
Precision Medicine in Systemic Mastocytosis

 ,
,  ,
,  and
and
Abstract
:1. Introduction
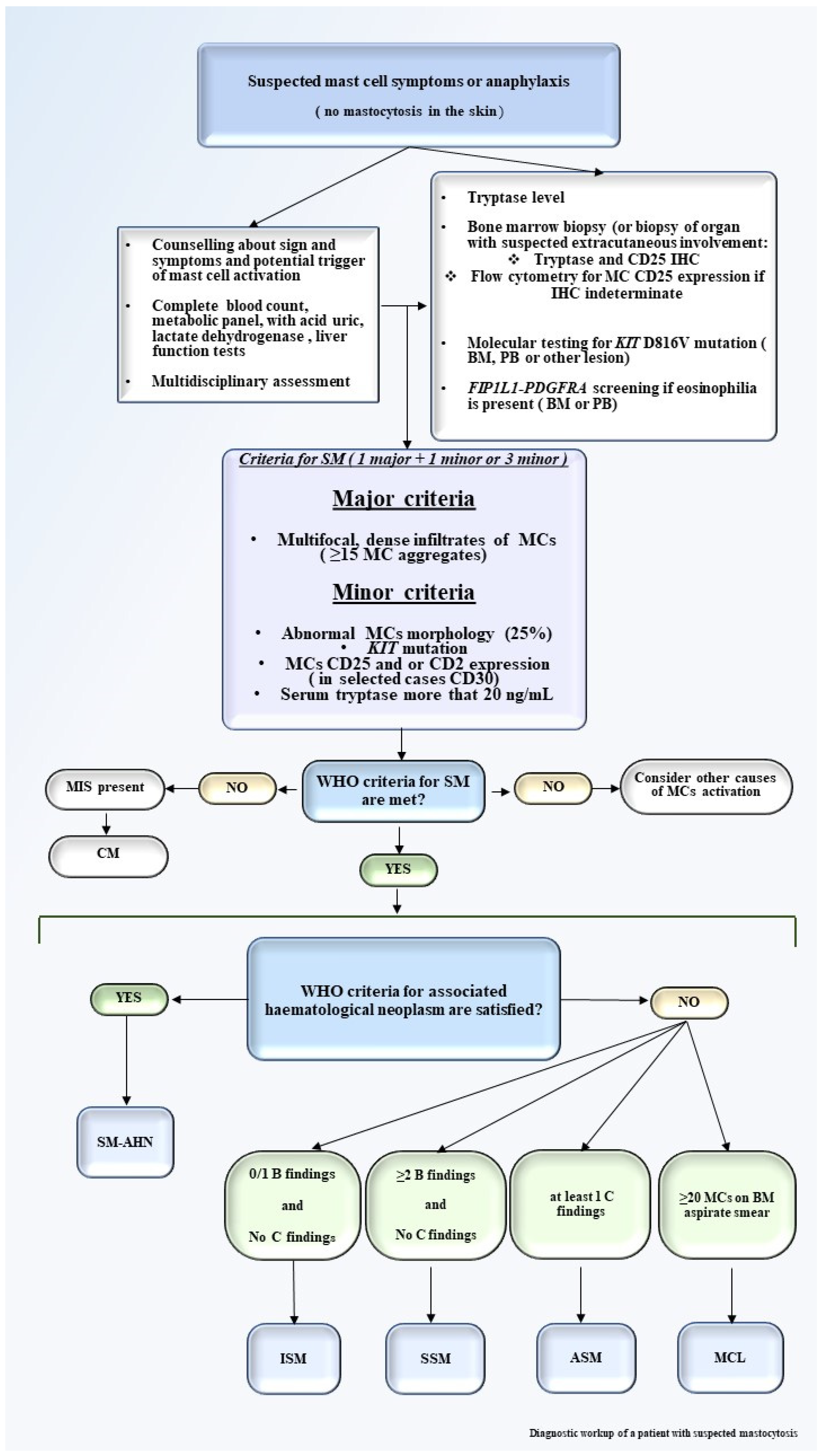
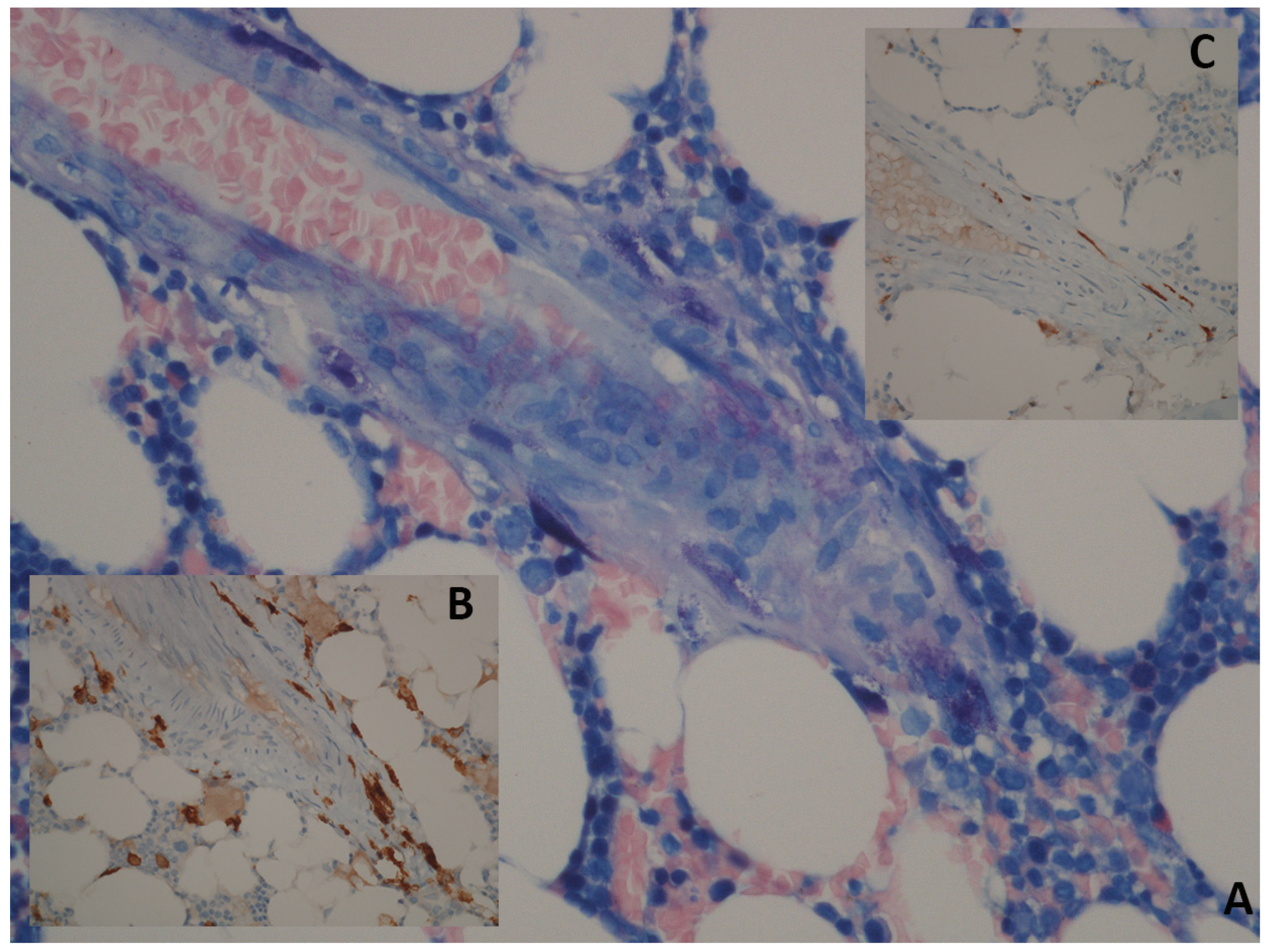
2. Clinical Presentation and Diagnostic Work-Up
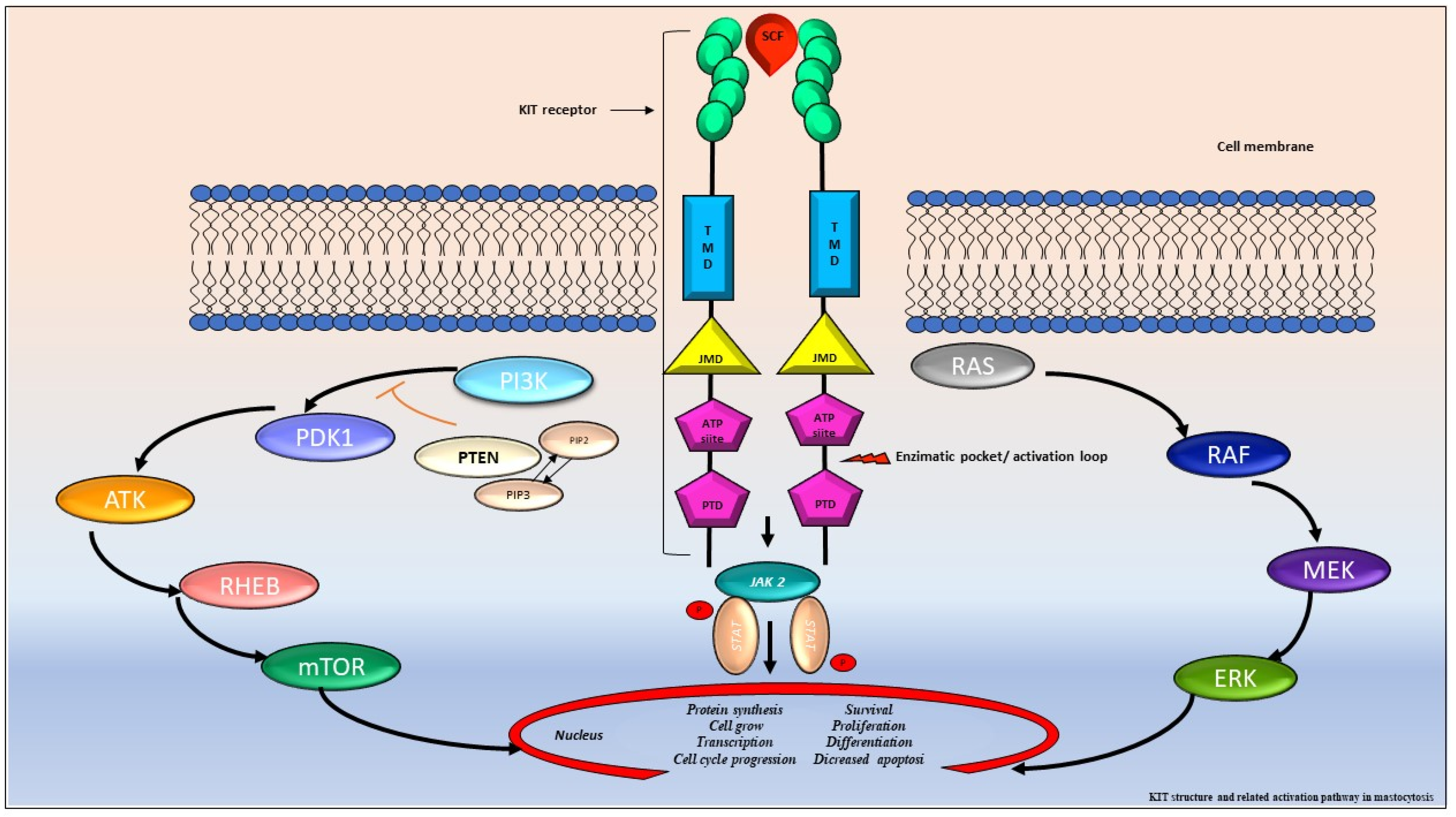
3. Molecular Aspects: KIT and beyond as Biomarkers of the Disease
4. Cytogenetic Information
5. Risk Stratification
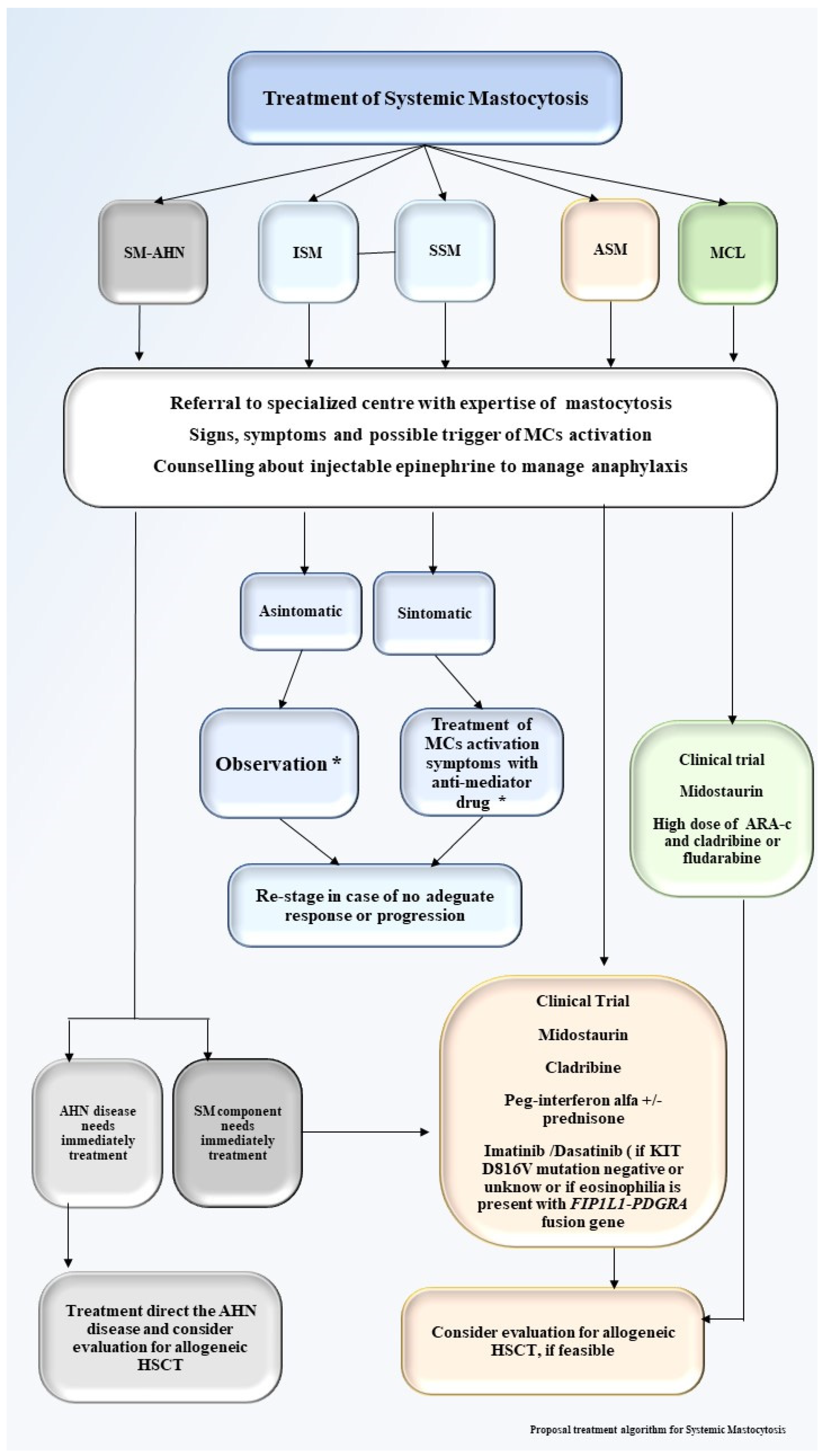
6. Treatment
6.1. BLC-2 Inhibitors
6.2. Future Perspectives
7. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.M. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leuke-mia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Brockow, K. Epidemiology, Prognosis, and Risk Factors in Mastocytosis. Immunol. Allergy Clin. N. Am. 2014, 34, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Caplan, R.M. The Natural Course of Urticaria Pigmentosa. Arch. Dermatol. 1963, 87, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Azaña, J.M.; Torrelo, A.; Mediero, I.G.; Zambrano, A. Urticaria Pigmentosa: A Review of 67 Pediatric Cases. Pediatr. Dermatol. 1994, 11, 102–106. [Google Scholar] [CrossRef]
- Lim, K.-H.; Tefferi, A.; Lasho, T.L.; Finke, C.; Patnaik, M.; Butterfield, J.H.; McClure, R.F.; Li, C.-Y.; Pardanani, A. Systemic mastocytosis in 342 consecutive adults: Survival studies and prognostic factors. Blood 2009, 113, 5727–5736. [Google Scholar] [CrossRef] [Green Version]
- Valent, P.; Akin, C.; Gleixner, K.V.; Sperr, W.R.; Reiter, A.; Arock, M.; Triggiani, M. Multidisciplinary Challenges in Mastocytosis and How to Address with Personalized Medicine Approaches. Int. J. Mol. Sci. 2019, 20, 2976. [Google Scholar] [CrossRef] [Green Version]
- Valent, P.; Spanblöchl, E.; Sperr, W.R.; Sillaber, C.; Zsebo, K.M.; Agis, H.; Strobl, H.; Geissler, K.; Bettelheim, P.; Lechner, K. Induction of differentiation of human mast cells from bone marrow and peripheral blood mononuclear cells by recombinant human stem cell factor/kit-ligand in long-term culture. Blood 1992, 80, 2237–2245. [Google Scholar] [CrossRef] [Green Version]
- Jawhar, M.; Schwaab, J.; Horny, H.-P.; Sotlar, K.; Naumann, N.; Fabarius, A.; Valent, P.; Cross, N.; Hofmann, W.-K.; Metzgeroth, G.; et al. Impact of centralized evaluation of bone marrow histology in systemic mastocytosis. Eur. J. Clin. Investig. 2016, 46, 392–397. [Google Scholar] [CrossRef]
- Muñoz-González, J.I.; Jara-Acevedo, M.; Alvarez-Twose, I.; Merker, J.D.; Teodosio, C.; Hou, Y.; Henriques, A.; Roskin, K.M.; Sanchez-Muñoz, L.; Tsai, A.; et al. Impact of somatic and germline mutations on the outcome of systemic mastocytosis. Blood Adv. 2018, 2, 2814–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martelli, M.; Monaldi, C.; De Santis, S.; Bruno, S.; Mancini, M.; Cavo, M.; Soverini, S. Recent Advances in the Molecular Biology of Systemic Mastocytosis: Implications for Diagnosis, Prognosis, and Therapy. Int. J. Mol. Sci. 2020, 21, 3987. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J.; Kluin-Nelemans, J.C.; George, T.I.; Akin, C.; Sotlar, K.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Sternberg, D.W.; et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N. Engl. J. Med. 2016, 374, 2530–2541. [Google Scholar] [CrossRef] [PubMed]
- DeAngelo, D.; George, T.; Linder, A.; Langford, C.; Perkins, C.; Ma, J.; Westervelt, P.; Merker, J.D.; Berube, C.; Coutre, S.; et al. Efficacy and safety of midostaurin in patients with advanced systemic mastocytosis: 10-year median follow-up of a phase II trial. Leukemia 2018, 32, 470–478. [Google Scholar] [CrossRef]
- Ustun, C.; Reiter, A.; Scott, B.L.; Nakamura, R.; Damaj, G.; Kreil, S.; Shanley, R.; Hogan, W.J.; Perales, M.-A.; Shore, T.; et al. Hematopoietic Stem-Cell Transplantation for Advanced Systemic Mastocytosis. J. Clin. Oncol. 2014, 32, 3264–3274. [Google Scholar] [CrossRef] [Green Version]
- Ustun, C.; Gotlib, J.; Popat, U.; Artz, A.; Litzow, M.; Reiter, A.; Nakamura, R.; Kluin-Nelemans, J.C.; Srdan Verstovsek, M.D.; Gajewski, J.; et al. Consensus Opinion on Allogeneic Hematopoietic Cell Transplantation in Advanced Systemic Mastocytosis. Biol. Blood Marrow Transplant. 2016, 22, 1348–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valent, P.; Horny, H.-P.; Escribano, L.; Longley, B.; Li, C.Y.; Schwartz, L.B.; Marone, G.; Nuñez, R.; Akin, C.; Sotlar, K.; et al. Diagnostic criteria and classification of mastocytosis: A consensus proposal. Leuk. Res. 2001, 25, 603–625. [Google Scholar] [CrossRef]
- González-De-Olano, D.; Alvarez-Twose, I. Insights in Anaphylaxis and Clonal Mast Cell Disorders. Front. Immunol. 2017, 8, 792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escribano, L.; Diaz-Agustin, B.; López, A.; López, R.N.; García-Montero, A.; Almeida, J.; Prados, A.; Angulo, M.; Herrero, S.; Orfao, A.; et al. Immunophenotypic analysis of mast cells in mastocytosis: When and how to do it. Proposals of the Spanish Network on Mastocytosis (REMA). Cytom. Part B Clin. Cytom. 2004, 58B, 1–8. [Google Scholar] [CrossRef]
- Pardanani, A. Systemic mastocytosis in adults: 2019 update on diagnosis, risk stratification and management. Am. J. Hematol. 2018, 94, 363–377. [Google Scholar] [CrossRef] [Green Version]
- Gotlib, J.; Gerds, A.T.; Bose, P.; Castells, M.C.; Deininger, M.W.; Gojo, I.; Gundabolu, K.; Hobbs, G.; Jamieson, C.; McMahon, B.; et al. Systemic Mastocytosis, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2018, 16, 1500–1537. [Google Scholar] [CrossRef] [Green Version]
- Scherber, R.M.; Borate, U. How we diagnose and treat systemic mastocytosis in adults. Br. J. Haematol. 2017, 180, 11–23. [Google Scholar] [CrossRef]
- Sotlar, K.; Cerny-Reiterer, S.; Petat-Dutter, K.; Hessel, H.; Berezowska, S.; Müllauer, L.; Valent, P.; Horny, H.-P. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod. Pathol. 2010, 24, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Horny, H.-P.; Parwaresch, M.; Lennert, K. Bone marrow findings in systemic mastocytosis. Hum. Pathol. 1985, 16, 808–814. [Google Scholar] [CrossRef]
- Horny, H.P.; Sotlar, K.; Sperr, W.R.; Valent, P. Systemic mastocytosis with associated clonal haematological non-mast cell lineage diseases: A histopatho-logical challenge. J. Clin. Pathol. 2004, 57, 604–608. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.; Reeder, T.; Li, C.-Y.; Tefferi, A. Eosinophils are derived from the neoplastic clone in patients with systemic mastocytosis and eosinophil-ia. Leuk. Res. 2003, 27, 883–885. [Google Scholar] [CrossRef]
- Taylor, M.L.; Sehgal, D.; Raffeld, M.; Obiakor, H.; Akin, C.; Mage, R.G.; Metcalfe, D.D. Demonstration That Mast Cells, T Cells, and B Cells Bearing the Activating Kit Mutation D816V Occur in Clusters within the Marrow of Patients with Mastocytosis. J. Mol. Diagn. 2004, 6, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.; Kimlinger, T.; Reeder, T.; Li, C.-Y.; Tefferi, A. Bone marrow mast cell immunophenotyping in adults with mast cell disease: A prospective study of 33 patients. Leuk. Res. 2004, 28, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Hartmann, K.; Nilsson, G.; Reiter, A.; Hermine, O.; Sotlar, K.; Sperr, W.R.; Escribano, L.; George, T.I.; et al. Advances in the Classification and Treatment of Mastocytosis: Current Status and Outlook toward the Fu-ture. Cancer Res. 2017, 77, 1261–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardanani, A.; Lim, K.-H.; Lasho, T.L.; Finke, C.M.; McClure, R.F.; Li, C.-Y.; Tefferi, A. WHO subvariants of indolent mastocytosis: Clinical details and prognostic evaluation in 159 consecutive adults. Blood 2010, 115, 150–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Shah, S.; Reichard, K.K.; Hanson, C.A.; Pardanani, A. Smoldering mastocytosis: Survival comparisons with indolent and aggressive mastocytosis. Am. J. Hematol. 2019, 94, E1–E2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valent, P. Mast cell leukemia—A rare form of myeloid leukemia. Wien. Klin. Wochenschr. 2002, 114, 173–174. [Google Scholar]
- Tefferi, A.; Shah, S.; Lasho, T.L.; Patnaik, M.M.; Reichard, K.K.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A. Practice-relevant demarcation of systemic mastocytosis associated with another hematologic neoplasm. Am. J. Hematol. 2018, 93, E383–E386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Olano, D.G.; Caballer, B.D.L.H.; López, R.N.; Muñoz, L.S.; Agustín, M.C.; Dieguez, M.C.; Álvarez Twose, I.; Castells, M.C.; Mora, L.E. Prevalence of allergy and anaphylactic symptoms in 210 adult and pediatric patients with mastocytosis in Spain: A study of the Spanish network on mastocytosis (REMA). Clin. Exp. Allergy 2007, 37, 1547–1555. [Google Scholar] [CrossRef]
- Sokol, H.; Georgin-Lavialle, S.; Canioni, D.; Barete, S.; Damaj, G.; Soucie, E.; Bruneau, J.; Chandesris, M.-O.; Suarez, F.; Launay, J.-M.; et al. Gastrointestinal manifestations in mastocytosis: A study of 83 patients. J. Allergy Clin. Immunol. 2013, 132, 866–873.e3. [Google Scholar] [CrossRef] [PubMed]
- Moura, D.S.; Georgin-Lavialle, S.; Gaillard, R.; Hermine, O. Neuropsychological Features of Adult Mastocytosis. Immunol. Allergy Clin. N. Am. 2014, 34, 407–422. [Google Scholar] [CrossRef]
- Barete, S.; Assous, N.; de Gennes, C.; Grandpeix, C.; Feger, F.; Palmerini, F.; Dubreuil, P.; Arock, M.; Roux, C.; Launay, J.M.; et al. Systemic mastocytosis and bone involvement in a cohort of 75 patients. Ann. Rheum. Dis. 2010, 69, 1838–1841. [Google Scholar] [CrossRef]
- Wang, S.A.; Hutchinson, L.; Tang, G.; Chen, S.S.; Miron, P.M.; Huh, Y.O.; Jones, D.M.; Bueso-Ramos, C.; Verstovsek, S.; Medeiros, L.J.; et al. Systemic mastocytosis with associated clonal hematological non-mast cell lineage disease: Clinical signifi-cance and comparison of chomosomal abnormalities in SM and AHNMD components. Am. J. Hematol. 2013, 88, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.; Lim, K.-H.; Lasho, T.L.; Finke, C.; McClure, R.F.; Li, C.-Y.; Tefferi, A. Prognostically relevant breakdown of 123 patients with systemic mastocytosis associated with other myeloid malignancies. Blood 2009, 114, 3769–3772. [Google Scholar] [CrossRef] [PubMed]
- Galura, G.M.; Cherukuri, S.V.; Hakim, N.; Gaur, S.; Orazi, A. Acute aleukemic mast cell leukemia: Report of a case and review of the literature. Leuk. Res. Rep. 2020, 14, 100230. [Google Scholar] [CrossRef]
- Leguit, R.; Hebeda, K.; Kremer, M.; Van Der Walt, J.; Gianelli, U.; Tzankov, A.; Orazi, A. The Spectrum of Aggressive Mastocytosis: A Workshop Report and Literature Review. Pathobiol. 2019, 87, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Reiter, A.; George, T.I.; Gotlib, J. New developments in diagnosis, prognostication, and treatment of advanced systemic mas-tocytosis. Blood 2020, 135, 1365–1376. [Google Scholar] [CrossRef]
- Kirshenbaum, A.S.; Kessler, S.W.; Goff, J.P.; Metcalfe, D.D. Demonstration of the origin of human mast cells from CD34+ bone marrow progenitor cells. J. Immunol. 1991, 146, 1410–1415. [Google Scholar] [PubMed]
- Cruse, G.; Metcalfe, D.D.; Olivera, A. Functional deregulation of KIT: Link to mast cell proliferative diseases and other neo-plasms. Immunol. Allergy Clin. N. Am. 2014, 34, 219–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenbark, G.R.; DeCastro, C.M.; Taylor, H.; Dew-Knight, S.; Kaufman, R.E. Cloning and structural analysis of the human c-kit gene. Oncogene 1992, 7, 1259–1266. [Google Scholar] [PubMed]
- Giebel, L.B.; Strunk, K.M.; Holmes, S.A.; Spritz, R.A. Organization and nucleotide sequence of the human KIT (mast/stem cell growth factor receptor) pro-to-oncogene. Oncogene 1992, 7, 2207–2217. [Google Scholar] [PubMed]
- Nagata, H.; Worobec, A.S.; Oh, C.K.; Chowdhury, B.A.; Tannenbaum, S.; Suzuki, Y.; Metcalfe, D.D. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc. Natl. Acad. Sci. USA 1995, 92, 10560–10564. [Google Scholar] [CrossRef] [Green Version]
- Bibi, S.; Langenfeld, F.; Jeanningros, S.; Brenet, F.; Soucie, E.; Hermine, O.; Damaj, G.; Dubreuil, P.; Arock, M. Molecular defects in mastocytosis: KIT and beyond KIT. Immunol. Allergy Clin. N. Am. 2014, 34, 239–262. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Orfao, A.; Garcia-Montero, A.C.; Sanchez, L.; Escribano, L. Recent advances in the understanding of mastocytosis: The role of KIT mutations. Br. J. Haematol. 2007, 138, 12–30. [Google Scholar] [CrossRef]
- Kristensen, T.; Vestergaard, H.; Bindslev-Jensen, C.; Møller, M.B.; Broesby-Olsen, S.; Centre, M.; MastOUH. Sensitive KIT D816V mutation analysis of blood as a diagnostic test in mastocytosis. Am. J. Hematol. 2014, 89, 493–498. [Google Scholar] [CrossRef]
- Schwaab, J.; Schnittger, S.; Sotlar, K.; Walz, C.; Fabarius, A.; Pfirrmann, M.; Kohlmann, A.; Grossmann, V.; Meggendorfer, M.; Horny, H.-P.; et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood 2013, 122, 2460–2466. [Google Scholar] [CrossRef] [Green Version]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Sotlar, K.; Horny, H.-P.; Metzgeroth, G.; Müller, N.; Schneider, S.; Naumann, N.; Walz, C.; et al. Molecular profiling of myeloid progenitor cells in multi-mutated advanced systemic mastocytosis identifies KIT D816V as a distinct and late event. Leukemia 2015, 29, 1115–1122. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Meggendorfer, M.; Pfirrmann, M.; Sotlar, K.; Horny, H.-P.; Metzgeroth, G.; Kluger, S.; Naumann, N.; et al. Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of patients with KIT D816V+ advanced systemic mastocytosis. Leukemia 2016, 30, 136–143. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.; Elala, Y.; Wassie, E.; Finke, C.; Reichard, K.K.; Chen, D.; Hanson, C.A.; Ketterling, R.; Tefferi, A. Next-generation sequencing in systemic mastocytosis: Derivation of a mutation-augmented clinical prognostic model for survival. Am. J. Hematol. 2016, 91, 888–893. [Google Scholar] [CrossRef]
- Martinelli, G.; Mancini, M.; De Benedittis, C.; Rondoni, M.; Papayannidis, C.; Manfrini, M.; Meggendorfer, M.; Calogero, R.; Guadagnuolo, V.; Fontana, M.C.; et al. SETD2 and histone H3 lysine 36 methylation deficiency in advanced systemic mastocytosis. Leukemia 2017, 32, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.; Vítor, A.; Sridhara, S.C.; Martins, F.B.; Raposo, A.C.; Desterro, J.; Ferreira, J.; De Almeida, S.F. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. eLife 2014, 3, e02482. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.-M. The Histone Mark H3K36me3 Regulates Human DNA Mismatch Repair through Its Interaction with MutSα. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swolin, B.; Rödjer, S.; Roupe, G. Cytogenetic Studies in Patients with Mastocytosis. Cancer Genet. Cytogenet. 2000, 120, 131–135. [Google Scholar] [CrossRef]
- Gupta, R.; Bain, B.J.; Knight, C.L. Cytogenetic and Molecular Genetic Abnormalities in Systemic Mastocytosis. Acta Haematol. 2002, 107, 123–128. [Google Scholar] [CrossRef]
- Naumann, N.; Jawhar, M.; Schwaab, J.; Kluger, S.; Lübke, J.; Metzgeroth, G.; Popp, H.D.; Khaled, N.; Horny, H.-P.; Sotlar, K.; et al. Incidence and prognostic impact of cytogenetic aberrations in patients with systemic mastocytosis. Genes Chromosom. Cancer 2018, 57, 252–259. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; Pardanani, A.; Elala, Y.C.; Lasho, T.L.; Patnaik, M.M.; Reichard, K.K.; Hanson, C.A.; Ketterling, R.P.; Tefferi, A. Cytogenetic abnormalities in systemic mastocytosis: WHO subcategory-specific incidence and prognostic im-pact among 348 informative cases. Am. J. Hematol. 2018, 93, 1461–1466. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.; Shah, S.; Mannelli, F.; Elala, Y.C.; Guglielmelli, P.; Lasho, T.L.; Patnaik, M.M.; Gangat, N.; Ketterling, R.P.; Reichard, K.K.; et al. Mayo alliance prognostic system for mastocytosis: Clinical and hybrid clinical-molecular models. Blood Adv. 2018, 2, 2964–2972. [Google Scholar] [CrossRef] [Green Version]
- Jawhar, M.; Schwaab, J.; Álvarez-Twose, I.; Shoumariyeh, K.; Naumann, N.; Lübke, J.; Perkins, C.; Muñoz-González, J.I.; Meggendorfer, M.; Kennedy, V.; et al. MARS: Mutation-Adjusted Risk Score for Advanced Systemic Mastocytosis. J. Clin. Oncol. 2019, 37, 2846–2856. [Google Scholar] [CrossRef]
- Sperr, W.R.; Kundi, M.; Alvarez-Twose, I.; van Anrooij, B.; Elberink, J.N.G.O.; Górska, A.; Niedoszytko, M.; Gleixner, K.V.; Hadzijusufovic, E.; Zanotti, R.; et al. International prognostic scoring system for mastocytosis (IPSM): A retrospective cohort study. Lancet Haematol. 2019, 6, e638–e649. [Google Scholar] [CrossRef]
- Castells, M.; Butterfield, J. Mast Cell Activation Syndrome and Mastocytosis: Initial Treatment Options and Long-Term Man-agement. J. Allergy Clin. Immunol. Pract. 2019, 7, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Valent, P.; Akin, C. Mast Cells, Mastocytosis, and Related Disorders. N. Engl. J. Med. 2015, 373, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Jendoubi, F.; Gaudenzio, N.; Gallini, A.; Negretto, M.; Paul, C.; Livideanu, C.B. Omalizumab in the treatment of adult patients with mastocytosis: A systematic review. Clin. Exp. Allergy 2020, 50, 654–661. [Google Scholar] [CrossRef]
- Distler, M.; Maul, J.-T.; Steiner, U.C.; Jandus, P.; Kolios, A.G.; Murer, C.; Graf, N.; Seebach, J.D.; Pichler, W.J.; Navarini, A.A.; et al. Efficacy of Omalizumab in Mastocytosis: Allusive Indication Obtained from a Prospective, Double-Blind, Multicenter Study (XOLMA Study). Dermatology 2020, 236, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Jennings, S.V.; Slee, V.M.; Zack, R.M.; Verstovsek, S.; George, T.; Shi, H.; Lee, P.; Castells, M.C. Patient Perceptions in Mast Cell Disorders. Immunol. Allergy Clin. N. Am. 2018, 38, 505–525. [Google Scholar] [CrossRef] [PubMed]
- Lamb, N.Y.; Deeks, E.D. Sarilumab: A Review in Moderate to Severe Rheumatoid Arthritis. Drugs 2018, 78, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.A.; Brock, E.C.; Leung, J.; Chang, A.T.; Bebbington, C.; Tomasevic, N. AK002, a Novel Humanized Monoclonal Antibody to Siglec-8, Inhibits Mast Cell Activity and De-pletes Eosinophils in Ex Vivo Bone Marrow Tissue from Patients with Systemic Mastocytosis. Blood 2018, 132 (Suppl. S1), 1104. [Google Scholar] [CrossRef]
- Hochhaus, A.; Baccarani, M.; Giles, F.J.; Le Coutre, P.D.; Müller, M.C.; Reiter, A.; Santanastasio, H.; Leung, M.; Novick, S.; Kantarjian, H.M. Nilotinib in patients with systemic mastocytosis: Analysis of the phase 2, open-label, single-arm nilotinib registration study. J. Cancer Res. Clin. Oncol. 2015, 141, 2047–2060. [Google Scholar] [CrossRef] [Green Version]
- Verstovsek, S.; Tefferi, A.; Cortes, J.; O’Brien, S.; Garcia-Manero, G.; Pardanani, A.; Akin, C.; Faderl, S.; Manshouri, T.; Thomas, D.; et al. Phase II Study of Dasatinib in Philadelphia Chromosome–Negative Acute and Chronic Myeloid Diseases, Including Systemic Mastocytosis. Clin. Cancer Res. 2008, 14, 3906–3915. [Google Scholar] [CrossRef] [Green Version]
- Broderick, V.; Waghorn, K.; Langabeer, S.E.; Jeffers, M.; Cross, N.C.; Hayden, P.J. Molecular response to imatinib in KIT F522C-mutated systemic mastocytosis. Leuk. Res. 2018, 77, 28–29. [Google Scholar] [CrossRef]
- Alvarez-Twose, I.; Matito, A.; Morgado, J.M.; Sanchez-Muñoz, L.; Jara-Acevedo, M.; Garcia-Montero, A.C.; Mayado, A.; Caldas, C.; Teodosio, C.; Muñoz-González, J.I.; et al. Imatinib in systemic mastocytosis: A phase IV clinical trial in patients lacking exon 17 KIT mutations and review of the literature. Oncotarget 2016, 8, 68950–68963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, B.; Winter, G.; Blatt, K.; Bennett, K.L.; Stefanzl, G.; Rix, U.; Eisenwort, G.; Hadzijusufovic, E.; Gridling, M.; Dutreix, C.; et al. Target interaction profiling of midostaurin and its metabolites in neoplastic mast cells predicts distinct effects on activation and growth. Leukemia 2016, 30, 464–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleixner, K.V.; Mayerhofer, M.; Aichberger, K.J.; Derdak, S.; Sonneck, K.; Böhm, A.; Gruze, A.; Samorapoompichit, P.; Manley, P.W.; Fabbro, D.; et al. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: Comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood 2006, 107, 752–759. [Google Scholar] [CrossRef] [Green Version]
- Chandesris, M.-O.; Damaj, G.; Canioni, D.; Brouzes, C.; Lhermitte, L.; Hanssens, K.; Frenzel, L.; Cherquaoui, Z.; Durieu, I.; Durupt, S.; et al. Midostaurin in Advanced Systemic Mastocytosis. N. Engl. J. Med. 2016, 374, 2605–2606. [Google Scholar] [CrossRef]
- Grifoni, F.I.; Sciumé, M.; Pravettoni, V.; Ulivieri, F.M.; Muratori, S.; Fracchiolla, N.; Tagliaferri, E.; Gianelli, U.; Migliorini, A.C.; Cro, L.; et al. A case report of systemic mastocytosis associated with multiple hematologic non–mast cell lineage diseases. Hematol. Oncol. 2019, 37, 205–211. [Google Scholar] [CrossRef]
- Barete, S.; Lortholary, O.; Damaj, G.; Hirsch, I.; Chandesris, M.-O.; Elie, C.; Hamidou, M.; Durieu, I.; Suarez, F.; Grosbois, B.; et al. Long-term efficacy and safety of cladribine (2-CdA) in adult patients with mastocytosis. Blood 2015, 126, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.H.; Pardanani, A.; Butterfield, J.H.; Li, C.-Y.; Tefferi, A. Cytoreductive therapy in 108 adults with systemic mastocytosis: Outcome analysis and response prediction during treatment with interferon-alpha, hydroxyurea, imatinib mesylate or 2-chlorodeoxyadenosine. Am. J. Hematol. 2009, 84, 790–794. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Hartmann, K.; George, T.; Sotlar, K.; Peter, B.; Gleixner, K.; Blatt, K.; Sperr, W.; Manley, P.; et al. Midostaurin: A magic bullet that blocks mast cell expansion and activation. Ann. Oncol. 2017, 28, 2367–2376. [Google Scholar] [CrossRef]
- van Anrooij, B.; Elberink, J.N.G.O.; Span, L.F.R.; de Monchy, J.G.R.; Rosati, S.; Mulder, A.B.; Kluin-Nelemans, J.C. Midostaurin in patients with indolent systemic mastocytosis: An open-label phase 2 trial. J. Allergy Clin. Immunol. 2018, 142, 1006–1008.e7. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J. Activity of the tyrosine kinase inhibitor PKC412 in a patient with mast cell leukemia with the D816V KIT mutation. Blood 2005, 106, 2865–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szudy-Szczyrek, A.; Bachanek-Mitura, O.; Gromek, T.; Chromik, K.; Mital, A.; Szczyrek, M.; Krupski, W.; Szumiło, J.; Kanduła, Z.; Helbig, G.; et al. Real-World Efficacy of Midostaurin in Aggressive Systemic Mastocytosis. J. Clin. Med. 2021, 10, 1109. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.K.; Gardino, A.K.; Kim, J.L.; Hodous, B.L.; Shutes, A.; Davis, A.; Zhu, X.J.; Schmidt-Kittler, O.; Wilson, D.; Wilson, K.; et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci. Transl. Med. 2017, 9, eaao1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotlib, J.; Pardanani, A.; Akin, C.; Reiter, A.; George, T.; Hermine, O.; Kluin-Nelemans, H.; Hartmann, K.; Sperr, W.R.; Brockow, K.; et al. International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) & Europe-an Competence Network on Mastocytosis (ECNM) consensus response criteria in advanced systemic mastocytosis. Blood 2013, 121, 2393–2401. [Google Scholar] [PubMed]
- Smith, B.D.; Kaufman, M.D.; Lu, W.-P.; Gupta, A.; Leary, C.B.; Wise, S.C.; Rutkoski, T.J.; Ahn, Y.M.; Al-Ani, G.; Bulfer, S.L.; et al. Ripretinib (DCC-2618) Is a Switch Control Kinase Inhibitor of a Broad Spectrum of Oncogenic and Drug-Resistant KIT and PDGFRA Variants. Cancer Cell 2019, 35, 738–751.e9. [Google Scholar] [CrossRef]
- Lortholary, O.; Chandesris, M.O.; Livideanu, C.B.; Paul, C.; Guillet, G.; Jassem, E.; Niedoszytko, M.; Barete, S.; Verstovsek, S.; Grattan, C.; et al. Masitinib for treatment of severely symptomatic indolent systemic mastocytosis: A randomised, placebo-controlled, phase 3 study. Lancet 2017, 389, 612–620. [Google Scholar] [CrossRef]
- Kelly, C.M.; Sainz, L.G.; Chi, P. The management of metastatic GIST: Current standard and investigational therapeutics. J. Hematol. Oncol. 2021, 14, 1–12. [Google Scholar] [CrossRef]
- Escribano, L.; Bravo, P.; Herrera, P.; Vargas, M.; Orfao, A.; Cerveró, C.; Miguel, J.S.; Díaz-Agustín, B.; Villarrubia, J.; Garcia-Sanz, R.; et al. Expression of Bcl-2 by human bone marrow mast cells and its overexpression in mast cell leukemia. Am. J. Hematol. 1999, 60, 191–195. [Google Scholar] [CrossRef]
- Aichberger, K.J.; Mayerhofer, M.; Gleixner, K.V.; Krauth, M.-T.; Gruze, A.; Pickl, W.F.; Wacheck, V.; Selzer, E.; Müllauer, L.; Agis, H.; et al. Identification of MCL1 as a novel target in neoplastic mast cells in systemic mastocytosis: Inhibition of mast cell survival by MCL1 antisense oligonucleotides and synergism with PKC412. Blood 2007, 109, 3031–3041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konopleva, M.; Watt, J.; Contractor, R.; Tsao, T.; Harris, D.; Estrov, Z.; Bornmann, W.; Kantarjian, H.; Viallet, J.; Samudio, I.; et al. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obato-clax). Cancer Res. 2008, 68, 3413–3420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, B.; Cerny-Reiterer, S.; Hadzijusufovic, E.; Schuch, K.; Stefanzl, G.; Eisenwort, G.; Gleixner, K.V.; Hoermann, G.; Mayerhofer, M.; Kundi, M.; et al. The pan-Bcl-2 blocker obatoclax promotes the expression of Puma, Noxa, and Bim mRNA and induces apoptosis in neoplastic mast cells. J. Leukoc. Biol. 2013, 95, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worobec, A.S.; Kirshenbaum, A.S.; Schwartz, L.B.; Metcalfe, D.D. Treatment of Three Patients with Systemic Mastocytosis with Interferon Alpha-2b. Leuk. Lymphoma 1996, 22, 501–508. [Google Scholar] [CrossRef]
- Weide, R.; Ehlenz, K.; Lorenz, W.; Walthers, E.; Klausmann, M.; Pflüger, K.H. Successful treatment of osteoporosis in systemic mastocytosis with interferon alpha-2b. Ann. Hematol. 1996, 72, 41–43. [Google Scholar] [CrossRef]
- Takasaki, Y.; Tsukasaki, K.; Jubashi, T.; Tomonaga, M.; Kamihira, S.; Makiyama, K. Systemic mastocytosis with extensive polypoid lesions in the intestines; successful treatment with interfer-on-alpha. Intern. Med. 1998, 37, 484–488. [Google Scholar] [CrossRef] [Green Version]
- Hübner, C.; Wedding, U.; Sträter, J.; Limberg, B.; Stremmel, W. Clinical stable systemic mastocytosis with interferon alpha-2b therapy. J. Intern. Med. 2007, 241, 529–533. [Google Scholar] [CrossRef]
- Hauswirth, A.W.; Simonitsch-Klupp, I.; Uffmann, M.; Koller, E.; Sperr, W.R.; Lechner, K.; Valent, P. Response to therapy with interferon alpha-2b and prednisolone in aggressive systemic mastocytosis: Report of five cases and review of the literature. Leuk. Res. 2004, 28, 249–257. [Google Scholar] [CrossRef]
- Pardanani, A.; Reichard, K.K.; Zblewski, D.; Abdelrahman, R.A.; Wassie, E.A.; Ii, W.G.M.; Brooks, C.L.; Grogg, K.L.; Hanson, C.A.; Tefferi, A.; et al. CD123 immunostaining patterns in systemic mastocytosis: Differential expression in disease subgroups and potential prognostic value. Leukemia 2016, 30, 914–918. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Lane, A.A.; Sweet, K.L.; Stein, A.S.; Vasu, S.; Blum, W.; Rizzieri, D.A.; Wang, E.S.; Duvic, M.; Sloan, J.M.; et al. Tagraxofusp in Blastic Plasmacytoid Dendritic-Cell Neoplasm. N. Engl. J. Med. 2019, 380, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- Toledo, M.A.S.; Gatz, M.; Sontag, S.; Gleixner, K.V.; Eisenwort, G.; Feldberg, K.; Hamouda, A.E.I.; Kluge, F.; Guareschi, R.; Rossetti, G.; et al. Nintedanib targets KIT D816V neoplastic cells derived from induced pluripotent stem cells of systemic mastocytosis. Blood 2021, 137, 2070–2084. [Google Scholar] [CrossRef] [PubMed]
- Dorrance, A. Mastering drug discovery with iPSCs. Blood 2021, 137, 1993–1994. [Google Scholar] [CrossRef]
- Efficace, F.; Gaidano, G.; Lo-Coco, F. Patient-reported outcomes in hematology: Is it time to focus more on them in clinical trials and hematology practice? Blood 2017, 130, 859–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulfer, S.; Ziehfreund, S.; Winkler, J.; Hindelang, B.; Biedermann, T.; Brockow, K.; Zink, A. Health-related quality of life and influencing factors in adults with non-advanced mastocytosis - a cross-sectional study and qualitative approach. J. Allergy Clin. Immunol. Pract. 2021, 9, 3166. [Google Scholar] [CrossRef]
- Efficace, F.; Cottone, F.; Oswald, L.B.; Cella, D.; Patriarca, A.; Niscola, P.; Breccia, M.; Platzbecker, U.; Palumbo, G.A.; Caocci, G.; et al. The IPSS-R more accurately captures fatigue severity of newly diagnosed patients with myelodysplastic syndromes compared with the IPSS index. Leukemia 2020, 34, 2451–2459. [Google Scholar] [CrossRef]
- Efficace, F.; Stagno, F.; Iurlo, A.; Breccia, M.; Cottone, F.; Bonifacio, M.; Abruzzese, E.; Castagnetti, F.; Caocci, G.; Crugnola, M.; et al. Health-related quality of life of newly diagnosed chronic myeloid leukemia patients treated with first-line dasatinib versus imatinib therapy. Leukemia 2020, 34, 488–498. [Google Scholar] [CrossRef]
- Gamper, E.M.; Cottone, F.; Sommer, K.; Norman, R.; King, M.; Breccia, M.; Caocci, G.; Patriarca, A.; Palumbo, G.A.; Stauder, R.; et al. The EORTC QLU-C10D was more efficient in detecting clinical known group differences in myelodys-plastic syndromes than the EQ-5D-3L. J. Clin. Epidemiol. 2021, 137, 31–44. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cutaneous Mastocytosis (CM) |
|---|
|
| Systemic Mastocytosis (SM) |
|
| Mast Cell Sarcoma (MCS) |
| Major criterion | Multifocal, dense infiltrate of atypical MCs (≥15 MCs in aggregates) in BM biopsy and/or in section of other extracutaneous organs |
| Minor criteria |
|
| B—Findings |
|
|
|
| C—Findings |
|
|
|
|
|
|
| MARS [64] | MAPS [63] | IPSS for No AdvSM [65] | IPSS for AdvSM | ||||
|---|---|---|---|---|---|---|---|
| Prognostic Variable and Points | |||||||
| Age > 60 years | 1 | Age > 60 years | 1 | Age > 60 years | 1 | Age > 60 years | 1 |
| Hemoglobin < 10 g/dL | 1 | Advanced SM vs. ISM/SSM | 2 | ALP ≥ 100 U/L | 1 | Tryptase ≥ 125 ng/mL | 1 |
| Platelets < 100 × 109/L | 1 | Platelets < 150 × 109/L | 1 | Leukocytes ≥ 16 × 109/L | 1 | ||
| One S/A/R (SFRS2, ASXL, or RUNX1) mutation | 1 | Serum ALP > normal range | 1 | Hemoglobin ≤ 11 g/dL | 1 | ||
| ≥2 S/A/R mutation | 2 | Adverse mutation (ASXL1, RUNX1 and NRAS) | 1 | Platelets <150 × 109/L | 1 | ||
| Skin involvement | −1 | ||||||
| Risk Group and Points | |||||||
| Low | 0–1 | Low | ≤2 | Low | 0 | AdvSM-1 | −1 to 0 |
| Intermediate | 2 | Intermediate- 1 | 3 | Intermediate-1 | 1 | AdvSM-2 | 1 |
| High | 3–5 | Intermediate-2 | 4 | Intermediate-2 | 2 | AdvSM-3 | 2 |
| High | ≥5 | AdvSM-4 | 2–3 | ||||
| AdvSM-5 | 4–5 | ||||||
| Clinicaltrials.Gov Identifier | Intervention | Title | Primary Outcome Measures |
|---|---|---|---|
| NCT03770273 | Sarilimus | A Phase 2 Randomized Double-Blinded Placebo-Controlled Study to Evaluate the Safety and Efficacy of Subcutaneous Sarilumab in Improving the Quality of Life in Subjects with Indolent Systemic Mastocytosis | Frequency and severity of adverse events (AEs); mastocytosis Quality of Life Questionnaire (MC-QoL) |
| NCT04333108 | Masitinib | Phase 3 Study to Compare Oral Masitinib to Placebo in Treatment of Patients with Smouldering or Indolent Severe Systemic Mastocytosis, Unresponsive to Optimal Symptomatic Treatment | Cumulative response in at least one of three severe baseline symptoms of mast cell mediator release (pruritus, flushes, or depression). |
| NCT04910685 | BLU-263 | A Randomized, Double-Blind, Placebo-Controlled Phase 2/3 Study of BLU-263 in Indolent Systemic Mastocytosis | Recommended Dose (RD) in patients with ISM; response rate in patients with ISM; long-term safety and tolerability of BLU-263 as assessed by the number of adverse events and serious adverse events; mean change in Indolent Systemic Mastocytosis, Symptom Assessment Form (ISM-SAF) Total Symptom Score (TSS) |
| NCT03731260 | Avapritinib (BLU-285) | A 3-Part, Randomized, Double-Blind, Placebo-Controlled Phase 2 Study to Evaluate Safety and Efficacy of Avapritinib (BLU-285), a Selective KIT Mutation-Targeted Tyrosine Kinase Inhibitor, in Indolent and Smoldering Systemic Mastocytosis With Symptoms Inadequately Controlled With Standard Therapy | Recommended Phase 2 dose (RP2D) in patients with ISM; proportion of responders, defined as ≥30% reduction in ISM Symptom Assessment Form |
| NCT04996875 | Bezuclastinib (CGT9486; PLX9486) | A Phase 2 Open-Label, Multicenter Clinical Study of the Safety, Efficacy, Pharmacokinetic, and Pharmacodynamic Profiles of CGT9486 as a Single Agent in Patients With Advanced Systemic Mastocytosis | Determine the optimal dose of CGT9486 by safety assessments and response criteria; objective response rate according to modified IWG-MRT-ECNM response criteria |
| NCT03214666 | GTB-3550 | GTB-3550 (CD16/IL-15/CD33) Tri-Specific Killer Engager (TriKE™) for the Treatment of HighRisk Myelodysplastic Syndromes, Refractory/Relapsed Acute Myeloid Leukemia and Advanced Systemic Mastocytosis | Maximum Tolerated Dose (MTD) of GTB-3550 TriKE™ finding; incidence of complete and partial remission due to GTB-3550 TriKE™ treatment |
| NCT04681105 | Flotetuzumab | A Phase 1 Trial to Evaluate the Safety of Single Agent Flotetuzumab in Advanced CD123-Positive Hematological Malignancies | Maximum tolerated dose (recommended phase 2 dose, RP2D) of flotetuzumab; evaluate the safety and tolerability of flotetuzumab in CD123-positive advanced ALL) (Cohort A) and other hematological malignancies (Cohort B), by evaluation of toxicities including: type, frequency, severity, attribution, and duration of the toxicity. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolosi, M.; Patriarca, A.; Andorno, A.; Mahmoud, A.M.; Gennari, A.; Boldorini, R.; Gaidano, G.; Crisà, E. Precision Medicine in Systemic Mastocytosis. Medicina 2021, 57, 1135. https://doi.org/10.3390/medicina57111135
Nicolosi M, Patriarca A, Andorno A, Mahmoud AM, Gennari A, Boldorini R, Gaidano G, Crisà E. Precision Medicine in Systemic Mastocytosis. Medicina. 2021; 57(11):1135. https://doi.org/10.3390/medicina57111135
Chicago/Turabian StyleNicolosi, Maura, Andrea Patriarca, Annalisa Andorno, Abdurraouf Mokhtar Mahmoud, Alessandra Gennari, Renzo Boldorini, Gianluca Gaidano, and Elena Crisà. 2021. "Precision Medicine in Systemic Mastocytosis" Medicina 57, no. 11: 1135. https://doi.org/10.3390/medicina57111135
APA StyleNicolosi, M., Patriarca, A., Andorno, A., Mahmoud, A. M., Gennari, A., Boldorini, R., Gaidano, G., & Crisà, E. (2021). Precision Medicine in Systemic Mastocytosis. Medicina, 57(11), 1135. https://doi.org/10.3390/medicina57111135