Tanjungides A and B: New Antitumoral Bromoindole Derived Compounds from Diazona cf formosa. Isolation and Total Synthesis


 ,
,
Abstract
:1. Introduction
2. Results and Discussion
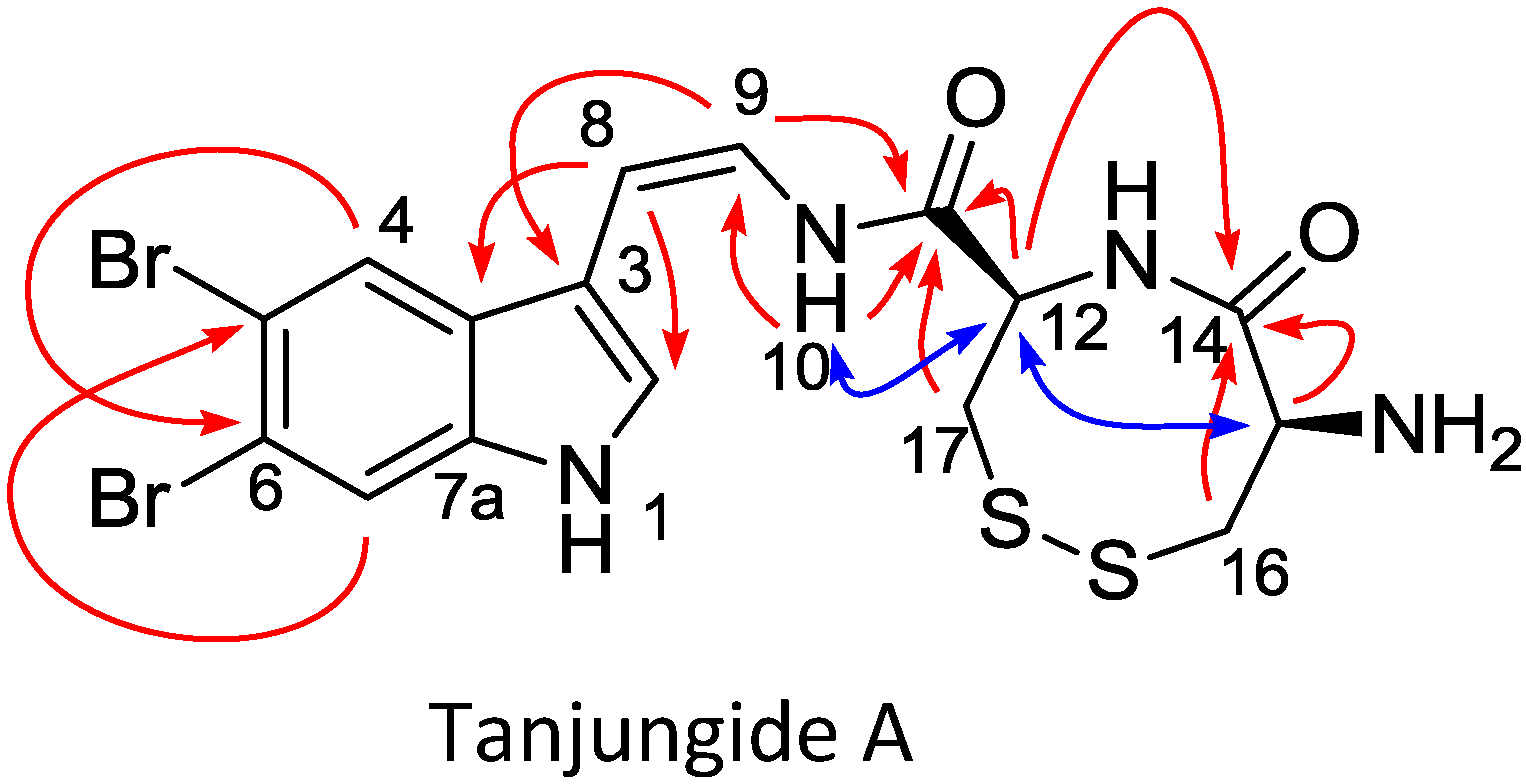
2.1. Isolation and Structure Elucidation

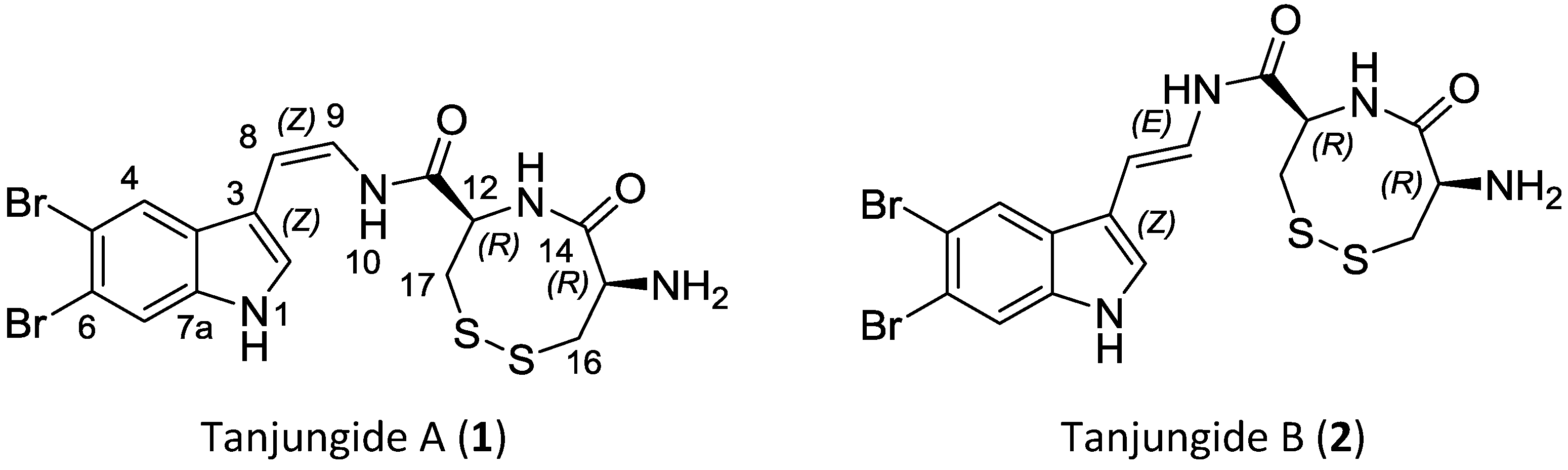{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Tanjungide A (1) | Tanjungide B (2) | ||
|---|---|---|---|---|
| δH (m, J in Hz) | δC, mult. | δH (m, J in Hz) | δC, mult. | |
| 1 | 11.78 (d, 2.6) | - | - | - |
| 2 | 7.78 (d, 2.4) | 126.8, d | 7.35 (s) | 126.7, d |
| 3 | - | 109.2, s | - | 112.9, s |
| 3a | - | 127.6, s | - | 127.4, s |
| 4 | 8.01 (s) | 123.0, d | 8.04 (s) | 124.6, d |
| 5 | - | 113.5, s | - | 117.5, s |
| 6 | - | 115.7, s | - | 115.5, s |
| 7 | 7.81 (s) | 116.3, d | 7.72 (s) | 117.4, d |
| 7a | - | 135.4, s | - | 138.3, s |
| 8 | 6.06 (d, 9.4) | 103.8, d | 6.50 (d, 14.8) | 109.2, d |
| 9 | 6.68 (dd, 9.4, 9.8) | 118.9, d | 7.34 (d, 14.8) | 120.7, d |
| 10 | 9.60 (d, 9.8) | - | - | - |
| 11 | - | 167.1, s | - | 167.8, s |
| 12 | 5.02 (ddd, 11.8, 11.5, 3.6) | 52.5, d | 4.93 (m) | 54.2, d |
| 13 | 8.44 (br s) | - | - | - |
| 14 | - | 169.9, s | - | 169.0, s |
| 15 | 4.65 (m) | 51.2, d | 4.60 (m) | 53.7, d |
| 16 | 2.94 (dd, 14.2, 11.5) 3.17 (m) | 39.7, t | 3.11 (m) | 41.8, t |
| 17 | 2.86 (dd, 13.3, 11.6) 3.40 (m) | 41.6, t | 2.90 (dd, 12.4, 12.4) 3.47 (m) | 43.2, t |


2.2. Biological Activities of Tanjungides A and B
| Compound | Lung-NSCLC | Colon | Breast |
|---|---|---|---|
| A549 | HT29 | MDA-MB-231 | |
| GI50 | GI50 | GI50 | |
| Natural Tanjungide A | 0.33 | 0.19 | 0.23 |
| Synthetic Tanjungide A | 0.33 | 0.25 | 0.19 |
| Natural Tanjungide B | 2.50 | 2.31 | 1.63 |
| Synthetic Tanjungide B | 1.00 | 1.15 | 1.11 |
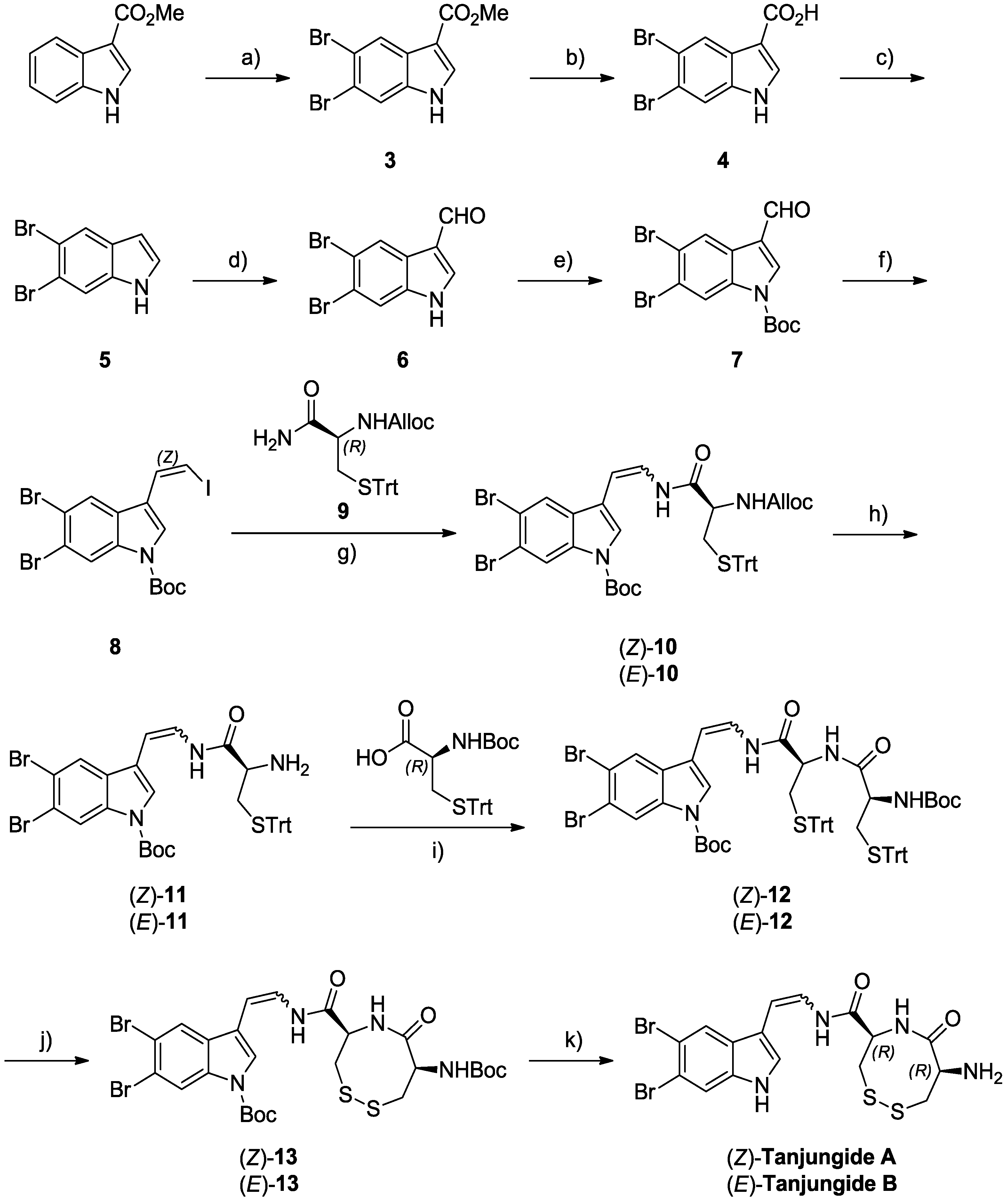
2.3. Total Synthesis of Tanjungides A and B


3. Experimental Section
3.1. General
3.2. Animal Material
3.3. Extraction and Isolation
3.4. Absolute Configuration of Cysteine Residues
3.5. Total Synthesis of Tanjungides A (1) and B (2)
3.5.1. 5,6-Dibromo-1H-indole-3-carboxylic Acid (4)
3.5.2. 5,6-Dibromo-1H-indole (5)
3.5.3. 5,6-Dibromo-1H-indole-3-carbaldehyde (6)
3.5.4. tert-Butyl 5,6-dibromo-3-formyl-1H-indole-1-carboxylate (7)
3.5.5. (Z)-tert-Butyl 5,6-dibromo-3-(2-iodovinyl)-1H-indole-1-carboxylate (8)
3.5.6. (R)-Allyl (1-amino-1-oxo-3-(tritylthio)propan-2-yl)carbamate (9)
3.5.7. (R,Z)-tert-Butyl 3-(2-(2-(((allyloxy)carbonyl)amino)-3-(tritylthio)propanamido)vinyl)-5,6-dibromo-1H-indole-1-carboxylate (Z-10)
3.5.8. (R,E)-tert-Butyl 3-(2-(2-(((allyloxy)carbonyl)amino)-3-(tritylthio)propanamido)vinyl)-5,6-dibromo-1H-indole-1-carboxylate (E-10)
3.5.9. (R,Z)-tert-Butyl 3-(2-(2-amino-3-(tritylthio)propanamido)vinyl)-5,6-dibromo-1H-indole-1-carboxylate (Z-11)
3.5.10. (R,E)-tert-Butyl 3-(2-(2-amino-3-(tritylthio)propanamido)vinyl)-5,6-dibromo-1H-indole-1-carboxylate (E-11)
3.5.11. tert-Butyl 5,6-dibromo-3-((6R,9R,Z)-2,2-dimethyl-4,7,10-trioxo-6,9-bis((tritylthio)methyl)-3-oxa-5,8,11-triazatridec-12-en-13-yl)-1H-indole-1-carboxylate (Z-12)
3.5.12. tert-Butyl 5,6-dibromo-3-((6R,9R,E)-2,2-dimethyl-4,7,10-trioxo-6,9-bis((tritylthio)methyl)-3-oxa-5,8,11-triazatridec-12-en-13-yl)-1H-indole-1-carboxylate (E-12)
3.5.13. tert-Butyl 5,6-dibromo-3-((Z)-2-((4R,7R)-7-((tert-butoxycarbonyl)amino)-6-oxo-1,2,5-dithiazocane-4-carboxamido)vinyl)-1H-indole-1-carboxylate (Z-13)
3.5.14. tert-Butyl 5,6-dibromo-3-((E)-2-((4R,7R)-7-((tert-butoxycarbonyl)amino)-6-oxo-1,2,5-dithiazocane-4-carboxamido)vinyl)-1H-indole-1-carboxylate (E-13)
3.5.15. Tanjungide A (1)
3.5.16. Tanjungide B (2)
3.6. Biological Activity
4. Conclusions
Supplementary Files
Acknowledgments
Conflicts of Interest
References
- Menna, M.; Fattorusso, E.; Imperatore, C. Alkaloids from marine ascidians. Molecules 2011, 16, 8694–8732. [Google Scholar] [CrossRef] [Green Version]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2013, 30, 237–323. [Google Scholar] [CrossRef]
- Vervoort, H.C.; Richards-Gross, S.E.; Fenical, W.; Lee, A.Y.; Clardy, J. Didemnimides A–D: Novel, predator-deterrent alkaloids from the caribbean mangrove ascidian Didemnum conchyliatum. J. Org. Chem. 1997, 62, 1486–1490. [Google Scholar] [CrossRef]
- Seldes, A.M.; Rodríguez, M.F.; Hernández, L.; Palermo, J.A. Identification of two meridianins from the crude extract of the tunicate Aplidium meridianum by tandem mass spectrometry. Nat. Prod. Res. 2007, 21, 555–563. [Google Scholar] [CrossRef]
- Bokesch, H.R.; Pannell, L.K.; McKee, T.C.; Boyd, M.R. Coscinamides A, B and C, three new bis indole alkaloids from the marine sponge Coscinoderma sp. Tetrahedron Lett. 2000, 41, 6305–6308. [Google Scholar] [CrossRef]
- Sato, H.; Tsuda, M.; Watanabe, K.; Kobayashi, J. Rhopaladins A–D, new indole alkaloids from marine tunicate Rhopalaea sp. Tetrahedron 1998, 54, 8687–8690. [Google Scholar] [CrossRef]
- Appleton, D.R.; Copp, B.R. Kottamide E, the first example of a natural product bearing the amino acid 4-amino-1,2-dithiolane-4-carboxylic acid (Adt). Tetrahedron Lett. 2003, 44, 8963–8965. [Google Scholar] [CrossRef]
- Parsons, T.B.; Spencer, N.; Tsang, C.W.; Grainger, R.S. Total synthesis of kottamide E. Chem. Commun. 2013, 49, 2296–2298. [Google Scholar]
- Reyes, F.; Fernández, R.; Rodríguez, A.; Francesch, A.; Taboada, S.; Ávila, C.; Cuevas, C. Aplicyanins A–F, new cytotoxic bromoindole derivatives from the marine tunicate Aplidium Cyaneum. Tetrahedron 2008, 64, 5119–5123. [Google Scholar] [CrossRef]
- Lindquist, N.; Fenical, W. Isolation and structure determination of Diazonamides A and B, unusual cytotoxic metabolites from the marine ascidian Diazona chinensis. J. Am. Chem. Soc. 1991, 11, 2303–2304. [Google Scholar] [CrossRef]
- Fernández, R.; Martín, M.J.; Rodríguez-Acebes, R.; Reyes, F.; Francesch, A.; Cuevas, C. Diazonamides C–E, new cytotoxic metabolites from the ascidian Diazona sp. Tetrahedon Lett. 2008, 49, 2283–2285. [Google Scholar] [CrossRef]
- Whitson, E.L.; Ratnayake, A.S.; Bugni, T.S.; Harper, M.K.; Ireland, C.M. Isolation, structure elucidation, and synthesis of Eudistomides A and B, lipopeptides from a fijian ascidian Eudistoma sp. J. Org. Chem. 2009, 74, 1156–1162. [Google Scholar] [CrossRef]
- Marfey, P. Determination of d-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commun. 1984, 49, 591–596. [Google Scholar]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Boyd, E.M.; Sperry, J. Synthesis of the selective neuronal nitric oxide synthase (nNOS) inhibitor 5,6-dibromo-2′-demethylaplysinopsin. Synlett 2011, 6, 826–830. [Google Scholar]
- Seyferth, S.; Heeren, J.K.; Singh, G.; Grim, S.O.; Hughes, W.B. Studies in phosphinemethylene chemistry: XIII. Routes to triphenylphosphine-halomethylenes and -dihalomethylenes. J. Organomet. Chem. 1966, 5, 267–274. [Google Scholar]
- Stork, G.; Zhao, K. A stereoselective synthesis of (Z)-1-iodo-1-alkenes. Tetrahedron Lett. 1989, 30, 2173–2174. [Google Scholar] [CrossRef]
- Jiang, L.; Job, G.E.; Klapars, A.; Buchwald, S.L. Copper-catalyzed coupling of amides and carbamates with vinyl halides. Org. Lett. 2003, 5, 3667–3669. [Google Scholar] [CrossRef]
- Kamber, B.; Hartmann, A.; Eisler, K.; Riniker, B.; Rink, H.; Sieber, P.; Rittel, W. The synthesis of cystine peptides by iodine oxidation of S-trityl-cysteine and S-acetamidomethyl-cysteine peptides. Helv. Chim. Acta 1980, 63, 899–915. [Google Scholar] [CrossRef]
- Boger, D.L.; Ichikawa, S.; Tse, W.C.; Hedrick, M.P.; Jin, F.Q. Total synthesis of thiocoraline and BE-22179 and assessment of their DNA binding and biological properties. J. Am. Chem. Soc. 2001, 123, 561–568. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Murcia, C.; Coello, L.; Fernández, R.; Martín, M.J.; Reyes, F.; Francesch, A.; Munt, S.; Cuevas, C. Tanjungides A and B: New Antitumoral Bromoindole Derived Compounds from Diazona cf formosa. Isolation and Total Synthesis. Mar. Drugs 2014, 12, 1116-1130. https://doi.org/10.3390/md12021116
Murcia C, Coello L, Fernández R, Martín MJ, Reyes F, Francesch A, Munt S, Cuevas C. Tanjungides A and B: New Antitumoral Bromoindole Derived Compounds from Diazona cf formosa. Isolation and Total Synthesis. Marine Drugs. 2014; 12(2):1116-1130. https://doi.org/10.3390/md12021116
Chicago/Turabian StyleMurcia, Carmen, Laura Coello, Rogelio Fernández, María Jesús Martín, Fernando Reyes, Andrés Francesch, Simon Munt, and Carmen Cuevas. 2014. "Tanjungides A and B: New Antitumoral Bromoindole Derived Compounds from Diazona cf formosa. Isolation and Total Synthesis" Marine Drugs 12, no. 2: 1116-1130. https://doi.org/10.3390/md12021116
APA StyleMurcia, C., Coello, L., Fernández, R., Martín, M. J., Reyes, F., Francesch, A., Munt, S., & Cuevas, C. (2014). Tanjungides A and B: New Antitumoral Bromoindole Derived Compounds from Diazona cf formosa. Isolation and Total Synthesis. Marine Drugs, 12(2), 1116-1130. https://doi.org/10.3390/md12021116