Another Facet to the Anticancer Response to Lamellarin D: Induction of Cellular Senescence through Inhibition of Topoisomerase I and Intracellular Ros Production

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
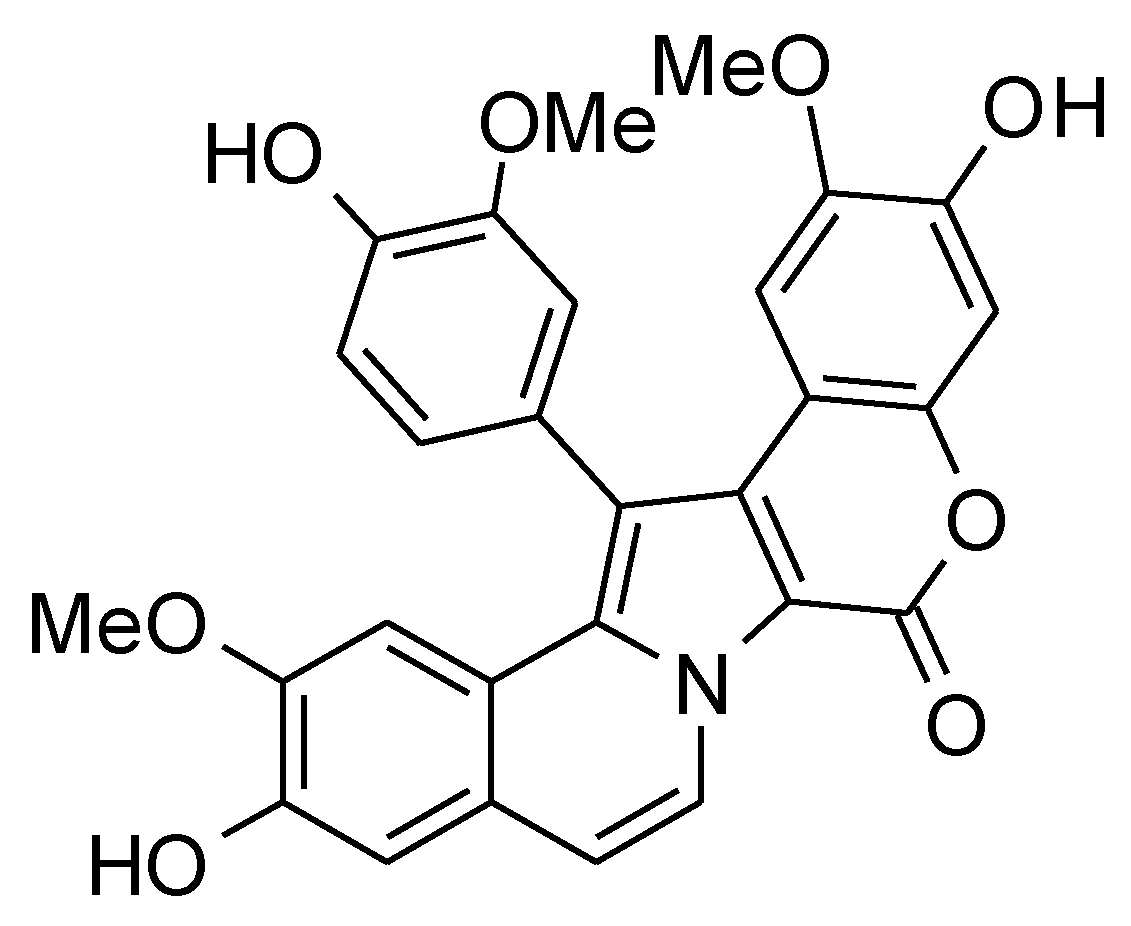
:1. Introduction

2. Results and Discussion
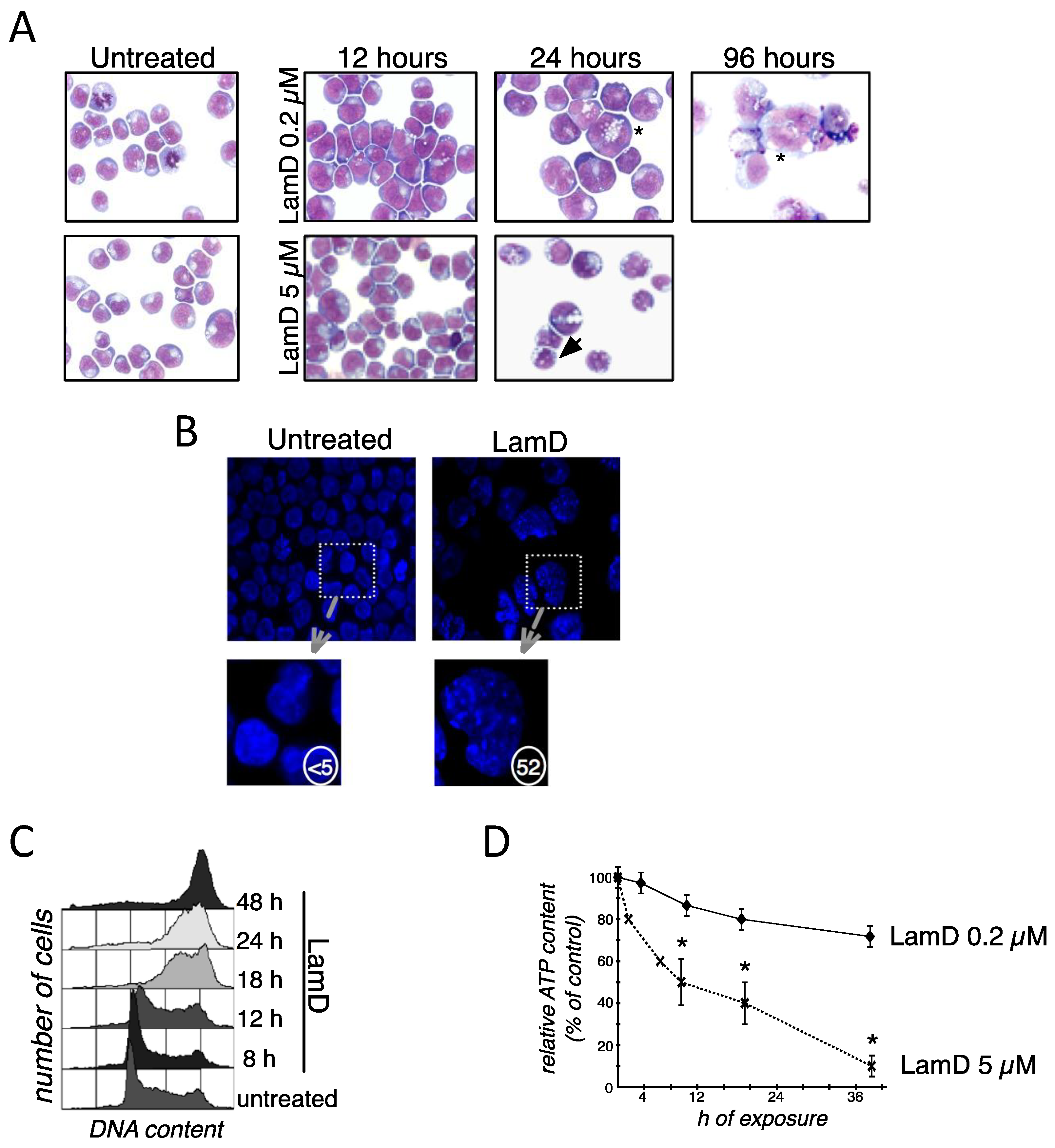
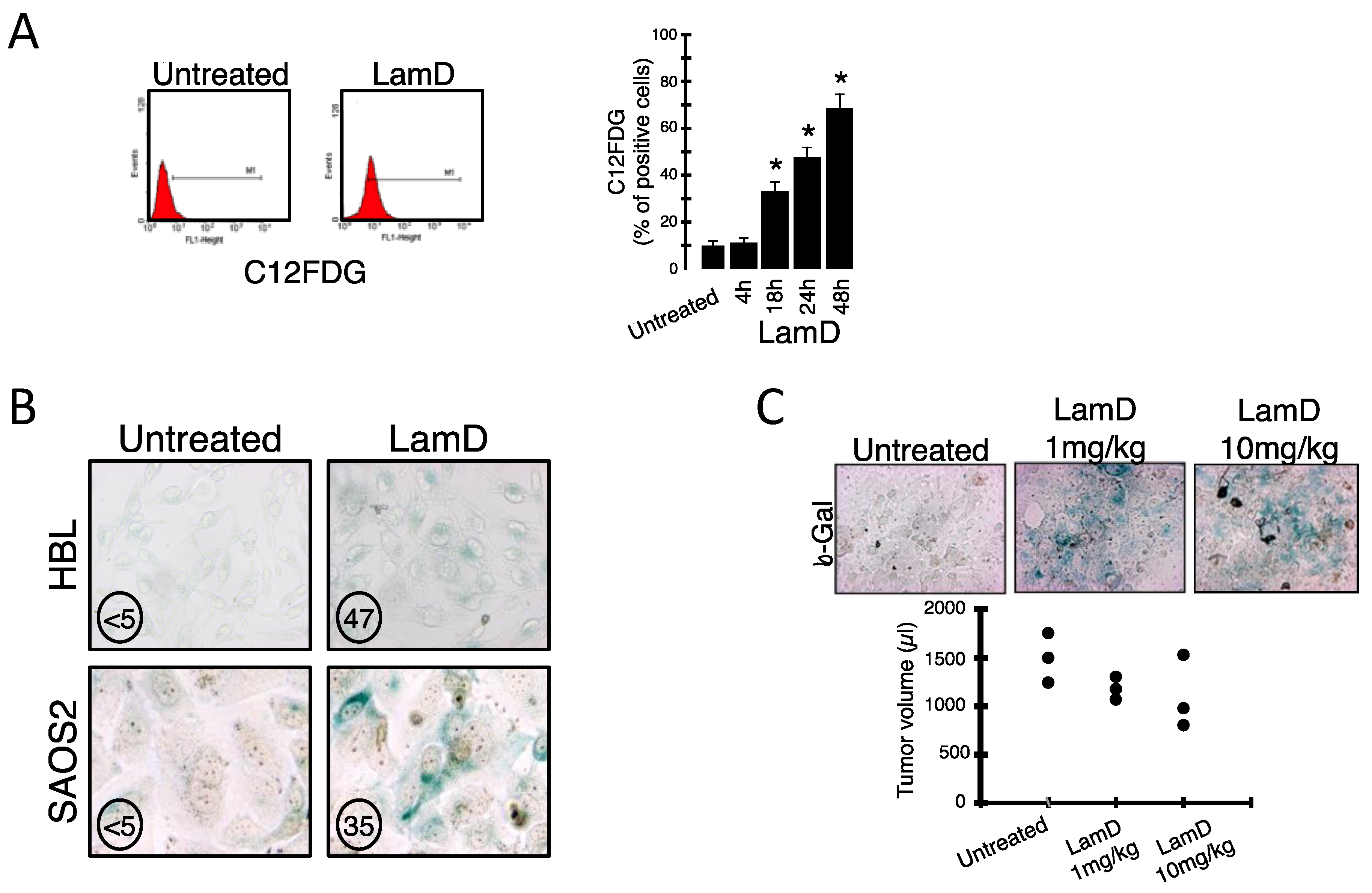
2.1. Lamellarin D Induces Senescence-Like Growth Arrest in Cancer Cells


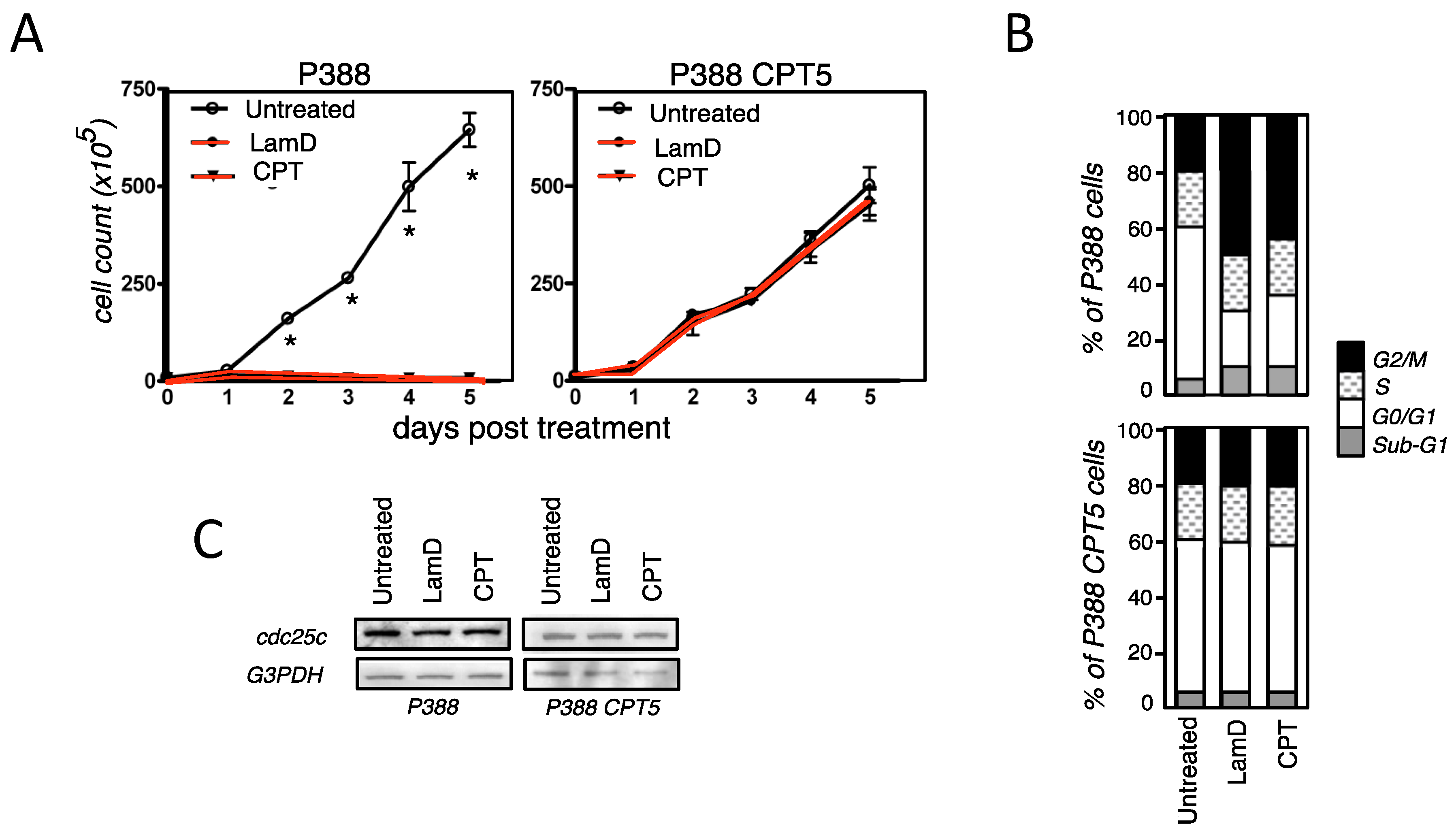
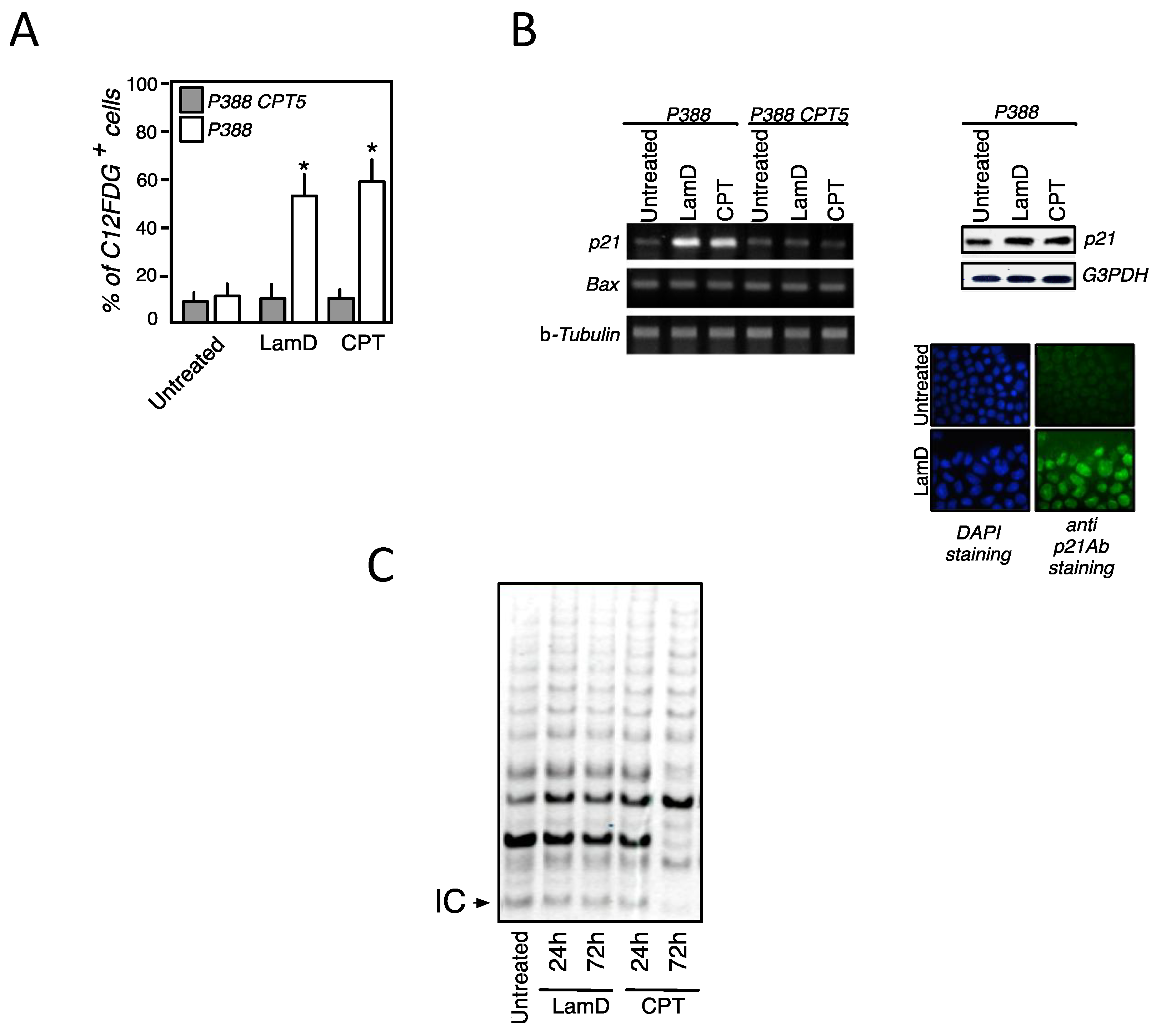
2.2. Senescence-Associated Growth Arrest Induced by Lamellarin D Is Dependent on Its Effect on Topoisomerase I


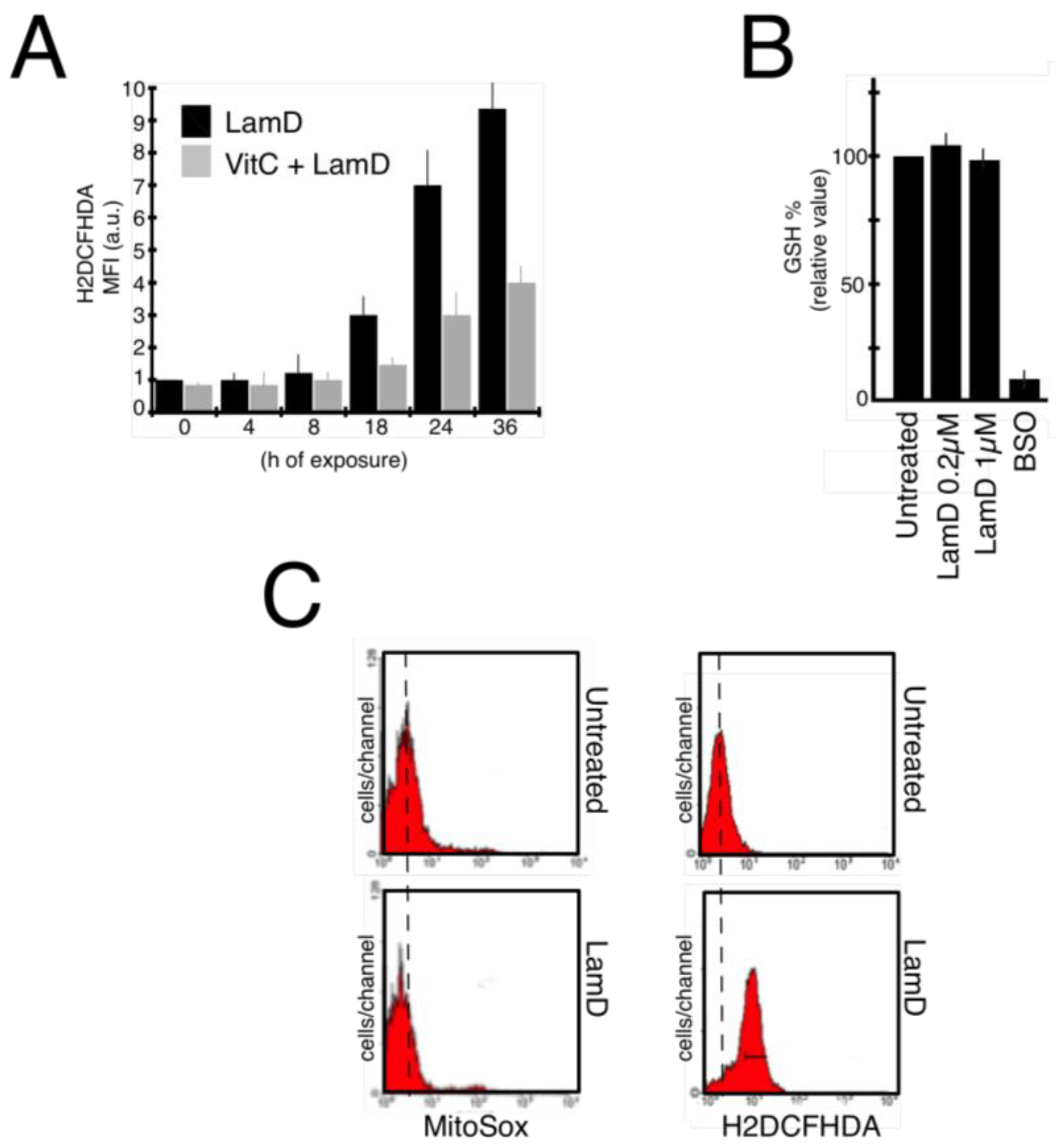
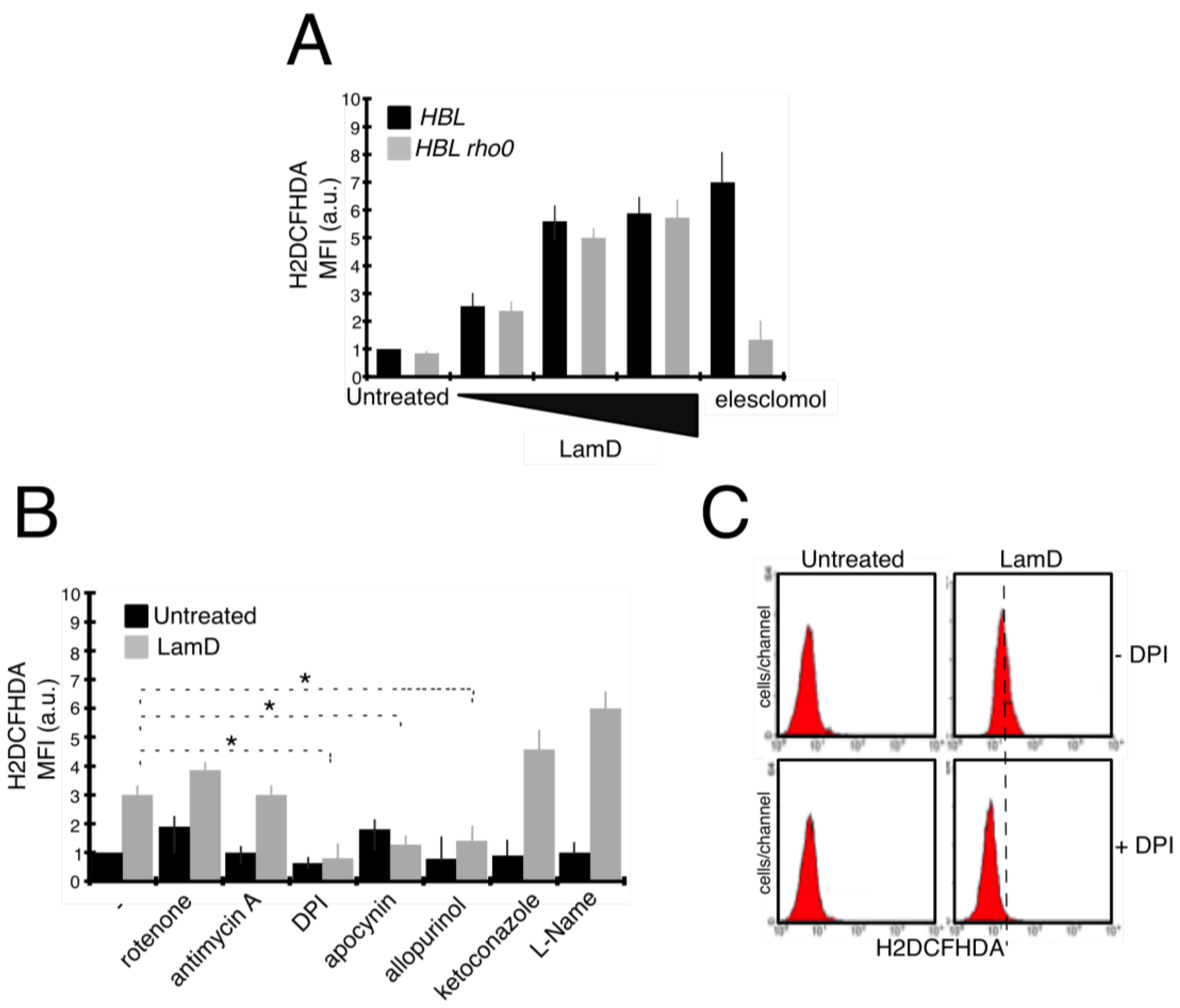
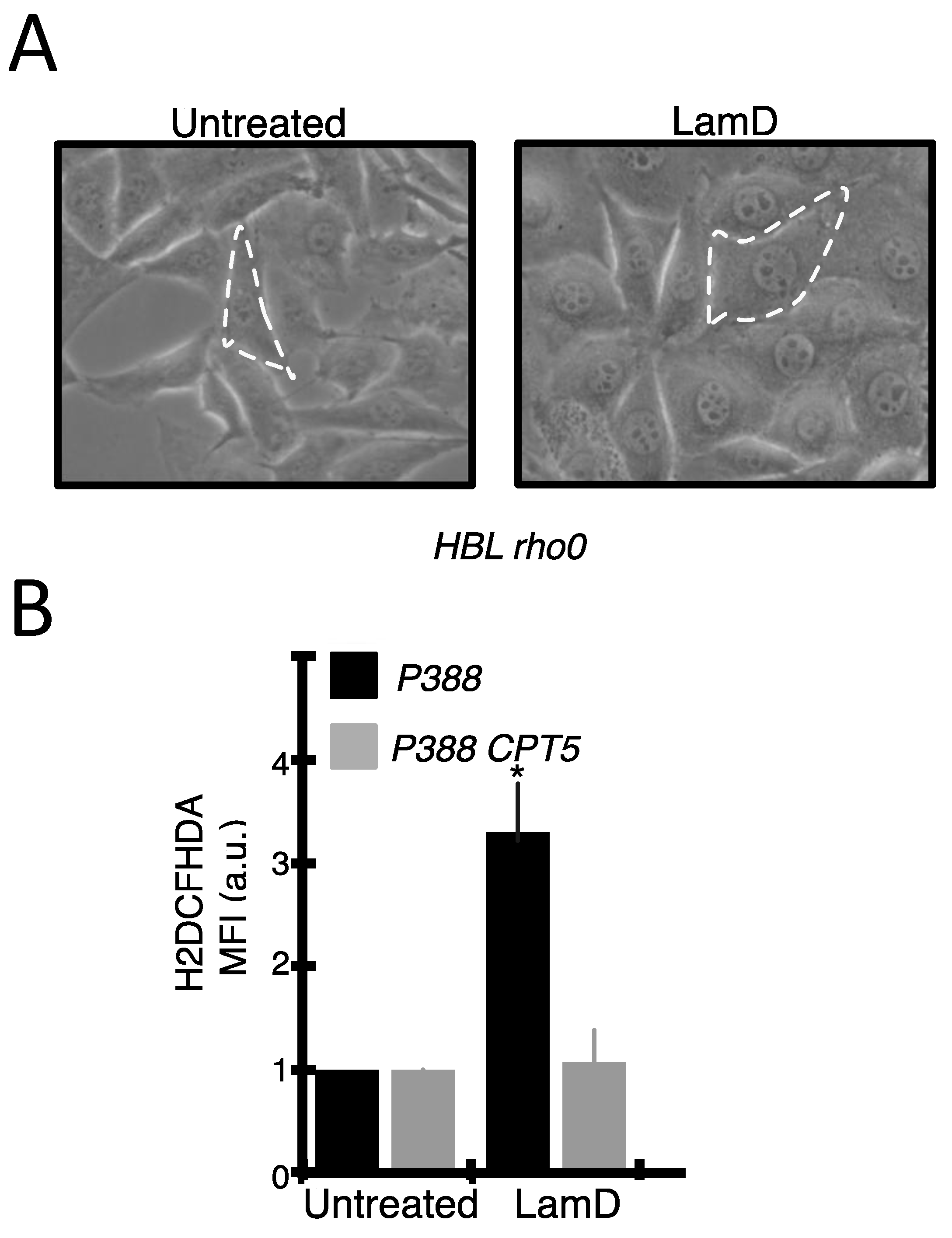
2.3. Lamellarin D Induced Extra-Mitochondrial ROS Accumulation in Senescent Cells


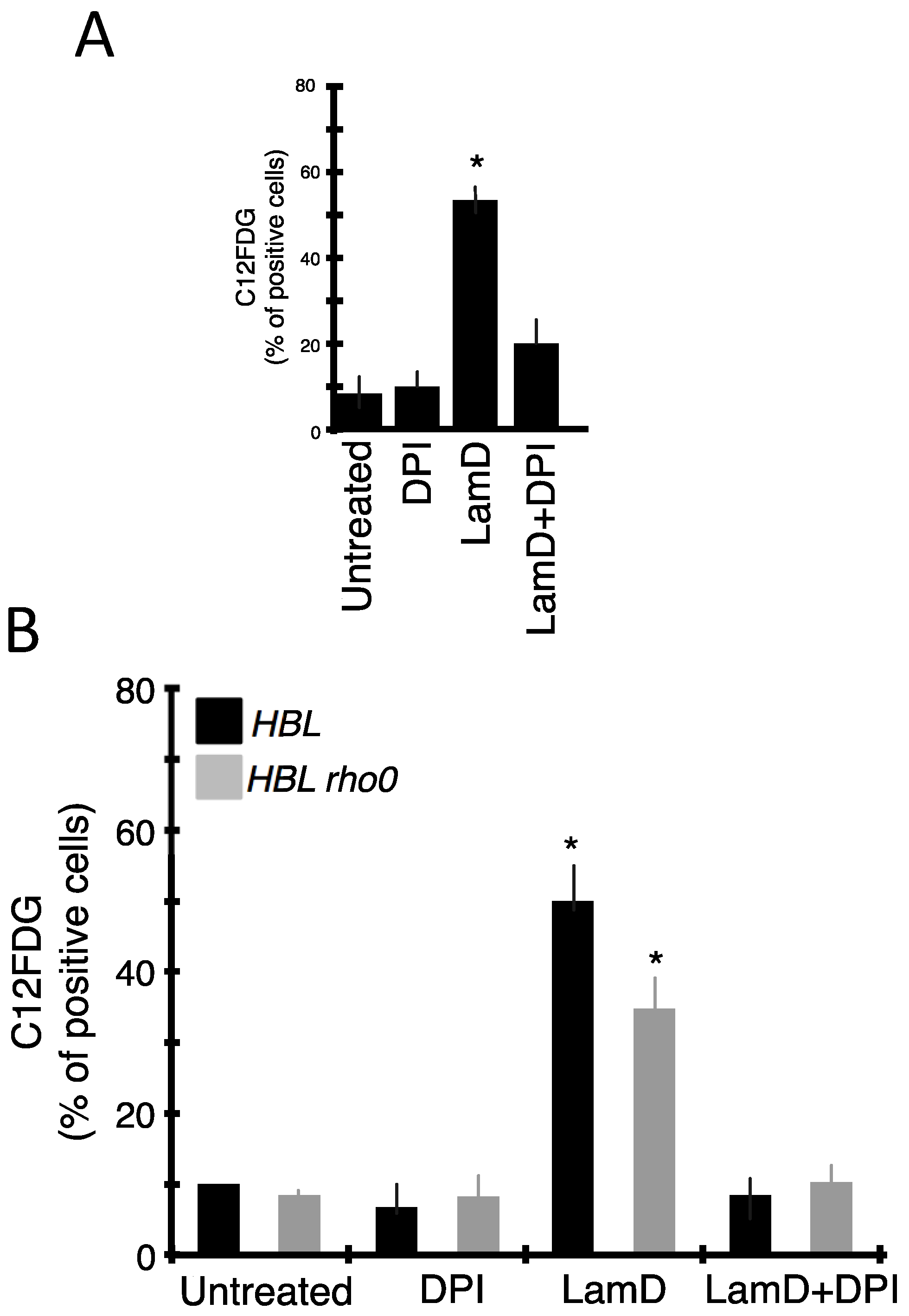
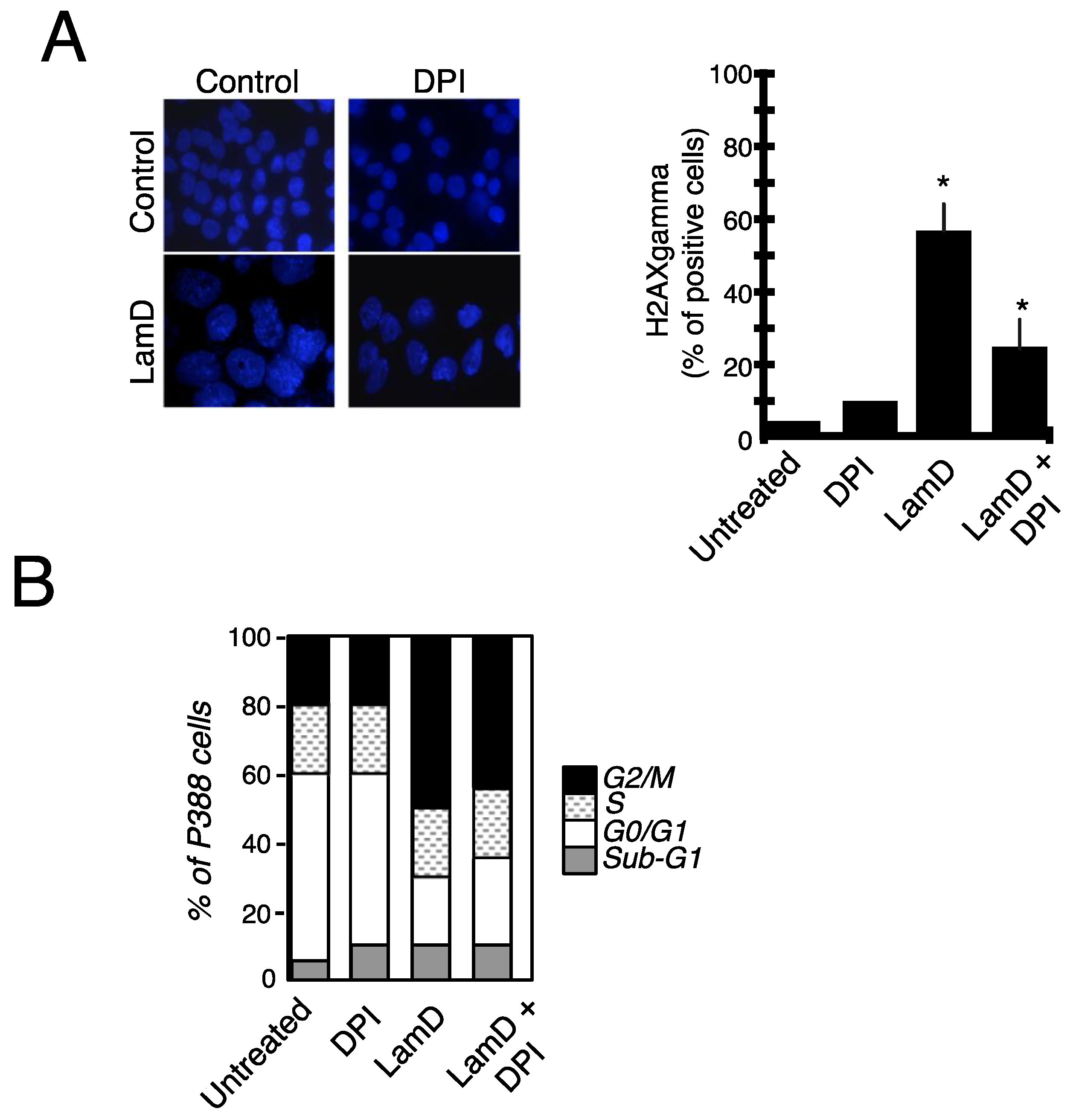
2.4. Role of ROS Production in Senescence and DNA Damage Induced by LamD



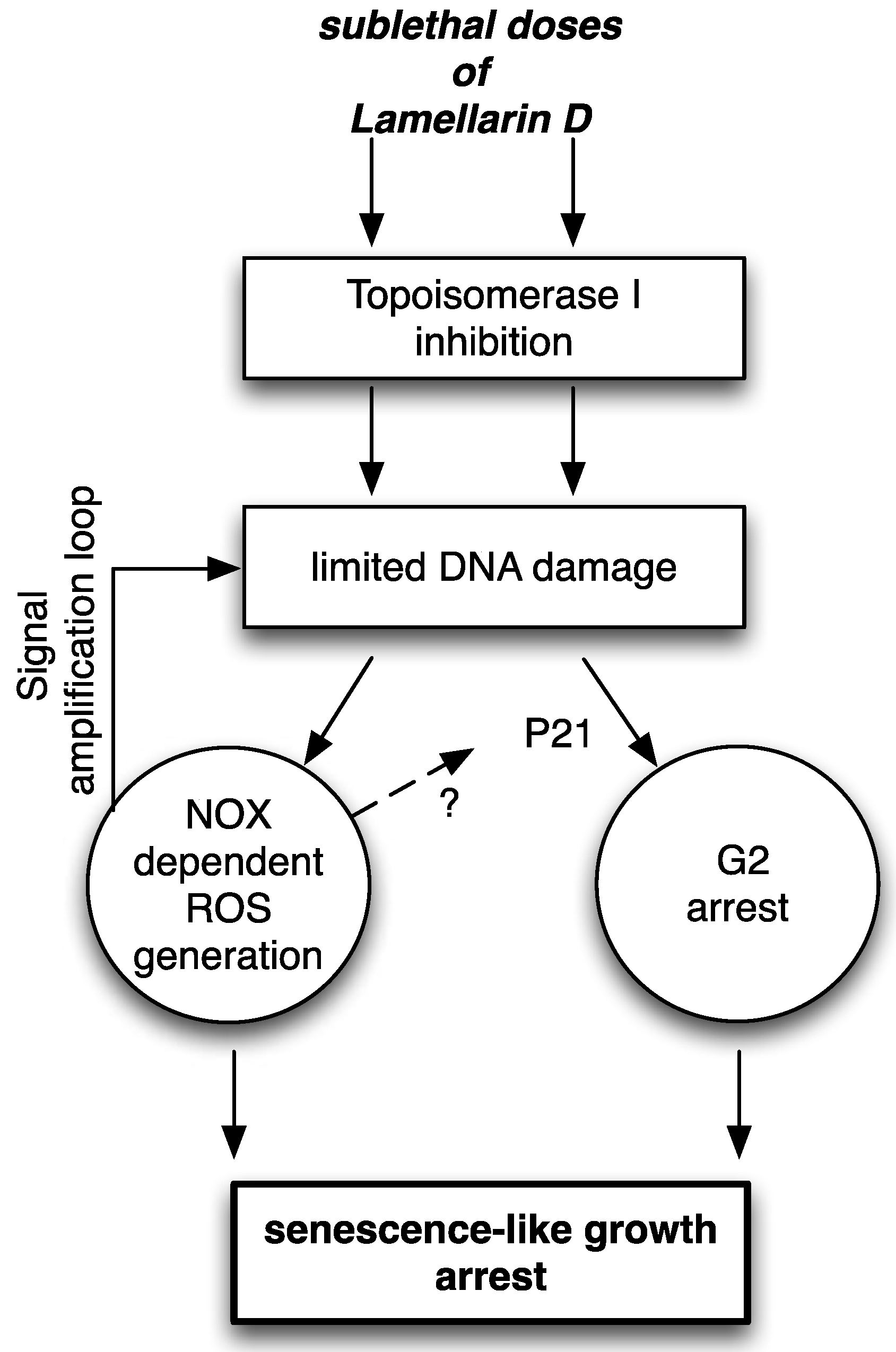
2.5. Proposed Mechanisms of LamD-Induced Senescence

3. Experimental Section
3.1. Chemicals
3.2. Cell Culture
3.3. Microscopic Analysis of Cells
3.4. Flow Cytometric Analysis
3.5. Determination of Intracellular Glutathione
3.6. RT-PCR Analysis
3.7. Immunoblot Analysis
3.8. Measurement of Intracellular ATP
3.9. Telomerase Repeat Amplification Protocol (TRAP) Assay
3.10. In Vivo Experiments
3.11. Statistics
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Simmons, T.L.; Andrianasolo, E.; McPhail, K.; Flatt, P.; Gerwick, W.H. Marine natural products as anticancer drugs. Mol. Cancer Ther. 2005, 4, 333–342. [Google Scholar]
- Xiong, Z.-Q.; Wang, J.-F.; Hao, Y.-Y.; Wang, Y. Recent advances in the discovery and development of marine microbial natural products. Mar. Drugs 2013, 11, 700–717. [Google Scholar] [CrossRef]
- Cioffi, A.; Italiano, A. Clinical and pharmacokinetic evaluation of trabectedin for the treatment of soft-tissue sarcoma. Expert Opin. Drug Metab. Toxicol. 2012, 8, 113–122. [Google Scholar] [CrossRef]
- Kasper, B.; Schmitt, T.; Wuchter, P.; Dimitrakopoulou-Strauss, A.; Ho, A.D.; Egerer, G. The use of positron emission tomography in soft tissue sarcoma patients under therapy with trabectedin. Mar. Drugs 2009, 7, 331–340. [Google Scholar] [CrossRef]
- Suarez, Y.; González, L.; Cuadrado, A.; Berciano, M.; Lafarga, M.; Muñoz, A. Kahalalide F, a new marine-derived compound, induces oncosis in human prostate and breast cancer cells. Mol. Cancer Ther. 2003, 2, 863–872. [Google Scholar]
- Kluza, J.; Marchetti, P.; Bailly, C. Lamellarin Alkaloids: Structure, Isolation, Synthesis and Biology; Fattorusso, E., Taglialatela-Scafati, O., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 171–187. [Google Scholar]
- Bailly, C. Lamellarins, from A to Z: A family of anticancer marine pyrrole alkaloids. Curr. Med. Chem. Anticancer Agents 2004, 4, 363–378. [Google Scholar] [CrossRef]
- Kluza, J.; Gallego, M.-A.; Loyens, A.; Beauvillain, J.-C.; Sousa-Faro, J.-M.; Cuevas, C.; Marchetti, P.; Bailly, C. Cancer cell mitochondria are direct proapoptotic targets for the marine antitumor drug lamellarin D. Cancer Res. 2006, 66, 3177–3187. [Google Scholar] [CrossRef]
- Facompré, M.; Tardy, C.; Bal-Mahieu, C.; Colson, P.; Perez, C.; Manzanares, I.; Cuevas, C.; Bailly, C. Lamellarin D: A novel potent inhibitor of topoisomerase I. Cancer Res. 2003, 63, 7392–7399. [Google Scholar]
- Baunbæk, D.; Trinkler, N.; Ferandin, Y.; Lozach, O.; Ploypradith, P.; Rucirawat, S.; Ishibashi, F.; Iwao, M.; Meijer, L. Anticancer alkaloid lamellarins inhibit protein kinases. Mar. Drugs 2008, 6, 514–527. [Google Scholar] [CrossRef]
- Ballot, C.; Kluza, J.; Lancel, S.; Martoriati, A.; Hassoun, S.M.; Mortier, L.; Vienne, J.-C.; Briand, G.; Formstecher, P.; Bailly, C.; et al. Inhibition of mitochondrial respiration mediates apoptosis induced by the anti-tumoral alkaloid lamellarin D. Apoptosis 2010, 15, 769–781. [Google Scholar] [CrossRef]
- Ballot, C.; Kluza, J.; Martoriati, A.; Nyman, U.; Formstecher, P.; Joseph, B.; Bailly, C.; Marchetti, P. Essential role of mitochondria in apoptosis of cancer cells induced by the marine alkaloid Lamellarin D. Mol. Cancer Ther. 2009, 8, 3307–3317. [Google Scholar] [CrossRef]
- Ballot, C.; Jendoubi, M.; Kluza, J.; Jonneaux, A.; Laine, W.; Formstecher, P.; Bailly, C.; Marchetti, P. Regulation by survivin of cancer cell death induced by F14512, a polyamine-containing inhibitor of DNA topoisomerase II. Apoptosis 2012, 17, 364–376. [Google Scholar] [CrossRef]
- Wang, Y.; Blandino, G.; Oren, M.; Givol, D. Induced p53 expression in lung cancer cell line promotes cell senescence and differentially modifies the cytotoxicity of anti-cancer drugs. Oncogene 1998, 17, 1923–1930. [Google Scholar]
- Dimri, G.P. What has senescence got to do with cancer? Cancer Cell. 2005, 7, 505–512. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Kluza, J.; Corazao-Rozas, P.; Touil, Y.; Jendoubi, M.; Maire, C.; Guerreschi, P.; Jonneaux, A.; Ballot, C.; Balayssac, S.; Valable, S.; et al. Inactivation of the HIF-1α/PDK3 signaling axis drives melanoma toward mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res. 2012, 72, 5035–5047. [Google Scholar] [CrossRef]
- Han, Z. Role of p21 in apoptosis and senescence of human colon cancer cells treated with camptothecin. J. Biol. Chem. 2002, 277, 17154–17160. [Google Scholar] [CrossRef]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial oxidative stress: Implications for cell death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef]
- Blackman, R.K.; Cheung-Ong, K.; Gebbia, M.; Proia, D.A.; He, S.; Kepros, J.; Jonneaux, A.; Marchetti, P.; Kluza, J.; Rao, P.E.; et al. Mitochondrial electron transport is the cellular target of the oncology drug elesclomol. PLoS One 2012, 7, e29798. [Google Scholar] [CrossRef]
- Chang, B.D.; Broude, E.V.; Dokmanovic, M.; Zhu, H.; Ruth, A.; Xuan, Y.; Kandel, E.S.; Lausch, E.; Christov, K.; Roninson, I.B. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999, 59, 3761–3767. [Google Scholar]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef]
- Gewirtz, D.A.; Holt, S.E.; Elmore, L.W. Accelerated senescence: An emerging role in tumor cell response to chemotherapy and radiation. Biochem. Pharmacol. 2008, 76, 947–957. [Google Scholar] [CrossRef]
- Pelicano, H.; Carney, D.A.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updat. 2004, 7, 97–110. [Google Scholar] [CrossRef]
- Achuthan, S.; Santhoshkumar, T.R.; Prabhakar, J.; Nair, S.A.; Pillai, M.R. Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J. Biol. Chem. 2011, 286, 37813–37829. [Google Scholar]
- Probin, V.; Wang, Y.; Zhou, D. Busulfan-induced senescence is dependent on ROS production upstream of the MAPK pathway. Free Radic. Biol. Med. 2007, 42, 1858–1865. [Google Scholar] [CrossRef]
- Lambeth, J.D.; Krause, K.H.; Clark, R.A. NOX enzymes as novel targets for drug development. Semin. Immunopathol. 2008, 30, 339–363. [Google Scholar] [CrossRef]
- Weyemi, U.; Lagente-Chevallier, O.; Boufraqech, M.; Prenois, F.; Courtin, F.; Caillou, B.; Talbot, M.; Dardalhon, M.; Al Ghuzlan, A.; Bidart, J.-M.; et al. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 2011, 31, 1117–1129. [Google Scholar]
- Luo, H.; Wang, L.; Schulte, B.A.; Yang, A.; Tang, S.; Wang, G.Y. Resveratrol enhances ionizing radiation-induced premature senescence in lung cancer cells. Int. J. Oncol. 2013, 43, 1999–2006. [Google Scholar]
- Kang, M.A.; So, E.-Y.; Simons, A.L.; Spitz, D.R.; Ouchi, T. DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death Dis. 2012, 3, e249. [Google Scholar] [CrossRef]
- Madelaine, I.; Prost, S.; Naudin, A.; Riou, G.; Lavelle, F.; Riou, J.F. Sequential modifications of topoisomerase I activity in a camptothecin-resistant cell line established by progressive adaptation. Biochem. Pharmacol. 1993, 45, 339–348. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated β-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Huang, X.; Darzynkiewicz, Z. Cytometric assessment of histone H2AX phosphorylation: A reporter of DNA damage. Methods Mol. Biol. 2006, 314, 73–80. [Google Scholar] [CrossRef]
- Castera, L.; Hatzfeld-Charbonnier, A.S.; Ballot, C.; Charbonnel, F.; Dhuiege, E.; Velu, T.; Formstecher, P.; Mortier, L.; Marchetti, P. Apoptosis-related mitochondrial dysfunction defines human monocyte-derived dendritic cells with impaired immuno-stimulatory capacities. J. Cell. Mol. Med. 2009, 13, 1321–1335. [Google Scholar] [CrossRef]
- Elmore, L.W.; Rehder, C.W.; Di, X.; McChesney, P.A.; Jackson-Cook, C.K.; Gewirtz, D.A.; Holt, S.E. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J. Biol. Chem. 2002, 277, 35509–35515. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ballot, C.; Martoriati, A.; Jendoubi, M.; Buche, S.; Formstecher, P.; Mortier, L.; Kluza, J.; Marchetti, P. Another Facet to the Anticancer Response to Lamellarin D: Induction of Cellular Senescence through Inhibition of Topoisomerase I and Intracellular Ros Production. Mar. Drugs 2014, 12, 779-798. https://doi.org/10.3390/md12020779
Ballot C, Martoriati A, Jendoubi M, Buche S, Formstecher P, Mortier L, Kluza J, Marchetti P. Another Facet to the Anticancer Response to Lamellarin D: Induction of Cellular Senescence through Inhibition of Topoisomerase I and Intracellular Ros Production. Marine Drugs. 2014; 12(2):779-798. https://doi.org/10.3390/md12020779
Chicago/Turabian StyleBallot, Caroline, Alain Martoriati, Manel Jendoubi, Sébastien Buche, Pierre Formstecher, Laurent Mortier, Jérome Kluza, and Philippe Marchetti. 2014. "Another Facet to the Anticancer Response to Lamellarin D: Induction of Cellular Senescence through Inhibition of Topoisomerase I and Intracellular Ros Production" Marine Drugs 12, no. 2: 779-798. https://doi.org/10.3390/md12020779
APA StyleBallot, C., Martoriati, A., Jendoubi, M., Buche, S., Formstecher, P., Mortier, L., Kluza, J., & Marchetti, P. (2014). Another Facet to the Anticancer Response to Lamellarin D: Induction of Cellular Senescence through Inhibition of Topoisomerase I and Intracellular Ros Production. Marine Drugs, 12(2), 779-798. https://doi.org/10.3390/md12020779