, M.T.; Ivankin, A.; Gidalevitz, D.; Zuckermann, R.N.; Barron, A.E. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Patch, J.A.; Barron, A.E. Helical peptoid mimics of magainin-2 amide. J. Am. Chem. Soc. 2003, 125, 12092–12093. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Eimerman, P.R.; Hardy, J.W.; Cirillo, J.D.; Contag, C.H.; Barron, A.E. Efficacy of antimicrobial peptoids against mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 3058–3062. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Wadman, M.W.; Dohm, M.T.; Czyzewski, A.M.; Spormann, A.M.; Barron, A.E. Antimicrobial peptoids are effective against pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2011, 55, 3054–3057. [Google Scholar] [CrossRef] [PubMed]
- Chongsiriwatana, N.P.; Wetzler, M.; Barron, A.E. Functional synergy between antimicrobial peptoids and peptides against gram-negative bacteria. Antimicrob. Agents Chemother. 2011, 55, 5399–5402. [Google Scholar] [CrossRef] [PubMed]
- Mojsoska, B.; Zuckermann, R.N.; Jenssen, H. Structure-activity relationship study of novel peptoids that mimic the structure of antimicrobial peptides. Antimicrob. Agents Chemother. 2015, 59, 4112–4120. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.B.; Yoo, B.; Todaro, L.J.; Kirshenbaum, K. Cyclic peptoids. J. Am. Chem. Soc. 2007, 129, 3218–3225. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Shin, S.B.; Benson, M.A.; Torres, V.J.; Kirshenbaum, K. A comparison of linear and cyclic peptoid oligomers as potent antimicrobial agents. ChemMedChem 2012, 7, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Benson, M.A.; Shin, S.B.Y.; Torres, V.J.; Kirshenbaum, K. Amphiphilic cyclic peptoids that exhibit antimicrobial activity by disrupting staphylococcus aureus membranes. Eur. J. Org. Chem. 2013, 3560–3566. [Google Scholar] [CrossRef]
- Gobbo, M.; Benincasa, M.; Bertoloni, G.; Biondi, B.; Dosselli, R.; Papini, E.; Reddi, E.; Rocchi, R.; Tavano, R.; Gennaro, R. Substitution of the arginine/leucine residues in apidaecin ib with peptoid residues: Effect on antimicrobial activity, cellular uptake, and proteolytic degradation. J. Med. Chem. 2009, 52, 5197–5206. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Lee, S.A.; Shin, S.; Lee, J.Y.; Jeong, K.W.; Nan, Y.H.; Park, Y.S.; Shin, S.Y.; Kim, Y. Structural flexibility and the positive charges are the key factors in bacterial cell selectivity and membrane penetration of peptoid-substituted analog of piscidin 1. Biochim. Biophys. Acta Biomembr. 2010, 1798, 1913–1925. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Knapp, K.M.; Yang, L.; Molin, S.; Franzyk, H.; Folkesson, A. High in vitro antimicrobial activity of beta-peptoid-peptide hybrid oligomers against planktonic and biofilm cultures of staphylococcus epidermidis. Int. J. Antimicrob. Agents 2013, 41, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.A.; Bonke, G.; Vedel, L.; Adsersen, A.; Witt, M.; Franzyk, H.; Jaroszewski, J.W. Alpha-peptide/beta-peptoid chimeras. Org. Lett. 2007, 9, 1549–1552. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.A.; Ziegler, H.L.; Nielsen, H.M.; Frimodt-Moller, N.; Jaroszewski, J.W.; Franzyk, H. Antimicrobial, hemolytic, and cytotoxic activities of beta-peptoid-peptide hybrid oligomers: Improved properties compared to natural amps. Chembiochem 2010, 11, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Bonke, G.; Vedel, L.; Witt, M.; Jaroszewski, J.W.; Olsen, C.A.; Franzyk, H. Dimeric building blocks for solid-phase synthesis of alpha-peptide-beta-peptoid chimeras. Synth. Stuttg. 2008, 2381–2390. [Google Scholar]
- Ryge, T.S.; Hansen, P.R. Novel lysine-peptoid hybrids with antibacterial properties. J. Pept. Sci. 2005, 11, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Jahnsen, R.D.; Haney, E.F.; Franzyk, H.; Hancock, R.E. Characterization of a proteolytically stable multifunctional host defense peptidomimetic. Chem. Biol. 2013, 20, 1286–1295. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, S.; Ifrah, D.; Lerche, S.; Gottlieb, C.T.; Cohn, M.T.; Hiasa, H.; Hansen, P.R.; Gram, L.; Ingmer, H.; Thomsen, L.E. The antimicrobial lysine-peptoid hybrid lp5 inhibits DNA replication and induces the sos response in staphylococcus aureus. BMC microbiology 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Di, L.; Zhang, X.; Yan, M.; Wan, D.; Li, L.; Zhang, Y.; Cai, J.; Dai, H.; Zhu, Q.; et al. Efficacy of oral etoposide in pretreated metastatic breast cancer: A multicenter phase 2 study. Medicine 2015, 94, e774. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Smith, C.; Wu, H.; Padhee, S.; Manoj, N.; Cardiello, J.; Qiao, Q.; Cao, C.; Yin, H.; Cai, J. Lipidated cyclic gamma-aapeptides display both antimicrobial and anti-inflammatory activity. ACS Chem. Biol. 2014, 9, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cai, J.; Zhou, X.; Chen, L.; Gong, Y.; Gao, Z.; Zhang, H.; Huang, W.; Zhou, H. Protective effect of spironolactone on endothelial-to-mesenchymal transition in huvecs via notch pathway. Cell. Physiol. Biochem. 2015, 36, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cui, H.; Cai, J.; Duan, Y.; Liu, Y. Development of fluorescence sensing material based on cdse/zns quantum dots and molecularly imprinted polymer for the detection of carbaryl in rice and chinese cabbage. J. Agric. Food Chem. 2015, 63, 4966–4972. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A.; Jankovic, B. Application of 'inductive' qsar descriptors for quantification of antibacterial activity of cationic polypeptides. Molecules 2004, 9, 1034–1052. [Google Scholar] [CrossRef] [PubMed]
- Taboureau, O.; Olsen, O.H.; Nielsen, J.D.; Raventos, D.; Mygind, P.H.; Kristensen, H.H. Design of novispirin antimicrobial peptides by quantitative structure-activity relationship. Chem. Biol. Drug Des. 2006, 68, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Juretic, D.; Vukicevic, D.; Ilic, N.; Antcheva, N.; Tossi, A. Computational design of highly selective antimicrobial peptides. J. Chem. Inf. Model. 2009, 49, 2873–2882. [Google Scholar] [CrossRef] [PubMed]
- Juretic, D.; Vukicevic, D.; Petrov, D.; Novkovic, M.; Bojovic, V.; Lucic, B.; Ilic, N.; Tossi, A. Knowledge-based computational methods for identifying or designing novel, non-homologous antimicrobial peptides. Eur. Biophys. J. 2011, 40, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Ilic, N.; Novkovic, M.; Guida, F.; Xhindoli, D.; Benincasa, M.; Tossi, A.; Juretic, D. Selective antimicrobial activity and mode of action of adepantins, glycine-rich peptide antibiotics based on anuran antimicrobial peptide sequences. Biochim. Biophys. Acta 2013, 1828, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A.; Hilpert, K.; Jenssen, H.; Fjell, C.D.; Waldbrook, M.; Mullaly, S.C.; Volkmer, R.; Hancock, R.E.W. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem. Biol. 2009, 4, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Jenssen, H.; Hilpert, K.; Cheung, W.A.; Pante, N.; Hancock, R.E.; Cherkasov, A. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J. Med. Chem. 2009, 52, 2006–2015. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Jenssen, H.; Cheung, W.A.; Hancock, R.E.; Cherkasov, A. Optimization of antibacterial peptides by genetic algorithms and cheminformatics. Chem. Biol. Drug Des. 2011, 77, 48–56. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mojsoska, B.; Jenssen, H. Peptides and Peptidomimetics for Antimicrobial Drug Design. Pharmaceuticals 2015, 8, 366-415. https://doi.org/10.3390/ph8030366
Mojsoska B, Jenssen H. Peptides and Peptidomimetics for Antimicrobial Drug Design. Pharmaceuticals. 2015; 8(3):366-415. https://doi.org/10.3390/ph8030366
Chicago/Turabian StyleMojsoska, Biljana, and Håvard Jenssen. 2015. "Peptides and Peptidomimetics for Antimicrobial Drug Design" Pharmaceuticals 8, no. 3: 366-415. https://doi.org/10.3390/ph8030366
APA StyleMojsoska, B., & Jenssen, H. (2015). Peptides and Peptidomimetics for Antimicrobial Drug Design. Pharmaceuticals, 8(3), 366-415. https://doi.org/10.3390/ph8030366