3.1. Materials and Synthetic Procedure
Melting points were determined using a Melt-Temp II melting point apparatus and are uncorrected. IR spectra were recorded using a Nicolet FT Magna-IR 750 spectrometer (Thermo, Madison, WI, USA). The 1H-NMR spectra were recorded using the Gemini-200 MHz Varian (Varian, Palo Alto, CA, USA) and a Jeol Eclipse 300 (Jeol Ltd., Tokyo, Japan), and samples were prepared in deuterated chloroform solutions containing tetramethylsilane or deuterated dimethylsulfoxide. 13C-NMR spectra were recorded at 75 MHz on the same instruments. The peak patterns are indicated as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; bs, broad singlet. The coupling constants (J) are reported in Hertz (Hz). Mass spectra were obtained using a Jeol JMS-AX505HA and Jeol 5X102A mass spectrometers (Jeol Ltd.) by electronic impact (EI) and fast atom bombardment (FAB+) ionization. The purification of compounds by column chromatography was performed on Silica Gel 60 F254 Merck (Merck, Darmstadt, Gemany). Commercial-grade reagents were used with further purification.
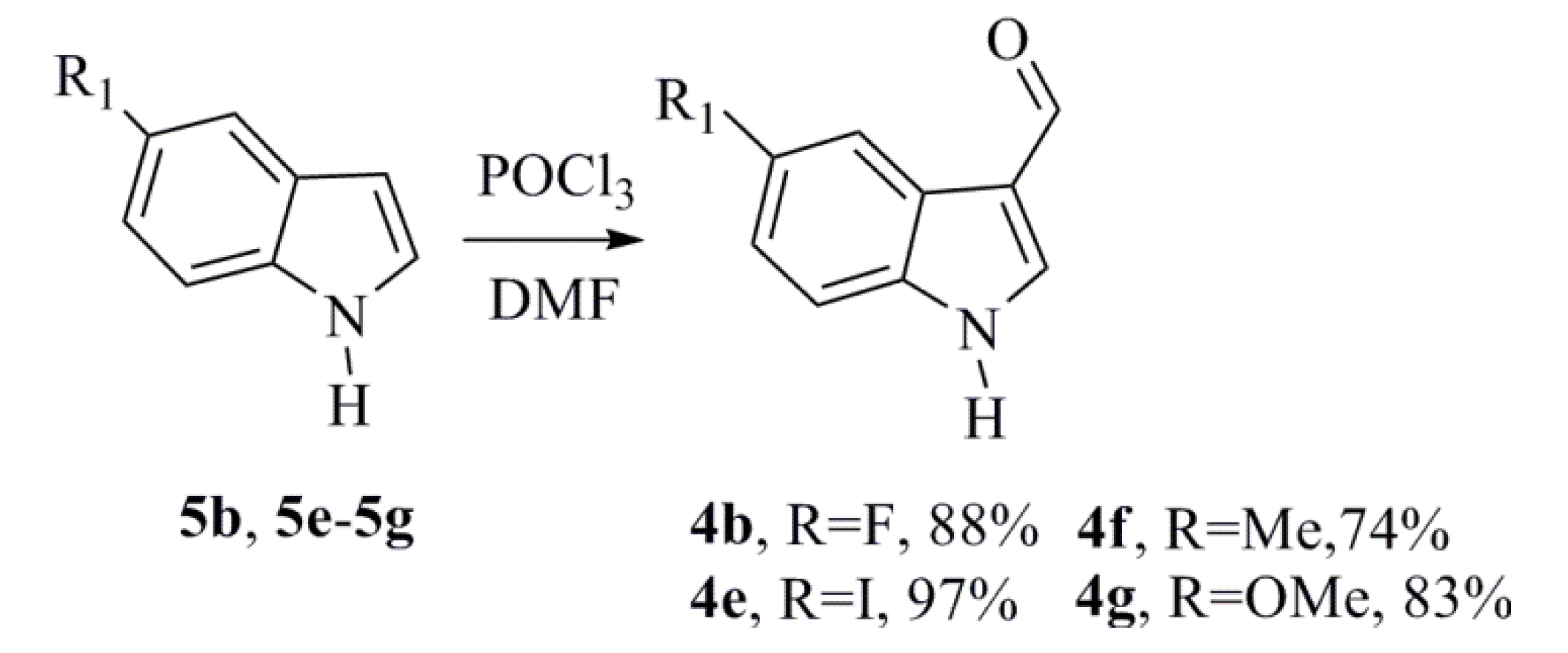
3.1.1. General Procedure for Synthesizing the Indoles-3-Carboxaldehydes (4b and 4e–4g)
Phosphorus oxychloride (0.42 g, 2.74 mmol) was added dropwise to a solution of the indole
5b,
5e–
5g (0.30 g, 2.29 mmol) in DMF (0.84 g, 11.4 mmol) at 0 °C for 30 min. The solution was then heated at 40 °C for 1 h. Ice was added to the reaction vessel, followed by a solution of sodium hydroxide (2 M). The solution was refluxed for 40 min. The mixture was cooled and extracted using ethyl acetate, and the organic phase was washed with brine. The organic extracts were combined, dried over Na
2SO
4, and concentrated. The crude residue was purified by chromatography on a silica gel column using hexane-ethyl acetate as an eluent to obtain the desired product [
19].
The indole derivatives 4a, 4c, and 4d were purchased from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO, USA).
5-Fluoro-1H-indole-3-carbaldehyde (4b)
Yellow solid; yield 88%; mp 158–160 °C;
1H-NMR (300 MHz, CDCl
3 + DMSO-
d6)
δ 11.72 (bs, 1H), 9.96 (s, 1H), 7.92 (d,
J = 3.0 Hz, 1H), 7.88 (dd,
J = 9.4, 2.4 Hz, 1H), 7.42 (dd,
J = 8.8, 4.2 Hz, 1H), 7.01 (dt,
J = 9.0, 2.4 Hz, 1H) [
24].
5-Iodo-1H-indole-3-carbaldehyde (4e)
Yellow solid; yield 97%; mp 192–194 °C;
1H-NMR (300 MHz, CDCl
3 + DMSO-
d6)
δ 11.72 (s, 1H), 9.95 (bs, 1H), 8.59 (d,
J = 1.8 Hz, 1H), 7.84 (d,
J = 3.0 Hz, 1H), 7.53 (dd,
J = 8.5, 1.8 Hz, 1H), 7.27 (d,
J = 8.4 Hz, 1H) [
25].
5-Methyl-1H-indole-3-carbaldehyde (4f)
Yellow solid; yield 74%; mp 144–146 °C;
1H-NMR (300 MHz, CDCl
3)
δ 10.02 (s, 1H), 9.21 (bs, 1H), 8.13 (s, 1H), 7.81 (d,
J = 3.3 Hz, 1H), 7.34 (d,
J = 8.4 Hz, 1H), 7.15 (d,
J = 8.4 Hz, 1H), 2.47 (s, 3H) [
26].
5-Methoxy-1H-indole-3-carbaldehyde (4g)
Light brown solid; yield 83%; mp 176–178 °C;
1H-NMR (200 MHz, CDCl
3 + DMSO-
d6)
δ 12.08 (bs, 1H), 9.93 (s, 1H), 8.25 (s, 1H), 7.62 (d,
J = 2.6 Hz, 1H), 7.44 (d,
J = 8.8 Hz, 1H), 6.92 (dd,
J = 8.8, 2.6 Hz, 1H),3.82 (s, 3H) [
27].
3.1.2. Procedure for Synthesizing the Xanthate of Diethyl Malonate (3)
To a solution of diethyl chloromalonate (1 g, 5.14 mmol) in acetonitrile (6.4 mL) was added potassium ethyl xanthogenate (0.99 g, 6.17 mmol) at 0 °C, and the mixture was stirred for 1 h. The crude product was then concentrated under reduced pressure, washed with brine, and extracted with CH2Cl2. The organic extracts were combined, dried over Na2SO4, and concentrated in vacuum. The crude residue was purified by chromatography on a silica gel column using hexane-ethyl acetate as the eluent to obtain the desired product.
Diethyl 2-(Ethoxycarbonothioylthio)malonate (3)
Yellow oil; yield 97%;
1H-NMR (300 MHz, CDCl
3)
δ 5.28 (s, 1H), 4.65 (q,
J = 7.2 Hz, 2H), 4.27 (q,
J = 7.2 Hz, 4H), 1.42 (t,
J = 7.2 Hz, 3H), 1.30 (t,
J = 7.2 Hz, 6H);
13C-NMR (75 MHz, CDCl
3)
δ 210.2, 165.1, 71.0, 62.8, 56.3, 13.9, 13.6 [
28].
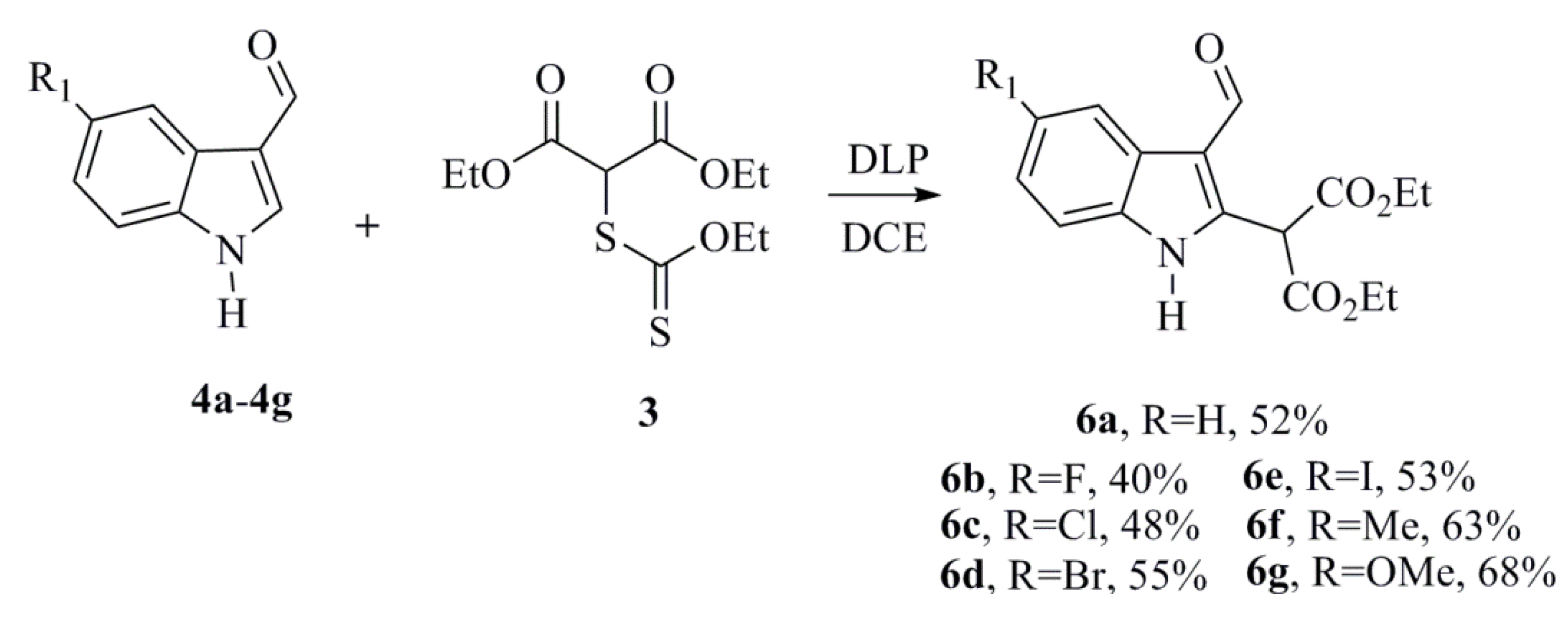
3.1.3. General Procedure for Synthesizing the Diethyl 2-(3-formyl-1H-indol-2-yl)malonate Derivatives (6a–6g)
A solution of the xanthate 3 (1.37 g, 4.87 mmol) and the corresponding indole (4a–4g) (0.353 g, 2.21 mmol) in degassed 1,2-dichloroethane (DCE) (9 mL) was heated at reflux, and dilauroyl peroxide (DLP) (2.21 g, 5.54 mmol) in solid form was added in several portions (0.92 mmol/h). The reaction was monitored by TLC and was stopped after 6 h. The solvent was removed under reduced pressure, and the crude residues were extracted with acetonitrile and washed with hexane. The polar phase was then concentrated under reduced pressure and purified by chromatography on a silica gel column using hexane-ethyl acetate as the eluent to obtain the desired products.
Diethyl 2-(3-Formyl-1H-indol-2-yl)malonate (6a)
Yellow solid; yield 52%; mp 70–72 °C; IR (KBr) νmax 3187, 1728, 1624 cm−1; 1H-NMR (300 MHz, CDCl3) δ 10.30 (s, 1H), 10.00 (bs, 1H), 8.20–8.17 (m, 1H), 7.46–7.26 (m, 3H), 5.74 (s, 1H), 4.35–4.19 (m, 4H), 1.29 (t, J = 7.2 Hz, 6H); 13C-NMR (75 MHz, CDCl3) δ 184.4, 166.3, 136.8, 135.3, 125.8, 124.2, 122.9, 120.2, 115.1, 111.8, 63.0, 49.0, 13.9; EIMS m/z 303 [M]+ (100), 157 (73).
Diethyl 2-(5-Fluoro-3-formyl-1H-indol-2-yl)malonate (6b)
Yellow solid; yield 40% (58% Based on Recovered Starting Material); mp 98–100 °C; IR (KBr) νmax 3149, 3108, 2983, 2933, 1738, 1637 cm−1; 1H-NMR (300 MHz, CDCl3) δ 10.23 (s, 1H), 10.10 (bs, 1H), 7.86 (dd, J = 9.3, 2.7 Hz, 1H), 7.37 (dd, J = 8.9, 4.2 Hz, 1H), 7.05 (dt, J = 9.0, 2.7 Hz, 1H), 5.69 (s, 1H), 4.37–4.20 (m, 4H), 1.30 (t, J = 6.9 Hz, 6H); 13C-NMR (75 MHz, CDCl3) δ 184.0, 166.2, 159.7 (d, JCF = 237.8 Hz, Ar-quat), 138.2, 131.7, 126.4 (d, JCF = 10.5 Hz, Ar-quat), 115.3 (d, JCF = 4.5 Hz, Ar-quat), 112.8 (d, JCF = 5.3 Hz, Ar-CH), 112.6 (d, JCF = 11.3 Hz, Ar-CH), 105.9 (d, JCF = 25 Hz, Ar-CH), 63.1, 49.0, 13.9; EIMS m/z 321 [M]+ (37), 181 (70), 115 (100); HRMS (FAB+) m/z calcd. for C16H17O5NF, 322.1091; found: 322.1082.
Diethyl 2-(5-Chloro-3-formyl-1H-indol-2-yl)malonate (6c)
Yellow solid; yield 48% (69% Based on Recovered Starting Material); mp 102–104 °C; IR (KBr) νmax 3139, 2979, 2826, 1734, 1633 cm−1; 1H-NMR (300 MHz, CDCl3) δ 10.24 (s, 1H), 10.00 (bs, 1H), 8.19 (d, J = 2.1 Hz, 1H), 7.36 (d, J = 8.7 Hz, 1H), 7.25 (dd, J = 8.7 Hz, 2.1 Hz, 1H), 5.68 (s, 1H), 4.37–4.20 (m, 4H), 1.31 (t, J = 7.2 Hz, 6H); 13C-NMR (75 MHz, CDCl3) δ 184.0, 166.1, 137.9, 133.6, 128.9, 126.7, 124.6, 120.1, 114.8, 112.8, 63.2, 48.9, 13.9; EIMS m/z 337 [M]+ (84), 338 (22), 339 (34), 340 (11); HRMS (FAB+) m/z calcd. for C16H17O5NCl, 338.0795; found: 338.0801.
Diethyl 2-(5-Bromo-3-formyl-1H-indol-2-yl)malonate (6d)
Yellow solid; yield 56%; mp 108–110 °C; IR (KBr) νmax 3159, 2989, 1732, 1634 cm−1; 1H-NMR (300 MHz, CDCl3) δ 10.23 (s, 1H), 10.08 (bs, 1H), 8.35 (d, J = 1.8 Hz, 1H), 7.39 (dd, J = 8.7, 1.8 Hz, 1H), 7.31 (d, J = 8.7 Hz, 1H), 5.68 (s, 1H), 4.37–4.20 (m, 4H), 1.31 (t, J = 7.2 Hz, 6H); 13C-NMR (75 MHz, CDCl3) δ 184.0, 166.1, 137.8, 133.9, 127.3, 123.1, 116.5, 114.7, 113.2, 63.2, 48.9, 13.9; EIMS m/z 381 [M]+ (66), 383 (65); HRMS (FAB+) m/z calcd. for C16H16O5NBr, 381.0212; found: 381.0213.
Diethyl 2-(3-Formyl-5-iodo-1H-indol-2-yl)malonate (6e)
Yellow solid; yield 53%; mp 90–92 °C; IR (KBr) νmax 3166, 2977, 2929, 1732, 1629 cm−1; 1H-NMR (300 MHz, CDCl3) δ 10.22 (s, 1H), 10.06 (bs, 1H), 8.56 (d, J = 1.5 Hz, 1H), 7.57 (dd, J = 8.5, 1.5 Hz, 1H), 7.22 (d, J = 8.4 Hz, 1H), 5.67 (s, 1H), 4.37–4.20 (m, 4H), 1.31 (t, J = 6.9 Hz, 6H); 13C-NMR (75 MHz, CDCl3) δ 184.0, 166.1, 137.4, 134.4, 132.8, 129.3, 127.9, 114.4, 113.6, 87.0, 63.2, 48.9, 13.9; EIMS m/z 429 [M]+ (100), 383 (87); HRMS (FAB+) m/z calcd. for C16H17O5NI, 430.0151; found: 430.0163.
Diethyl 2-(3-Formyl-5-methyl-1H-indol-2-yl)malonate (6f)
Yellow solid; yield 63%; mp 84–86 °C; IR (KBr) νmax 3218, 2986, 1734, 1636 cm−1; 1H-NMR (300 MHz, CDCl3) δ 10.27 (s, 1H), 9.84 (bs, 1H), 8.00 (s, 1H), 7.33 (d, J = 8.4 Hz, 1H), 7.13 (dd, J = 8.4, 1.5 Hz, 1H), 5.70 (s, 1H), 4.36–4.19 (m, 4H), 2.48 (s, 3H), 1.30 (t, J = 7.2 Hz, 6H); 13C-NMR (75 MHz, CDCl3) δ 184.3, 166.4, 136.8, 133.5, 132.7, 126.0, 125.8, 120.0, 114.8, 111.4, 63.0, 49.0, 21.5, 13.9; EIMS m/z 317 [M]+ (100), 271 (62); HRMS (FAB+) m/z calcd. for C17H20O5N, 318.1341; found: 318.1340.
Diethyl 2-(3-Formyl-5-methoxy-1H-indol-2-yl)malonate (6g)
Beige solid; yield 68%; mp 112–114 °C; IR (KBr) νmax 3176, 2980, 1732, 1628 cm−1; 1H-NMR (300 MHz, CDCl3) δ 10.24 (s, 1H), 9.94 (bs, 1H), 7.69 (d, J = 2.4 Hz, 1H), 7.32 (d, J = 8.7 Hz, 1H), 6.94 (dd, J = 8.9, 2.4 Hz, 1H), 5.64 (s, 1H), 4.36–4.19 (m, 4H), 3.88 (s, 3H), 1.30 (t, J = 7.2 Hz, 6H); 13C-NMR (75 MHz, CDCl3) δ 184.1, 166.3, 156.6, 137.1, 130.1, 126.4, 115.2, 114.7, 112.6, 102.0, 63.0, 55.8, 48.9, 13.9; EIMS m/z 333 [M]+ (64), 241 (100); HRMS (FAB+) m/z calcd. for C17H19O6N, 333.1212; found: 333.1211.
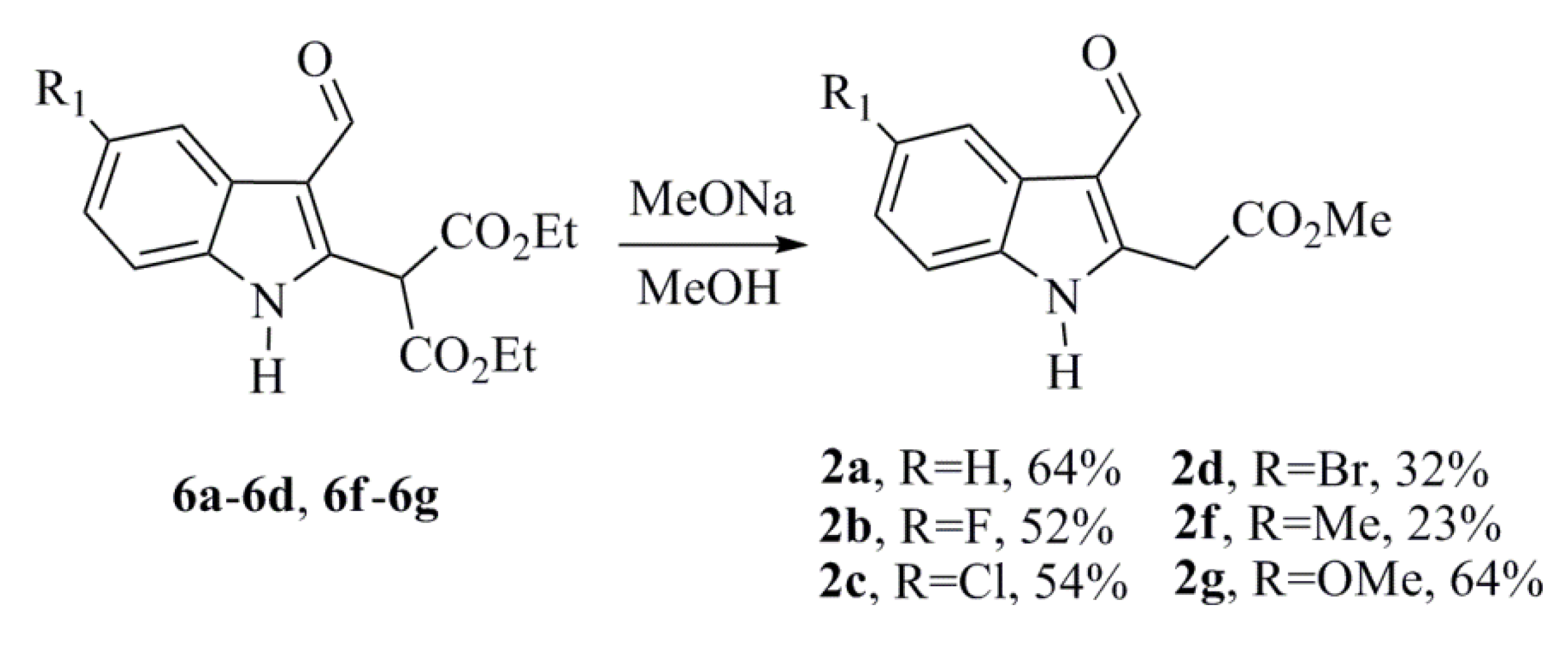
3.1.4. General Procedure for Synthesizing the Methyl 2-(3-Formyl-1H-indol-2-yl) Acetate Derivatives (2a–2d and 2f–2g). This Reaction Procedure Was Designed Based on the Method Described by Somei et al. [21], with Slight Changes
To a solution of NaOMe (prepared from sodium, 0.105 g, 4.56 mmol, and anhydrous MeOH, 5 mL) was added a solution containing the corresponding diethyl malonates (6a–6d and 6f–6g) (0.413 g, 1.30 mmol) in anhydrous MeOH (14 mL). The mixture was then refluxed for 1 h with stirring. After cooling, a saturated solution of NaHCO3 was added to the reaction mixture, and the crude product was extracted with CH2Cl2. The crude extract was washed with anhydrous Na2SO4 and evaporated under reduced pressure. The residue was purified by column chromatography on a silica gel column using hexane-ethyl acetate as the eluent to obtain the desired product.
Methyl 2-(3-Formyl-1H-indol-2-yl)acetate (2a)
Brown solid; yield 64%; mp 114–116 °C; IR (KBr) ν
max 3187, 2946, 1745, 1629 cm
−1;
1H-NMR (300 MHz, CDCl
3)
δ 10.22 (s, 1H), 10.03 (bs, 1H), 8.19–8.14 (m, 1H), 7.42–7.24 (m, 3H), 4.25 (s, 2H), 3.79 (s, 3H);
13C-NMR (75 MHz, CDCl
3)
δ 184.4, 170.3, 140.2, 135.1, 126.0, 123.7, 122.8, 120.0, 114.6, 111.5, 52.7, 31.3; EIMS
m/
z 217 [M]
+ (23), 185 (100) [
22].
Methyl 2-(5-Fluoro-3-formyl-1H-indol-2-yl)acetate (2b)
Yellow solid; yield 52%; mp 168–170 °C; IR (KBr) νmax 3159, 2952, 1746, 1623 cm−1; 1H-NMR (300 MHz, CDCl3) δ 11.78 (bs, 1H), 10.10 (s, 1H), 7.81 (dd, J = 9.6, 2.4 Hz, 1H), 7.37 (dd, J = 8.7, 4.5 Hz, 1H), 6.97 (dt, J = 9.0, 2.4 Hz, 1H), 4.17 (s, 2H), 3.75 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ 183.3, 168.4, 158.6 (d, JCF = 235.5 Hz, Ar-quat), 142.2, 131.5, 125.7 (d, JCF = 11.3 Hz, Ar-quat), 114.1 (d, JCF = 4.5 Hz, Ar-quat), 112.0 (d, JCF = 9.8 Hz, Ar-CH), 110.6 (d, JCF = 26.3 Hz, Ar-CH), 105.0 (d, JCF = 25 Hz, Ar-CH), 51.8, 31.4; EIMS m/z 235 [M]+ (100); HRMS (FAB+) m/z calcd. for C12H11O3NF, 236.0723; found: 236.0723.
Methyl 2-(5-Chloro-3-formyl-1H-indol-2-yl)acetate (2c)
Yellow solid; yield 54%; mp 160–162 °C; IR (KBr) νmax 3140, 2945, 1745, 1637 cm−1; 1H-NMR (300 MHz, CDCl3 + DMSO-d6) δ 11.60 (bs, 1H), 10.12 (s, 1H), 8.16 (d, J = 2.1 Hz, 1H), 7.35 (d, J = 8.7 Hz, 1H), 7.18 (dd, J = 8.7, 2.1 Hz, 1H), 4.16 (s, 2H), 3.75 (s, 3H); 13C-NMR (75 MHz, CDCl3 + DMSO-d6) δ 183.7, 168.9, 142.1, 133.8, 127.8, 126.6, 123.3, 119.6, 114.0, 112.5, 52.2, 31.7; EIMS m/z 251 [M]+ (85), 252 (20), 253 (34), 254 (7); HRMS (FAB+) m/z calcd. for C12H11O3NCl, 252.0427; found: 252.0422.
Methyl 2-(5-Bromo-3-formyl-1H-indol-2-yl)acetate (2d)
Yellow solid; yield 32%; mp 172–174 °C; IR (KBr) νmax 3132, 2945, 1745, 1637 cm−1; 1H-NMR (300 MHz, CDCl3 + DMSO-d6) δ 11.06 (bs, 1H), 10.15 (s, 1H), 8.33 (d, J = 1.8 Hz, 1H), 7.34 (dd, J = 8.5, 1.8 Hz, 1H), 7.28 (dd, J = 8.7, 0.6 Hz, 1H), 4.19 (s, 2H), 3.77 (s, 3H); 13C-NMR (75 MHz, CDCl3 + DMSO-d6) δ 183.9, 169.4, 141.6, 134.1, 127.5, 126.3, 122.9, 116.0, 114.1, 113.0, 52.6, 31.7; EIMS m/z 295 [M]+ (76), 297 (72); HRMS (FAB+) m/z calcd. for C12H11O3NBr, 295.9922; found: 295.9926.
Methyl 2-(3-Formyl-5-methyl-1H-indol-2-yl)acetate (2f)
Yellow solid; yield 23%; mp 124–126 °C; IR (KBr) νmax 3164, 2920, 1738, 1617 cm−1; 1H-NMR (300 MHz, CDCl3) δ 10.20 (s, 1H), 9.85 (bs, 1H), 7.98 (s, 1H), 7.28 (d, J = 8.1 Hz, 1H), 7.09 (d, J = 8.4 Hz, 1H), 4.24 (s, 2H), 3.80 (s, 3H), 2.46 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ 184.3, 170.4, 140.1, 133.4, 132.5, 126.2, 125.2, 119.9, 114.3, 111.1, 52.7, 31.3, 21.5; EIMS m/z 231 [M] + (100), 199 (85); HRMS (FAB+) m/z calcd. for C13H13O3N, 231.0895; found: 231.0898.
Methyl 2-(3-Formyl-5-methoxy-1H-indol-2-yl)acetate (2g)
Beige solid; yield 64%; mp 126–128 °C; IR (KBr) νmax 3314, 2950, 1727, 1641 cm−1; 1H-NMR (300 MHz, CDCl3) δ 10.16 (s, 1H), 9.92 (bs, 1H), 7.67 (d, J = 2.4 Hz, 1H), 7.27 (d, J = 9.0 Hz, 1H), 6.89 (dd, J = 8.8, 2.7 Hz, 1H), 4.21 (s, 2H), 3.87 (s, 3H), 3.79 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ 184.1, 170.2, 156.5, 140.5, 129.9, 126.7, 114.7, 113.9, 112.2, 102.1, 55.7, 52.7, 31.2; EIMS m/z 247 [M]+ (82), 215 (100); HRMS (FAB+) m/z calcd. for C13H13O4N, 247.0845; found: 247.0836.
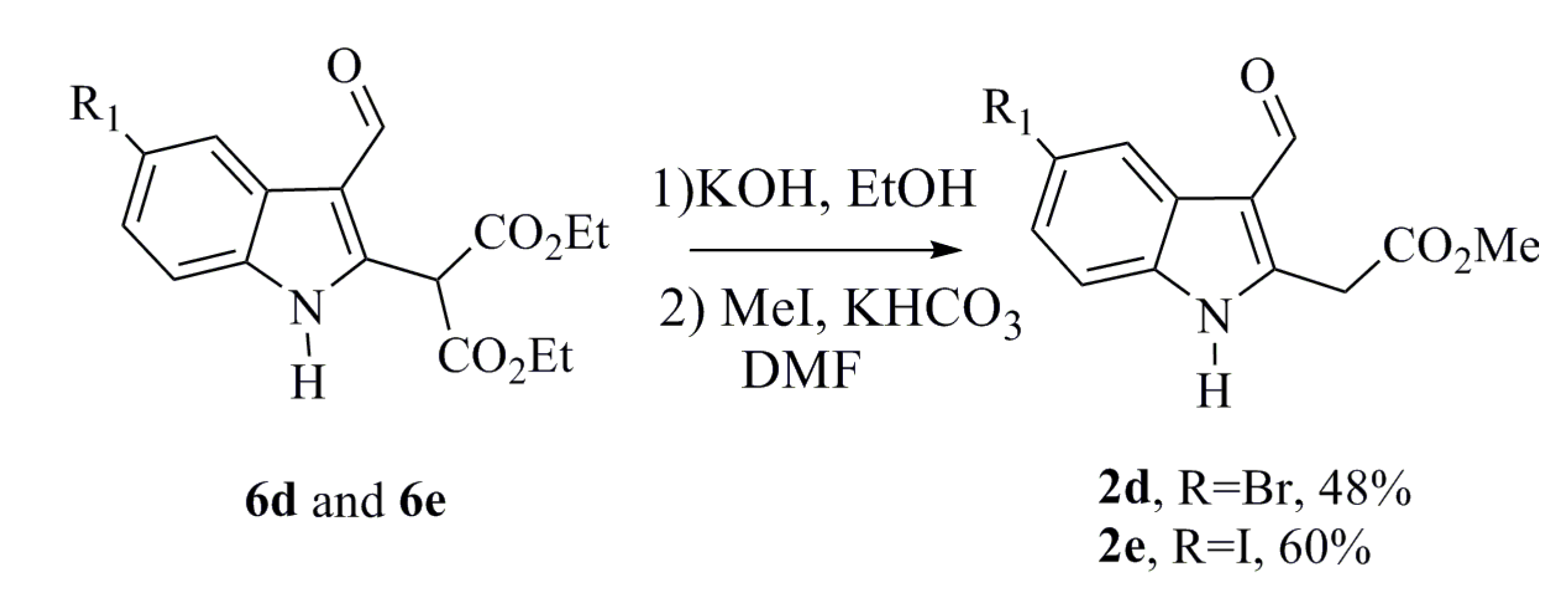
3.1.5. General Procedure for Synthesizing 2d and 2e
To a solution of the diethyl indole malonate (
6d or
6e) (0.350 g, 0.815 mmol) in ethanol (2.3 mL) was added a solution of KOH (2 M) (1.8 mL, 3.67 mmol). The solution was then stirred at room temperature for 21 h. A solution of HCl 2 N was then added and refluxed for 2 h. Subsequently, 20 mL of a saturated solution of Na
2CO
3 were added to the reaction mixture. The crude product was extracted with ethyl acetate, and the organic phases were combined and concentrated under reduced pressure. The esterification step was accomplished by hydrolyzing and decarboxylating the residue. This transformation was accomplished by dissolving crude acetic acid (0.213 g, 0.647 mmol) in DMF (1.5 mL). Potassium bicarbonate (KHCO
3) (0.097 g, 0.970 mmol) and MeI (0.137 g, 0.970 mmol) were added, and the reaction was stirred for 5 h at room temperature. To the reaction mixture was added 20 mL of a solution containing 1 M potassium bisulfate (KHSO
4), followed by the addition of ethyl acetate. The crude product was then extracted. The organic phases were combined and washed with an aqueous saturated solution of sodium bicarbonate (NaHCO
3) and brine. The organic extract was dried over Na
2SO
4 and concentrated under reduced pressure, and the crude product was purified by column chromatography using silica gel and a mixture hexane–ethyl acetate as the eluent to obtain the desired product [
22,
23].
Methyl 2-(5-Bromo-3-formyl-1H-indol-2-yl)acetate (2d)
Yellow solid; yield 48%; mp 172–174 °C.
Methyl 2-(3-Formyl-5-iodo-1H-indol-2-yl)acetate (2e)
Yellow solid; yield 60%; mp 168–170 °C; IR (KBr) νmax 3129, 2947, 2922, 1747, 1637 cm−1; 1H-NMR (300 MHz, CDCl3 + DMSO-d6) δ 11.73 (bs, 1H), 10.11 (s, 1H), 8.53 (s, 1H), 7.49 (dd, J = 8.5, 1.5 Hz, 1H), 7.22 (d, J = 8.4 Hz, 1H), 4.16 (s, 2H), 3.75 (s, 3H); 13C-NMR (75 MHz, CDCl3+ DMSO-d6) δ 183.6, 168.6, 141.6, 134.4, 131.2, 128.7, 127.6, 113.4, 113.3, 86.0, 52.1, 31.5; EIMS m/z 343 [M]+ (100), 311 (93); HRMS (FAB+) m/z calcd. for C12H11O3NI, 343.9784; found: 343.9786.
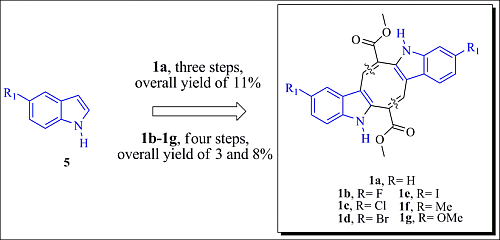
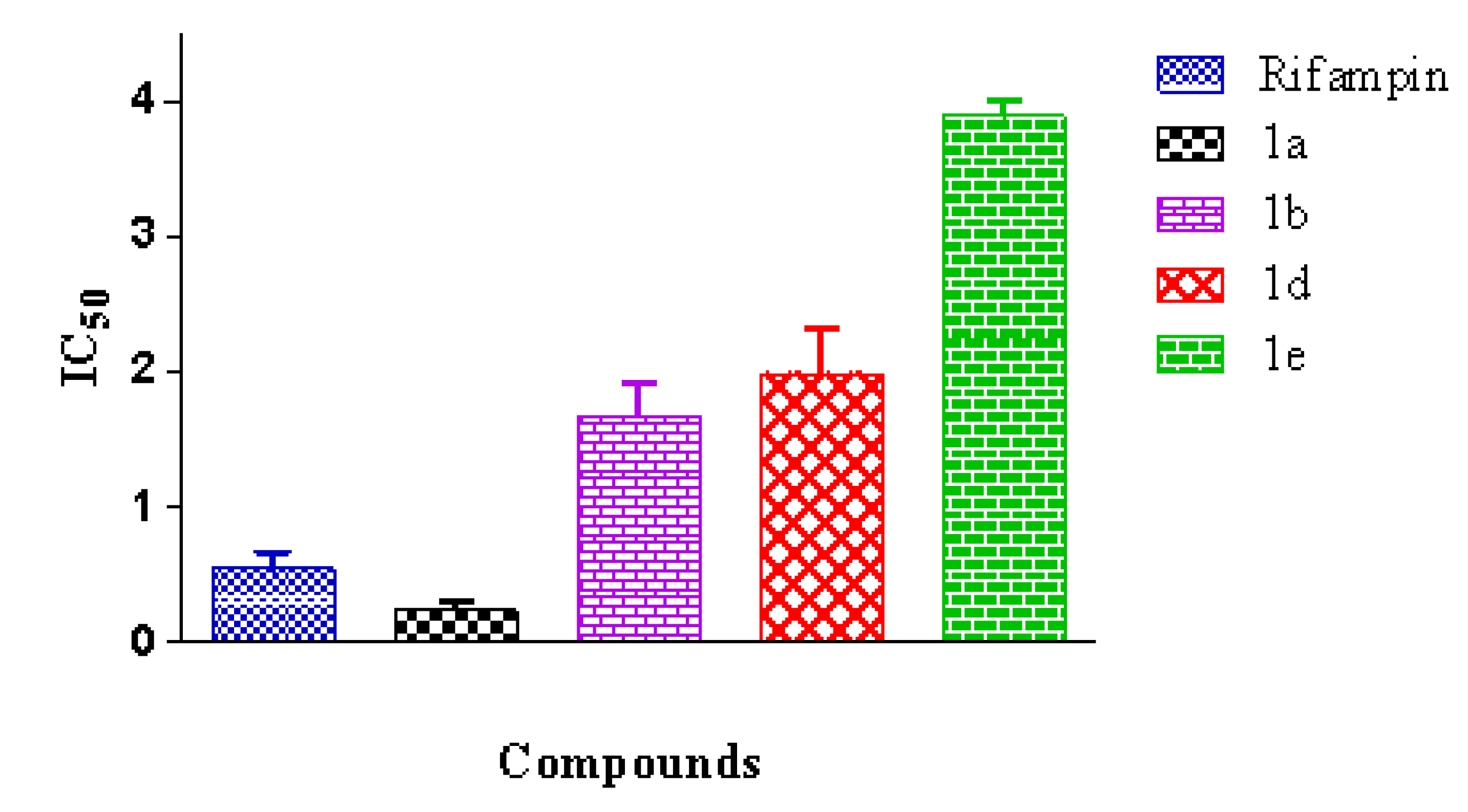
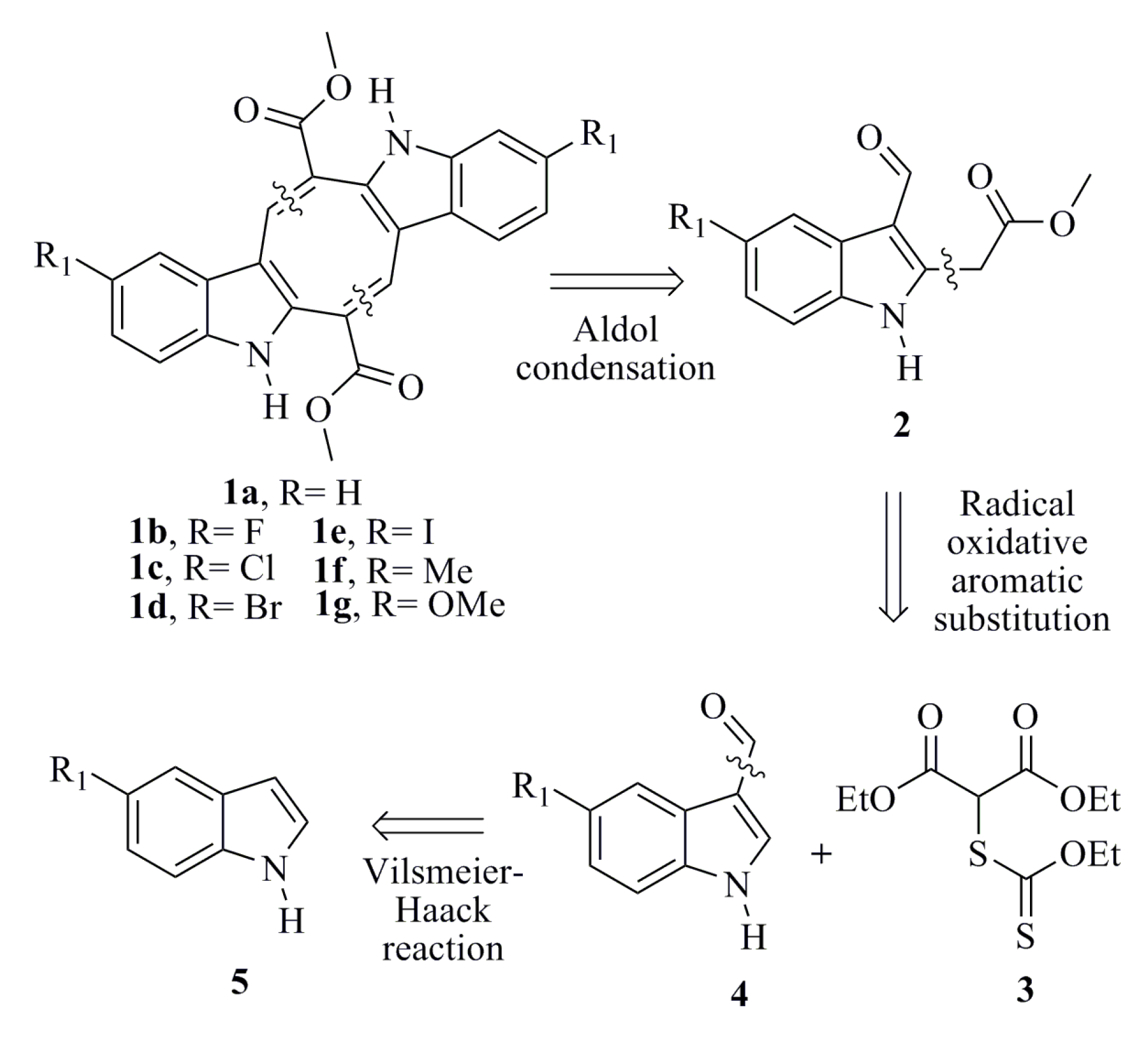
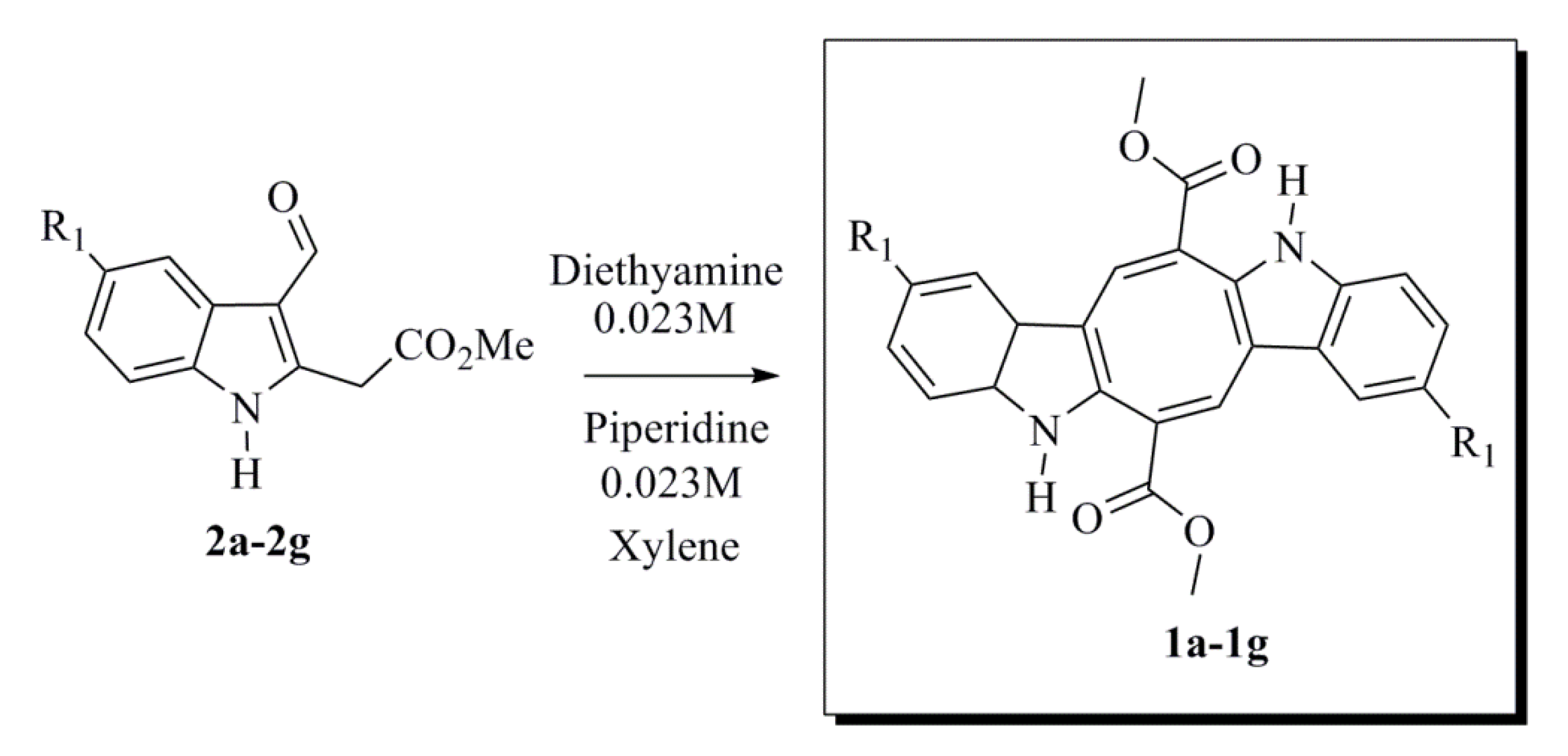
3.1.6. General Procedure for Synthesizing Caulerpin 1a and Its Analogues (1b–1g)
To a solution of the corresponding indole (2a–2g) (0.100 g, 0.432 mmol) in anhydrous xylene (14 mL/0.1mmol) were added piperidine (0.128 mL, 1.30 mmol) and diethylamine (0.134 mL, 1.30 mmol), and the mixture was refluxed during the removal of water using a Dean-Stark separator. After the starting material had been consumed, as indicated by TLC analysis, the solvent was concentrated in vacuum. The residue was purified by column chromatography using silica gel and a mixture of hexane-ethyl acetate as the eluent to obtain the desired product.
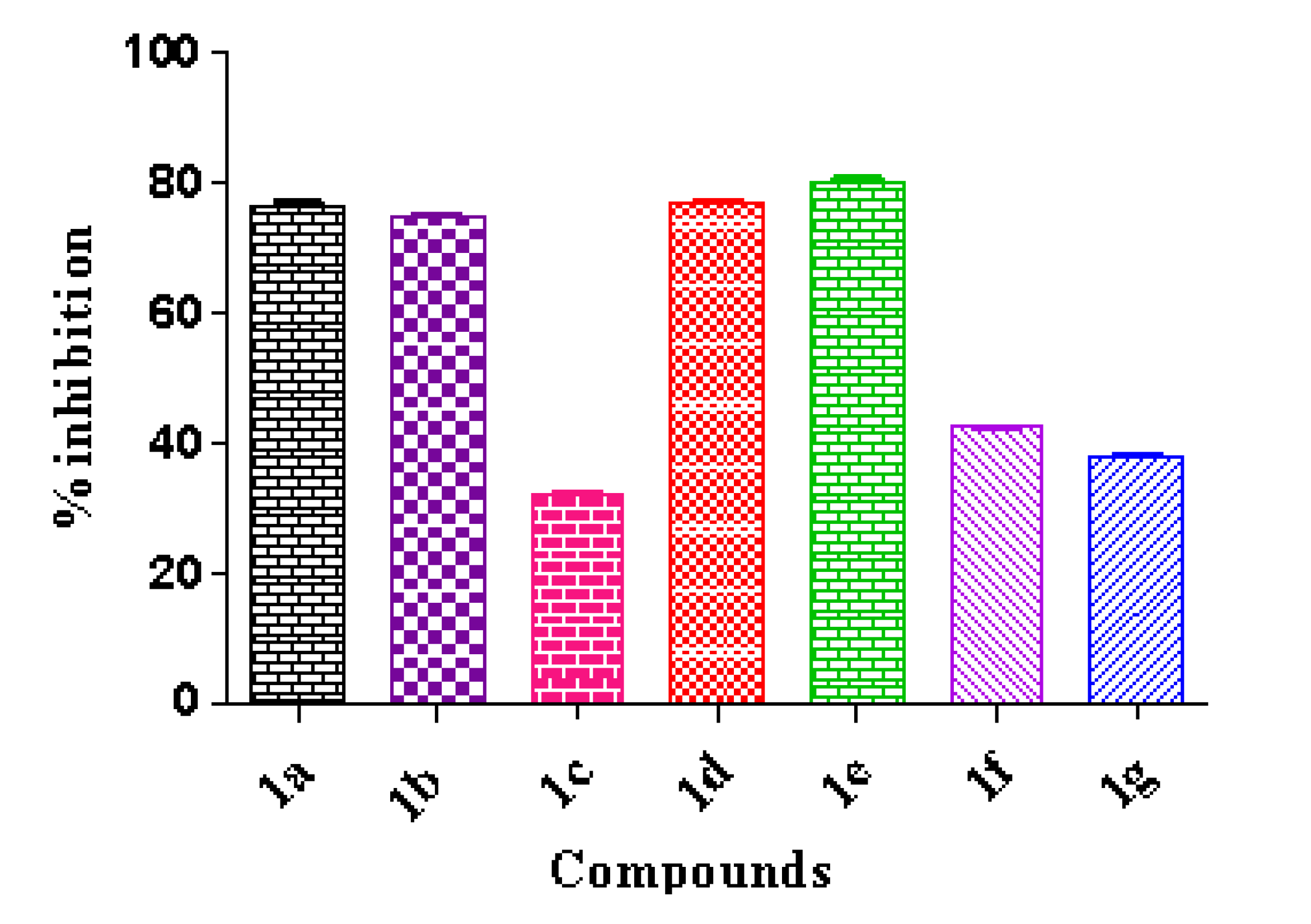
Dimethyl 5,12-Dihydrocycloocta[1,2-b:5,6-b′]-diindole-6,13-dicarboxylate (1a)
Red solid; yield 32%; mp 318 °C; IR (KBr) νmax 3377, 2917, 1681 cm−1; 1H-NMR (300 MHz, CDCl3 + DMSO-d6) δ 10.74 (bs, 2H), 8.18 (s, 2H), 7.42–7.32 (m, 4H), 7.12–7.03 (m, 4H), 3.84 (s, 6H); 13C-NMR (75 MHz, CDCl3 + DMSO-d6) δ 165.8, 141.7, 137.3, 132.5, 127.0, 125.3, 122.2, 119.6, 117.2, 111.3, 111.1, 51.7; EIMS m/z 398 [M]+ (100); HRMS (FAB+) m/z calcd. for C24H18O4N2, 398.1267; found: 398.1266.
Dimethyl 2,9-Difluoro-5,12-dihydrocycloocta[1,2-b:5,6-b′]-diindole-6,13-dicarboxylate (1b)
Red solid; yield 24%; mp 340–342 °C; IR (KBr) νmax 3373, 2952, 2918, 1678 cm−1; 1H-NMR (300 MHz, CDCl3 + DMSO-d6) δ 11.16 (bs, 2H), 8.09 (s, 2H), 7.32 (dd, J = 8.7, 4.5 Hz, 2H), 7.07 (dd, J = 9.3, 2.4 Hz, 2H), 6.90 (dt, J = 9.0, 2.4 Hz, 2H), 3.83 (s, 6H); 13C-NMR (75 MHz, CDCl3 + DMSO-d6) δ 164.5, 158.0, 154.9, 140.0, 133.0 (d, JCF = 15 Hz, Ar-quat), 126.0 (d, JCF = 9.8 Hz, Ar-quat), 124.3, 111.5 (d, JCF = 9 Hz, Ar-CH), 109.9 (d, JCF = 4.5 Hz, Ar-quat), 109.4 (d, JCF = 26 Hz, Ar-CH), 101.3 (d, JCF = 23.3 Hz, Ar-CH), 50.8; EIMS m/z 434 [M]+ (100); HRMS (FAB+) m/z calcd. for C24H16O4N2F2, 434.1078; found: 434.1081.
Dimethyl 2,9-Chloro-5,12-dihydrocycloocta[1,2-b:5,6-b′]-diindole-6,13-dicarboxylate (1c)
Red solid; yield 17%; mp 350–352 °C; IR (KBr) νmax 3378, 2955, 2922, 1686 cm−1; 1H-NMR (300 MHz, CDCl3 + DMSO-d6) δ 10.67 (bs, 2H), 8.09 (s, 2H), 7.38 (d, J = 2.1 Hz, 2H), 7.29 (d, J = 8.4 Hz, 2H), 7.08 (dd, J = 8.4, 2.1 Hz, 2H), 3.86 (s, 6H); 13C-NMR (75 MHz, CDCl3 + DMSO-d6) δ 165.6, 141.2, 135.7, 133.6, 127.9, 125.4, 125.2, 122.4, 116.7, 112.5, 110.7, 51.8; EIMS m/z 466 [M]+ (100), 467 (30), 468 (67), 469 (20), 470 (14); HRMS (FAB+) m/z calcd. for C24H16O4N2Cl2, 466.0487; found: 466.0484.
Dimethyl 2,9-Bromo-5,12-dihydrocycloocta[1,2-b:5,6-b′]-diindole-6,13-dicarboxylate (1d)
Red solid; yield 32%; mp 340–342 °C; IR (KBr) νmax 3374, 2919, 2850, 1679 cm−1; 1H-NMR (300 MHz, CDCl3 + DMSO-d6) δ 11.01 (bs, 2H), 8.10 (s, 2H), 7.53 (d, J = 1.5 Hz, 2H), 7.26 (d, J = 8.4 Hz, 2H), 7.19 (dd, J = 8.7, 1.8 Hz, 2H), 3.85 (s, 6H); 13C-NMR (75 MHz, CDCl3 + DMSO-d6) δ 164.9, 140.5, 135.5, 133.0, 127.9, 125.1, 124.4, 119.3, 112.6, 112.2, 110.0, 51.3; EIMS m/z 554 [M]+ (54), 556 (100), 558 (53); HRMS (FAB+) m/z calcd. for C24H16O4N279Br81Br, 555.9456; found: 555.9466.
Dimethyl 2,9-Iodo-5,12-dihydrocycloocta[1,2-b:5,6-b′]-diindole-6,13-dicarboxylate (1e)
Red solid; yield 10%; mp 318–320 °C; IR (KBr) νmax 3377, 2917, 2850, 1677 cm−1; 1H-NMR (300 MHz, CDCl3 + DMSO-d6) δ 11.03 (bs, 2H), 8.09 (s, 2H), 7.72 (s, 2H), 7.36 (dd, J = 8.4, 1.5 Hz, 2H), 7.16 (d, J = 8.7 Hz, 2H), 3.84 (s, 6H); 13C-NMR (75 MHz, CDCl3 + DMSO-d6) δ 164.8, 140.4, 135.8, 132.6, 129.7, 128.7, 125.5, 125.0, 113.0, 109.6, 82.5, 51.2; EIMS m/z 650 [M]+ (28), 649 (100); HRMS (FAB+) m/z calcd. for C24H16O4N2I2, 649.9200; found: 649.9208.
Dimethyl 2,9-Dimethyl-5,12-dihydrocycloocta[1,2-b:5,6-b′]-diindole-6,13-dicarboxylate (1f)
Red solid; yield 24%; mp 288–290 °C; IR (KBr) νmax 3382, 2913, 2852, 1680 cm−1; 1H-NMR (300 MHz, CDCl3 + DMSO-d6) δ 10.56 (bs, 2H), 8.13 (s, 2H), 7.22 (d, J = 8.1 Hz, 2H), 7.18 (s, 2H), 6.94 (dd, J = 8.3, 1.5 Hz, 2H), 3.83 (s, 6H), 2.39 (s, 6H); 13C-NMR (75 MHz, CDCl3 + DMSO-d6) δ 165.3, 141.1, 135.1, 131.9, 128.2, 126.7, 124.5, 123.3, 116.4, 110.5, 110.2, 51.1, 20.4; EIMS m/z 426 [M]+ (100); HRMS (FAB+) m/z calcd. for C26H22O4N2, 426.1580; found: 426.1573.
Dimethyl 2,9-Dimethoxy-5,12-dihydrocycloocta[1,2-b:5,6-b′]-diindole-6,13-dicarboxylate (1g)
Red solid; yield 23%; mp 306–308 °C; IR (KBr) νmax 3343, 2947, 2924, 1695 cm−1; 1H-NMR (300 MHz, CDCl3 + DMSO-d6) δ 10.43 (bs, 2H), 8.11 (s, 2H), 7.24 (d, J = 8.7 Hz, 2H), 6.83 (d, J = 2.4 Hz, 2H), 6.77 (dd, J = 8.7, 2.4 Hz, 2H), 3.85 (s, 6H), 3.82 (s, 6H); 13C-NMR (75 MHz, CDCl3 + DMSO-d6) δ 165.7, 153.8, 141.3, 132.6, 132.1, 127.2, 124.5, 112.3, 111.9, 110.6, 98.8, 54.9, 51.4; EIMS m/z 458 [M]+ (100); HRMS (FAB+) m/z calcd. for C26H22O6N2, 458.1478; found: 458.1479.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

