
Isolation and Synthesis of Laxaphycin B-Type Peptides: A Case Study and Clues to Their Biosynthesis


Abstract
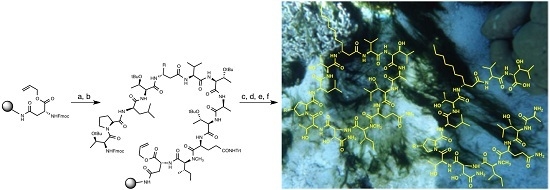
:
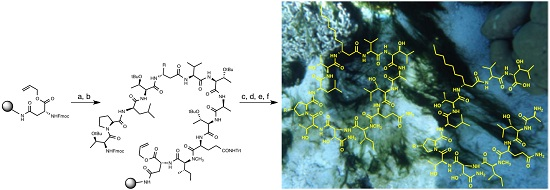
1. Introduction

2. Results and Discussion
2.1. Synthesis of Laxaphycin B Analogs

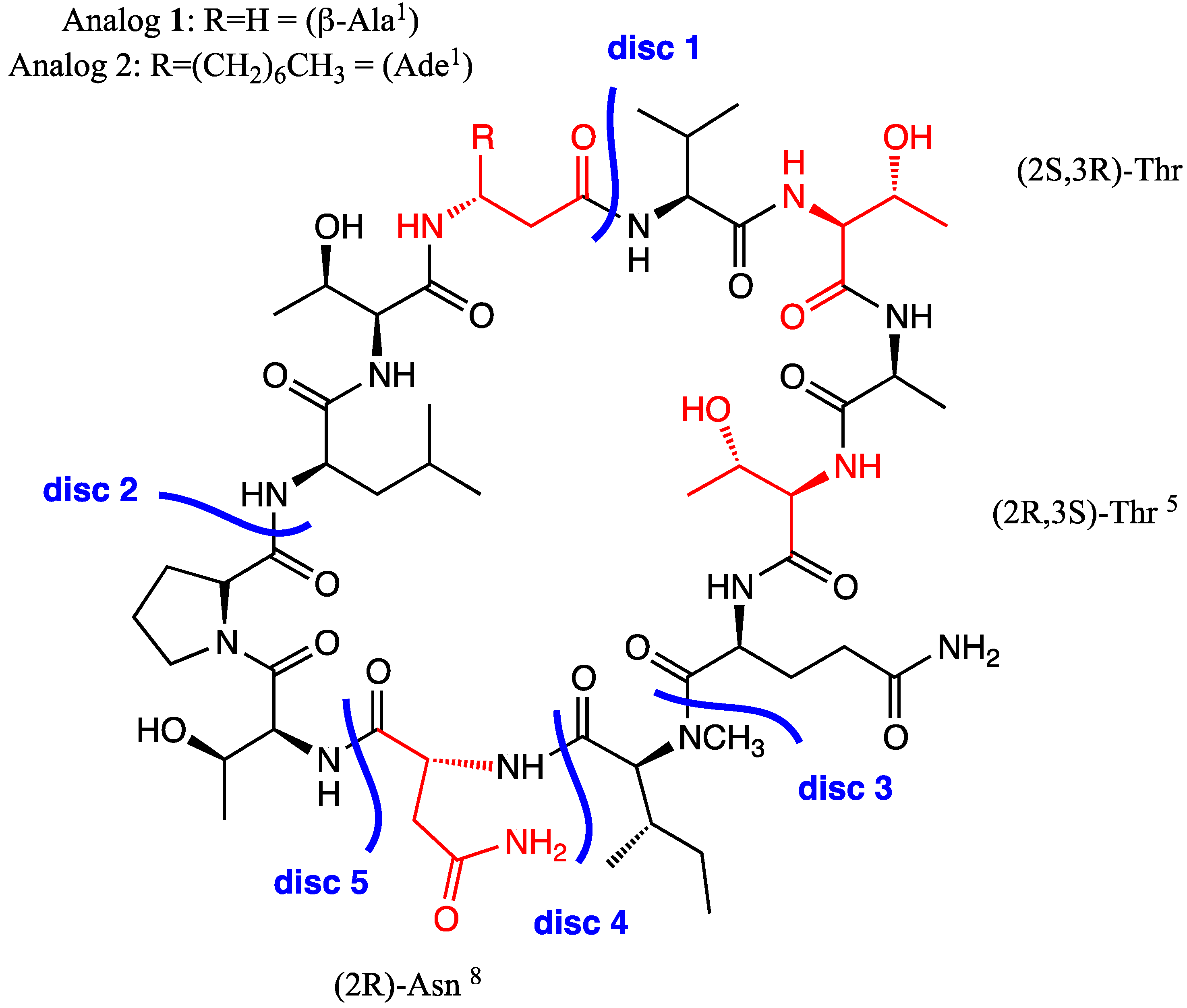
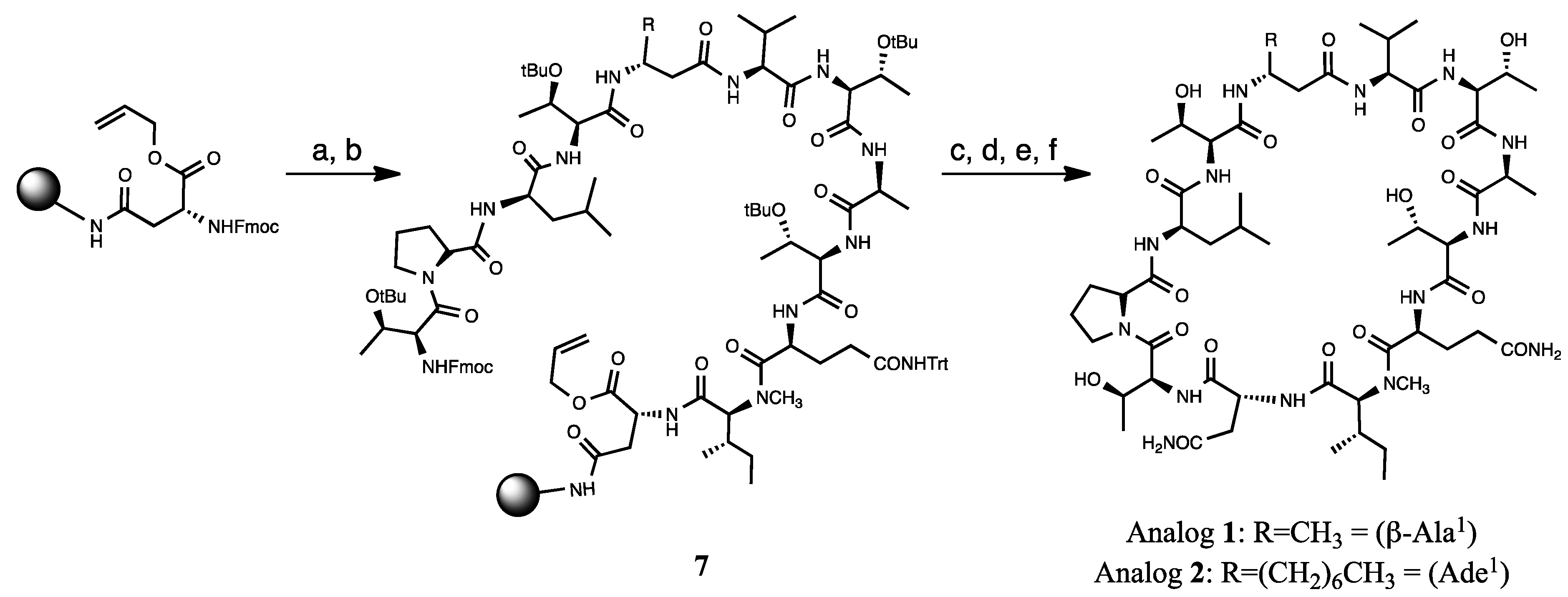
2.1.1. Retrosynthetic Analysis of the Laxaphycin B Analog 1

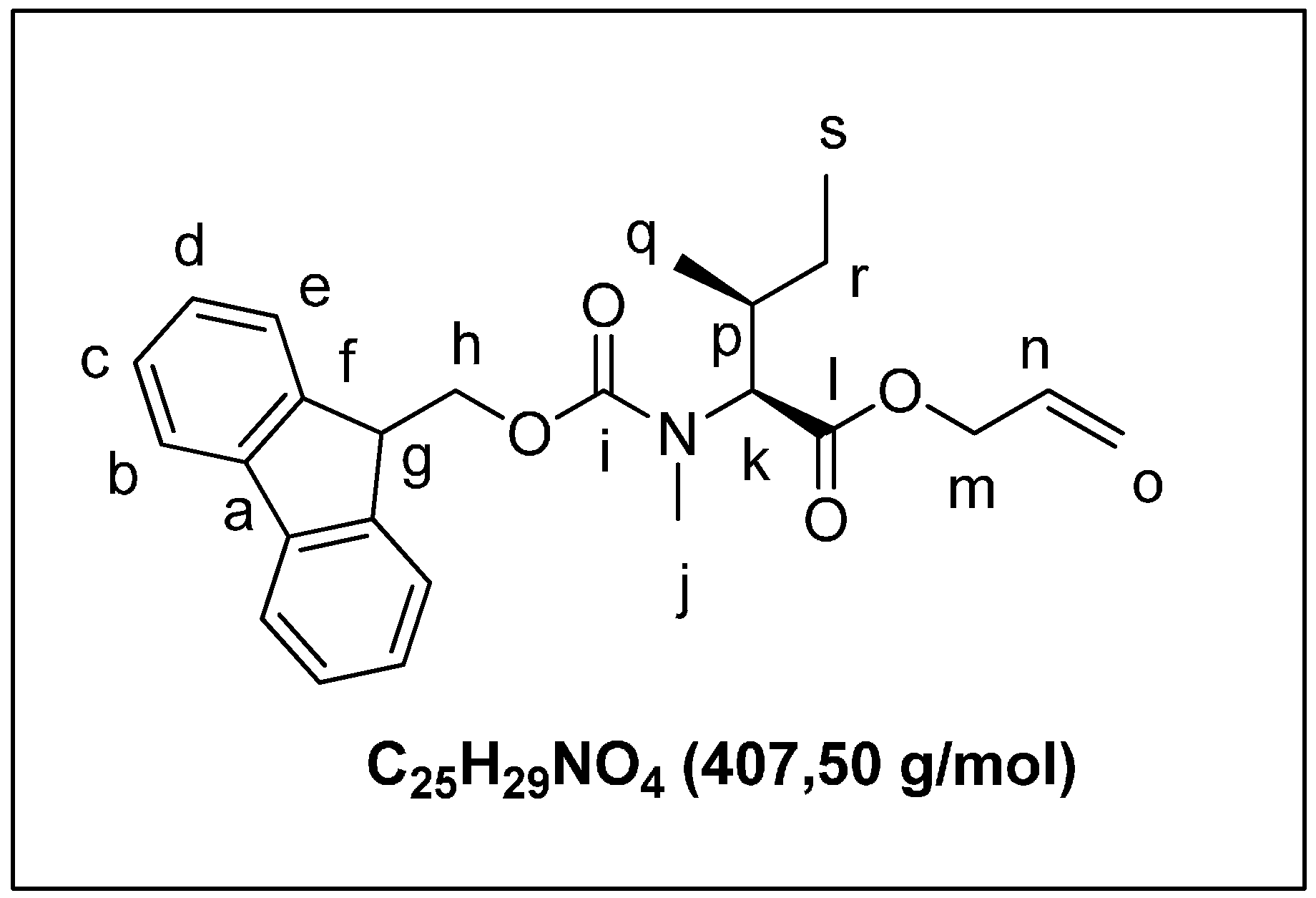
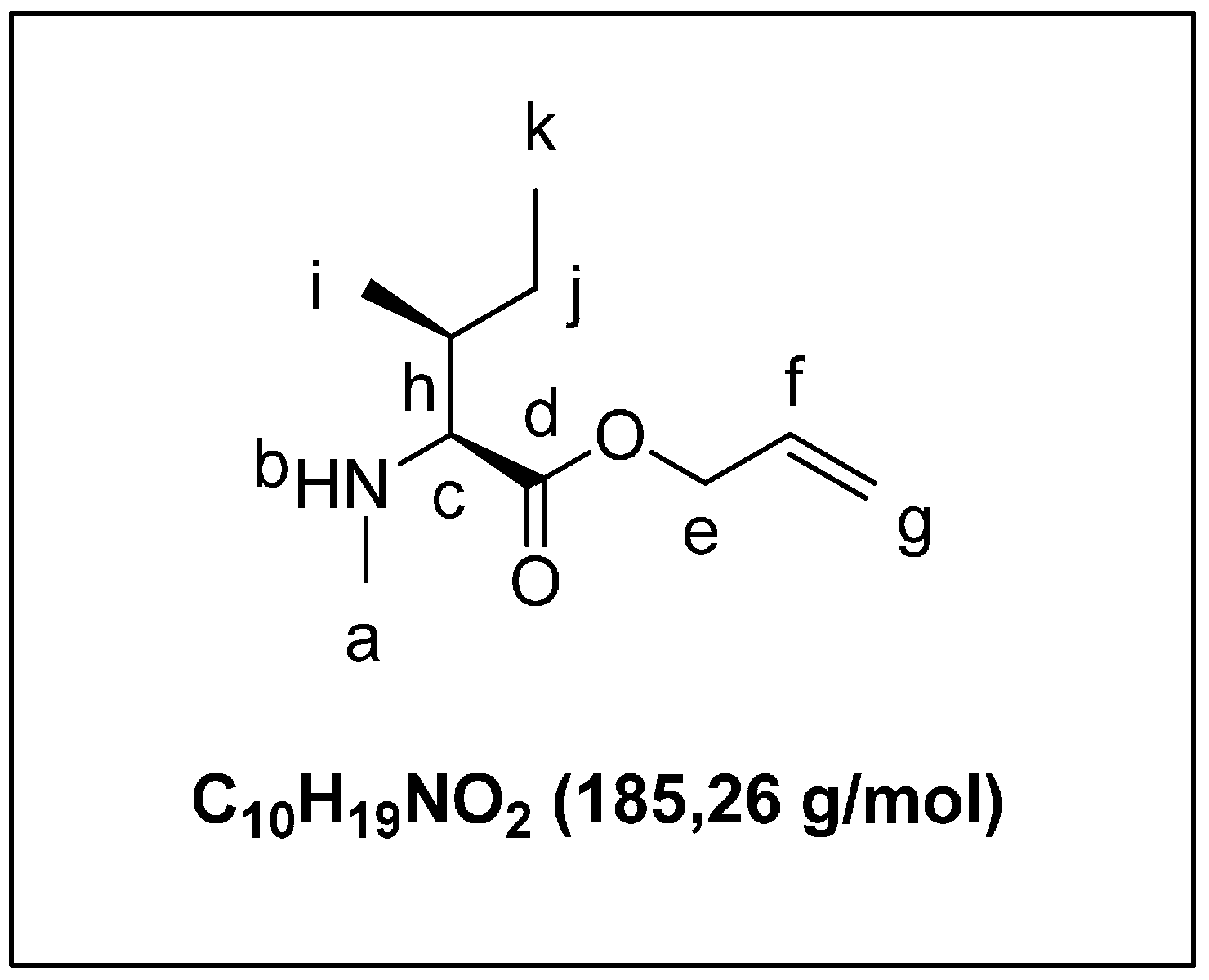
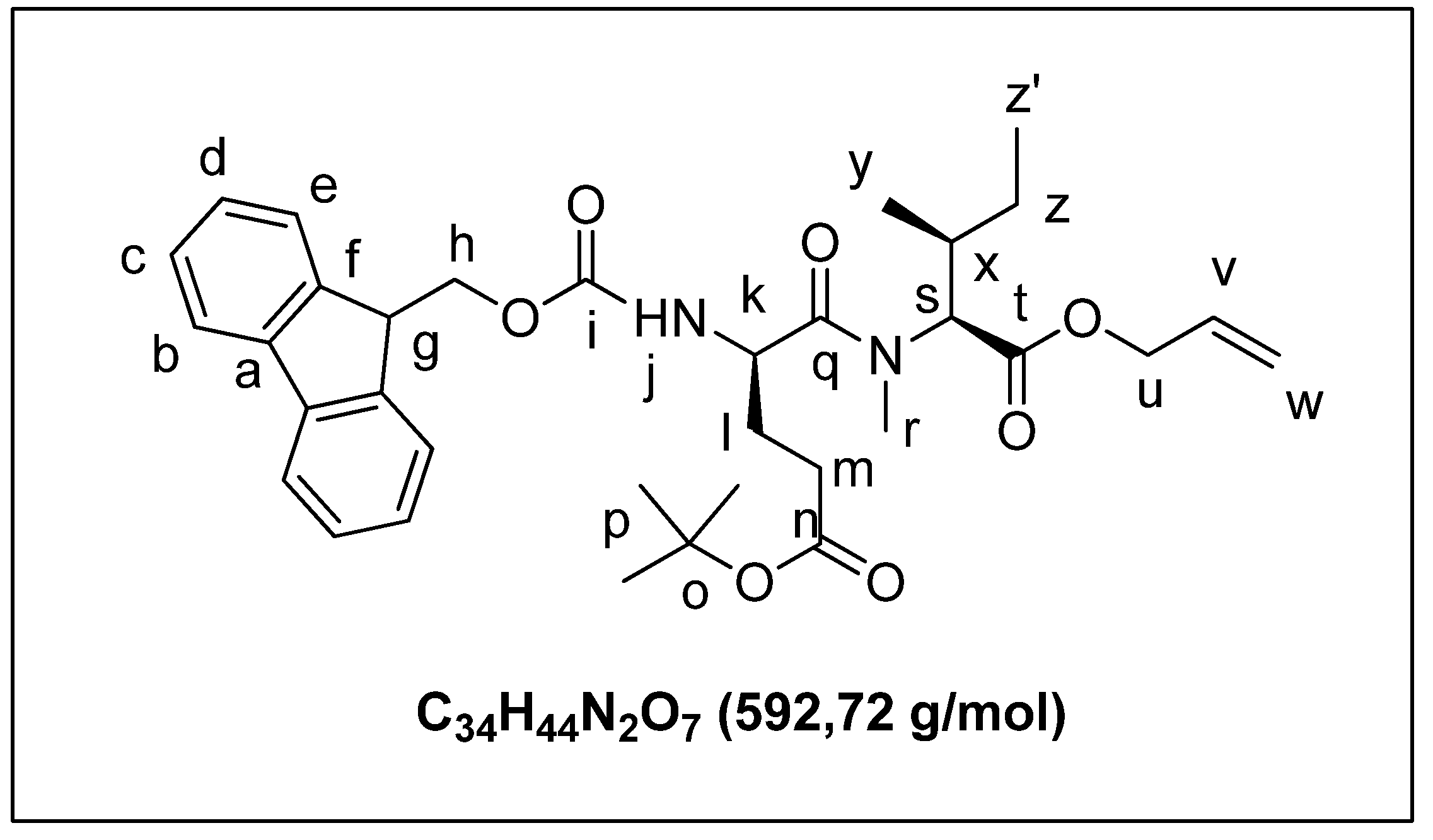
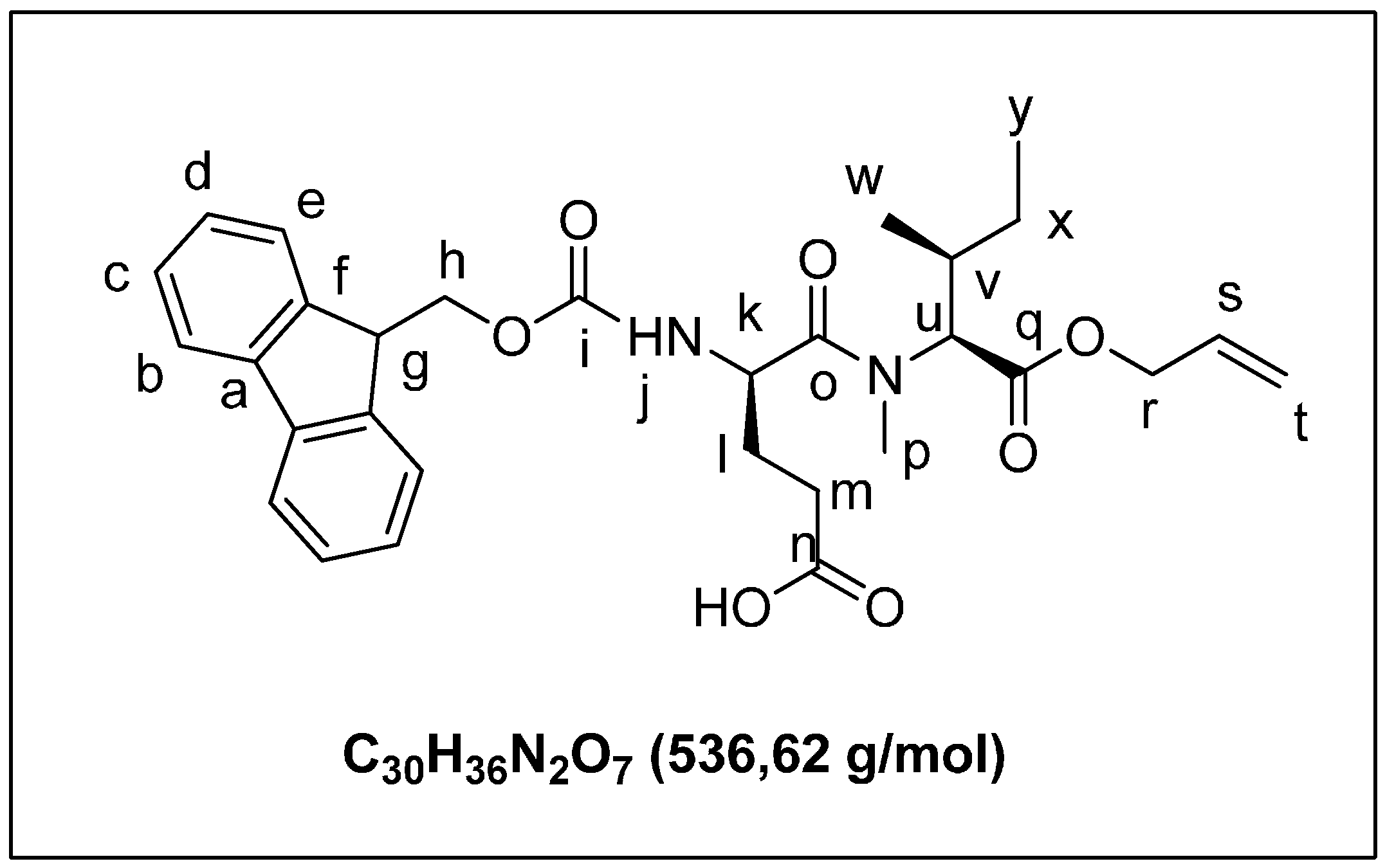
2.1.2. Solid Phase Synthesis Preliminary Assay of ((2S,3S)-Hle3)laxaphycin B Analogs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 | ((2 S,3S)-Hleu3)laxa B (8) | Laxaphycin B (9) |
|---|---|---|---|---|
| Mass Peaks | [M + H]+ m/z 1225 | [M + H]+ m/z 1323 | [M + H]+ m/z 1395 | [M + H]+ m/z 1395 |
| Rt (min) | 8.64 | 16.01 | 16.3 | Natural: 14.8 |
| Synthetic: 14.8 |
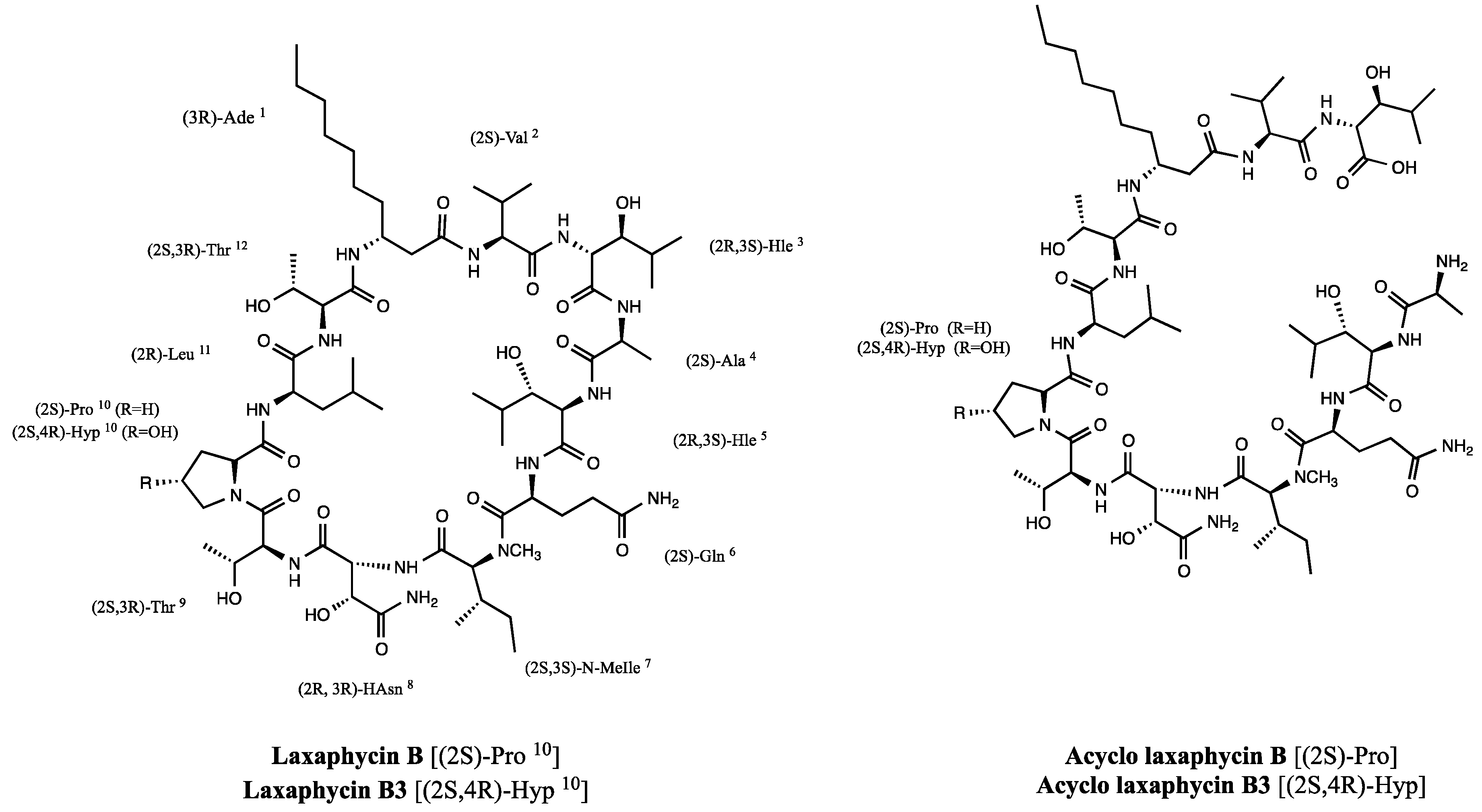
2.1.3. Laxaphycin B Revised Structure
2.2. New Natural Acyclolaxaphycin B Analogs
2.2.1. Acyclolaxaphycin B (11): Structure Elucidation
| Laxaphycin B | Acyclolaxaphycin B | Laxaphycin B3 | Acyclolaxaphycin B3 | |||||
|---|---|---|---|---|---|---|---|---|
| 13C | 1H | 13C | 1H | 13C | 1H | 13C | 1H | |
| β Ade1 | ||||||||
| NH | - | 7.58 | - | 7.53 | - | 7.52 | - | 7.53 |
| CαH2 | 40.28 | 2.33/2.40 | 40.52 | 2.27/2.38 | - | 2.30/2.44 | 40.42 | 2.28/2.40 |
| CβH | 45.93 | 4.11 | 46.27 | 4.05 | 45.92 | 4.08 | 46.13 | 4.05 |
| CγH2 | 33.45 | 1.29/1.40 | 33.62 | 1.34/1.40 | 33.41 | 1.40 | 33.49 | 1.33/1.40 |
| CδH2 | 28.67 * | 1.24 | 28.77 | 1.23 | 28.69 * | 1.24 | 28.69 | 1.21 |
| CεH2 | 28.47 * | 1.20 | 28.61 | 1.20 | 28.47 * | 1.20 | 28.53 | 1.21 |
| CζH2 | 25.18 * | 1.20 | 25.28 | 1.21 | 25.22 * | 1.20 | 25.20 | 1.22 |
| CηH2 | 31.11 * | 1.20 | 31.22 | 1.21 | 31.10 * | 1.20 | 31.15 | 1.20 |
| CθH2 | 21.92 * | 1.20 | 22.04 | 1.20 | 21.92 * | 1.20 | 21.97 | 1.24 |
| CιH3 | 13.79 | 0.84 | 13.91 | 0.85 | 13.79 | 0.82 | 13.81 | 0.83 |
| CO | 171.14 | - | 170.30 | - | 171.30 | - | 170.15 | - |
| Val2 | ||||||||
| NH | - | 8.18 | - | 7.89 | - | 8.10 | - | 7.89 |
| CαH | 59.03 | 4.09 | 57.64 | 4.30 | 58.89 | 4.12 | 57.50 | 4.31 |
| CβH2 | 29.33 | 1.97 | 30.59 | 2.02 | 29.37 | 1.98 | 30.51 | 2.02 |
| CγH3 | 18.80 | 0.91 | 18.85 | 0.93 | 18.56 | 0.88 | 18.89 | 0.93 |
| Cγ'H3 | 18.87 | 0.85 | 18.95 | 0.81 | 18.85 | 0.84 | 18.78 | 0.81 |
| CO | 171.05 | - | 171.27 | - | 171.30 | - | 171.15 | - |
| HLe3 | ||||||||
| NH | - | 7.94 | 7.69 | - | 7.90 | - | 7.70 | |
| CαH | 55.23 | 4.34 | 54.30 | 4.44 | 55.15 | 4.37 | 54.21 | 4.44 |
| CβH | 76.37 | 3.49 | 76.06 | 3.53 | 76.48 | 3.50 | 76.13 | 3.53 |
| OH | - | 4.94 | - | — | - | 4.90 | - | — |
| CγH | 30.54 | 1.58 | 30.68 | 1.51 | 30.57 | 1.60 | 30.84 | 1.52 |
| CδH3 | 19.22 * | 0.89 | 19.19 | 0.91 | 18.76 * | 0.89 | 19.23 | 0.91 |
| Cδ'H3 | 18.56 | 0.76 | 18.74 | 0.76 | 18.43 | 0.76 | 18.67 | 0.76 |
| CO | 171.35 | - | 172.40 | - | - | - | 172.34 | - |
| Ala4 | ||||||||
| NH/NH2 | - | 7.86 | - | 8.04 | - | 7.87 | - | 8.05 |
| CαH | 49.28 | 4.22 | 48.30 | 4.03 | 49.30 | 4.22 | 48.20 | 4.04 |
| CβH3 | 17.55 | 1.31 | 17.43 | 1.36 | 17.65 | 1.32 | 17.38 | 1.36 |
| CO | 172.33 | - | 170.02 | - | 172.47 | - | 169.87 | - |
| HLe5 | ||||||||
| NH | - | 7.69 | - | 8.34 | - | 7.61 | - | 8.37 |
| CαH | 55.52 | 4.28 | 55.40 | 4.44 | 55.64 | 4.28 | 55.27 | 4.46 |
| CβH | 75.80 | 3.49 | 76.21 | 3.53 | 75.78 | 3.48 | 75.94 | 3.53 |
| OH | - | 5.03 | - | — | - | 5.05 | - | — |
| CγH | 29.90 | 1.56 | 30.65 | 1.51 | 29.84 | 1.58 | 30.73 | 1.51 |
| CδH3 | 18.65 * | 0.89 | 17.58 | 0.82 | 18.69 * | 0.88 | 19.14 | 0.83 |
| Cδ'H3 | 18.56 | 0.76 | 19.28 | 0.81 | - | 0.74 | 17.52 | 0.83 |
| CO | 170.50 | - | 169.74 | - | 170.60 | - | 169.64 | - |
| Gln6 | ||||||||
| NH | - | 7.77 | - | 8.02 | - | 7.56 | - | 8.04 |
| CαH | 49.16 | 4.63 | 48.94 | 4.69 | 49.40 | 4.58 | 48.78 | 4.70 |
| CβH2 | 26.39 | 1.75/1.97 | 26.90 | 1.76/1.93 | - | 1.64/2.00 | 26.86 | 1.77/1.94 |
| CγH2 | 30.72 | 2.04/2.10 | 30.63 | 2.12 | - | 2.15/2.23 | 30.70 | 2.13 |
| CON | 174.60 | - | 174.38 | - | 174.74 | - | 174.31 | - |
| NH2 | - | 6.85/7.22 | - | 6.79/7.14 | - | 6.79/7.17 | - | 6.80/7.16 |
| CO | 172.49 | - | 172.45 | - | 172.64 | - | 172.27 | - |
| N-MeIle7 | ||||||||
| NCH3 | 30.03 | 2.97 | 30.24 | 2.97 | 30.15 | 3.01 | 30.20 | 2.98 |
| CαH | 59.85 | 4.72 | 59.94 | 4.71 | 59.87 | 4.73 | 59.79 | 4.73 |
| CβH | 31.56 | 1.90 | 31.50 | 1.91 | 31.80 | 1.90 | 31.41 | 1.92 |
| CγH2 | 23.88 | 0.89/1.29 | 23.98 | 0.87/1.28 | - | 0.74/1.27 | 23.92 | 0.87/1.27 |
| Cγ'H3 | 15.08 | 0.76 | 15.25 | 0.78 | 14.99 | 0.74 | 15.21 | 0.79 |
| CδH3 | 10.33 | 0.78 | 10.48 | 0.79 | 10.31 | 0.75 | 10.41 | 0.78 |
| CO | 170.02 | - | 169.66 | - | 170.10 | - | 169.47 | - |
| HAsn8 | ||||||||
| NH | - | 7.64 | - | 7.41 | - | 7.66 | - | 7.41 |
| CαH | 55.52 | 4.63 | 55.22 | 4.67 | 55.53 | 4.63 | 55.13 | 4.71 |
| CβH | 70.44 | 4.31 | 71.04 | 4.36 | 70.33 | 4.35 | 70.99 | 4.37 |
| OH | - | 5.79 | - | 5.78 | - | 5.70 | - | - |
| CON | 173.37 | - | 173.20 | - | 173.37 | - | 173.20 | - |
| NH2 | - | 7.27 | - | 7.26/7.30 | - | 7.17 | - | 7.27/7.32 |
| CO | 169.16 | - | 168.92 | - | 169.12 | - | 168.75 | - |
| Thr9 | ||||||||
| NH | - | 7.33 | - | 7.63 | - | 7.12 | - | 7.63 |
| CαH | 55.61 | 4.49 | 55.25 | 4.57 | 55.83 | 4.46 | 55.56 | 4.56 |
| CβH | 66.23 | 3.93 | 66.54 | 3.98 | 66.43 | 3.90 | 66.50 | 3.97 |
| OH | - | 4.94 | - | - | - | 4.89 | - | - |
| CγH3 | 18.87 | 1.05 | 18.64 | 1.05 | 18.85 * | 1.03 | 18.59 | 1.05 |
| CO | 168.58 | - | 168.87 * | - | 168.70 * | - | 169.04 * | - |
| Pro10/Hyp10 | ||||||||
| CαH | 59.60 | 4.33 | 59.90 | 4.37 | 58.62 | 4.43 | 58.87 | 4.44 |
| CβH2 | 29.08 | 1.82/2.04 | 28.77 | 1.83/2.03 | 37.73 | 1.84/2.01 | 37.45 | 1.89/2.05 |
| CγH2 | 24.00 | 1.80/1.90 | 24.16 | 1.83/1.90 | 68.50 | 4.32 | 68.48 | 4.31 |
| OH | 5.08 | - | ||||||
| CδH2 | 47.16 | 3.68 | 47.49 | 3.64/3.75 | 55.48 | 3.58/3.72 | 55.60 | 3.60/3.76 |
| CO | 171.21 | - | 171.42 | - | 171.47 ** | - | 171.33 | - |
| Leu11 | ||||||||
| NH | - | 7.89 | - | 7.77 | - | 7.86 | - | 7.84 |
| CαH | 51.36 | 4.31 | 51.44 | 4.30 | 51.31 | 4.35 | 51.36 | 4.29 |
| CβH2 | 40.82 | 1.47 | 40.44 | 1.47 | 41.24 | 1.47 | 40.51 | 1.46 |
| CγH | 24.06 | 1.53 | 24.09 | 1.58 | 24.12 | 1.52 | 24.06 | 1.58 |
| CδH3 | 22.71 | 0.87 | 22.96 | 0.86 | 22.75 | 0.86 | 22.83 | 0.86 |
| Cδ'H3 | 21.76 | 0.82 | 21.42 | 0.84 | 21.72 | 0.80 | 21.43 | 0.83 |
| CO | 171.67 | - | 171.83 | - | 171.41 ** | - | 171.33 | - |
| Thr12 | ||||||||
| NH | - | 7.74 | - | 7.57 | - | 7.68 | - | 7.59 |
| CαH | 57.85 | 4.11 | 58.13 | 4.10 | 58.17 | 4.10 | 58.12 | 4.10 |
| CβH | 66.19 | 4.00 | 66.52 | 3.97 | 66.35 | 3.97 | 66.50 | 3.97 |
| OH | - | 4.78 | - | - | - | 4.80 | - | - |
| CγH3 | 19.46 | 0.99 | 19.55 | 0.99 | 19.48 | 0.99 | 19.45 | 1.00 |
| CO | 168.67 | - | 168.87 * | - | 168.67 * | - | 168.99 * | - |

2.2.2. Acyclolaxaphycin B3 (12): Structure Elucidation
2.2.3. Acyclolaxaphycins B (11) and B3 (12): Clues to Their Biosynthesis
3. Experimental Section
3.1. Sampling Sites
3.2. Isolation Procedure
3.3. Mass and NMR Spectroscopies
3.4. Solid Phase Peptide Synthesis




4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Burja, A.M.; Banaigs, B.; Abou-Mansour, E.; Grant Burgess, J.; Wright, P.C. Marine cyanobacteria—A prolific source of natural products. Tetrahedron 2001, 57, 9347–9377. [Google Scholar] [CrossRef]
- Moore, R.E. Cyclic peptides and depsipeptides from cyanobacteria: A review. J. Ind. Microbiol. 1996, 16, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.T. Filamentous tropical marine cyanobacteria: A rich source of natural products for anticancer drug discovery. J. Appl. Phycol. 2010, 22, 659–676. [Google Scholar] [CrossRef]
- Condurso, H.L.; Bruner, S.D. Structure and noncanonical chemistry of nonribosomal peptide biosynthetic machinery. Nat. Prod. Rep. 2012, 29, 1099. [Google Scholar] [CrossRef] [PubMed]
- Sieber, S.A.; Marahiel, M.A. Molecular mechanisms underlying nonribosomal peptide synthesis: Approaches to new antibiotics. Chem. Rev. 2005, 105, 715–738. [Google Scholar] [CrossRef] [PubMed]
- Engene, N.; Choi, H.; Esquenazi, E.; Rottacker, E.C.; Ellisman, M.H.; Dorrestein, P.C.; Gerwick, W.H. Underestimated biodiversity as a major explanation for the perceived rich secondary metabolite capacity of the cyanobacterial genus Lyngbya. Environ. Microbiol. 2011, 13, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Banaigs, B.; Bonnard, I.; Witczak, A.; Inguimbert, N. Marine peptide secondary metabolites. In Outstanding Marine Molecules; Barre, S.L., Kornprobst, J.-M., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014; pp. 285–318. [Google Scholar]
- Frankmölle, W.P.; Knübel, G.; Moore, R.E.; Patterson, G.M. Antifungal cyclic peptides from the terrestrial blue-green alga Anabaena laxa. II. Structures of laxaphycins A, B, D and E. J. Antibiot. (Tokyo) 1992, 45, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, I.; Rolland, M.; Salmon, J.-M.; Debiton, E.; Barthomeuf, C.; Banaigs, B. Total structure and inhibition of tumor cell proliferation of Laxaphycins. J. Med. Chem. 2007, 50, 1266–1279. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, J.B.; Ernst-Russell, M.A.; de Ropp, J.S.; Molinski, T.F. Lobocyclamides A−C, Lipopeptides from a cryptic cyanobacterial mat containing Lyngbya confervoides. J. Org. Chem. 2002, 67, 8210–8215. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Krunic, A.; Kang, H.-S.; Chen, W.-L.; Woodard, J.L.; Fuchs, J.R.; Swanson, S.M.; Orjala, J. Trichormamides A and B with antiproliferative activity from the cultured freshwater cyanobacterium Trichormus sp. UIC 10339. J. Nat. Prod. 2014, 77, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Kang, H.-S.; Krunic, A.; Chen, W.-L.; Yang, J.; Woodard, J.L.; Fuchs, J.R.; Cho, S.H.; Franzblau, S.G.; Swanson, S.M.; Orjala, J. Trichormamides C and D, antiproliferative cyclic lipopeptides from the cultured freshwater cyanobacterium cf. Oscillatoria sp. UIC 10045. Bioorg. Med. Chem. 2015, 23, 3152–3162. [Google Scholar]
- Maru, N.; Ohno, O.; Uemura, D. Lyngbyacyclamides A and B, novel cytotoxic peptides from marine cyanobacteria Lyngbya sp. Tetrahedron Lett. 2010, 51, 6384–6387. [Google Scholar] [CrossRef]
- Zhaxybayeva, O.; Gogarten, J.P.; Charlebois, R.L.; Doolittle, W.F.; Papke, R.T. Phylogenetic analyses of cyanobacterial genomes: Quantification of horizontal gene transfer events. Genome Res. 2006, 16, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Pennings, S.C.; Pablo, S.R.; Paul, V.J. Chemical defenses of the tropical, benthic marine cyanobacterium Hormothamnion enteromorphoides: Diverse consumers and synergisms. Limnol. Oceanogr. 1997, 42, 911–917. [Google Scholar] [CrossRef]
- Boyaud, F.; Mahiout, Z.; Lenoir, C.; Tang, S.; Wdzieczak-Bakala, J.; Witczak, A.; Bonnard, I.; Banaigs, B.; Ye, T.; Inguimbert, N. First total synthesis and stereochemical revision of laxaphycin B and its extension to Lyngbyacyclamide A. Org. Lett. 2013, 15, 3898–3901. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.L.; Tofteng, A.P.; Malik, L.; Jensen, K.J. Microwave heating in solid-phase peptide synthesis. Chem. Soc. Rev. 2012, 41, 1826–1844. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.M.; Porter, K.A.; Singh, S.K.; Vanier, G.S. High-efficiency solid phase peptide synthesis (HE-SPPS). Org. Lett. 2014, 16, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Ben Haj Salah, K.; Inguimbert, N. Efficient microwave-assisted one shot synthesis of peptaibols using inexpensive coupling reagents. Org. Lett. 2014, 16, 1783–1785. [Google Scholar] [CrossRef] [PubMed]
- Ben Haj Salah, K.; Legrand, B.; Das, S.; Martinez, J.; Inguimbert, N. A straightforward strategy to substitute amide bonds by 1,2,3 triazoles in peptaibols analogs using Aibψ[Tz]-Xaa dipeptides. Pept. Sci. 2015, 104, 611–621. [Google Scholar] [CrossRef] [PubMed]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.S. The cyclization of peptides and depsipeptides. J. Pept. Sci. 2003, 9, 471–501. [Google Scholar] [CrossRef] [PubMed]
- Malesevic, M.; Strijowski, U.; Bächle, D.; Sewald, N. An improved method for the solution cyclization of peptides under pseudo-high dilution conditions. J. Biotechnol. 2004, 112, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Echalier, C.; Al-Halifa, S.; Kreiter, A.; Enjalbal, C.; Sanchez, P.; Ronga, L.; Puget, K.; Verdié, P.; Amblard, M.; Martinez, J.; Subra, G. Heating and microwave assisted SPPS of C-terminal acid peptides on trityl resin: The truth behind the yield. Amino Acids 2013, 45, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Kates, S.A.; Solé, N.A.; Johnson, C.R.; Hudson, D.; Barany, G.; Albericio, F. A novel, convenient, three-dimensional orthogonal strategy for solid-phase synthesis of cyclic peptides. Tetrahedron Lett. 1993, 34, 1549–1552. [Google Scholar] [CrossRef]
- El-Faham, A.; Funosas, R.S.; Prohens, R.; Albericio, F. COMU: A safer and more effective replacement for benzotriazole-based uronium coupling Reagents. Chem. Eur. J. 2009, 15, 9404–9416. [Google Scholar] [CrossRef] [PubMed]
- Del Fresno, M.; Alsina, J.; Royo, M.; Barany, G.; Albericio, F. Solid-phase synthesis of diketopiperazines, useful scaffolds for combinatorial chemistry. Tetrahedron Lett. 1998, 39, 2639–2642. [Google Scholar] [CrossRef]
- Subirós-Funosas, R.; Prohens, R.; Barbas, R.; El-Faham, A.; Albericio, F. Oxyma: An efficient additive for peptide synthesis to replace the benzotriazole-based HOBt and HOAt with a lower risk of explosion[1]. Chem. Eur. J. 2009, 15, 9394–9403. [Google Scholar] [CrossRef] [PubMed]
- Buron, F.; Turck, A.; Plé, N.; Bischoff, L.; Marsais, F. Towards a biomimetic synthesis of barrenazine A. Tetrahedron Lett. 2007, 48, 4327–4330. [Google Scholar] [CrossRef]
- Boyaud, F.; Viguier, B.; Inguimbert, N. Synthesis of a protected derivative of (2R,3R)-β-hydroxyaspartic acid suitable for Fmoc-based solid phase synthesis. Tetrahedron Lett. 2013, 54, 158–161. [Google Scholar]
- Hale, K.J.; Manaviazar, S.; Delisser, V.M. A practical new asymmetric synthesis of (2S,3S)- and (2R,3R)-3-hydroxyleucine. Tetrahedron 1994, 50, 9181–9188. [Google Scholar] [CrossRef]
- Zhang, C.; Kong, L.; Liu, Q.; Lei, X.; Zhu, T.; Yin, J.; Lin, B.; Deng, Z.; You, D. In vitro characterization of echinomycin biosynthesis: Formation and hydroxylation of l-tryptophanyl-S-enzyme and oxidation of (2S,3S) beta-hydroxytryptophan. PLoS ONE 2013, 8, e56772. [Google Scholar] [CrossRef] [PubMed]
- Cryle, M.J.; Meinhart, A.; Schlichting, I. Structural characterization of OxyD, a cytochrome P450 involved in beta-hydroxytyrosine formation in cancomycin biosynthesis. J. Biol. Chem. 2010, 285, 24562–24574. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, S.; Suessmuth, R.D.; Cryle, M.J. Cytochrome P450(sky) Interacts directly with the nonribosomal peptide synthetase to generate three amino acid precursors in skyllamycin biosynthesis. Acs Chem. Biol. 2013, 8, 2586–2596. [Google Scholar] [CrossRef] [PubMed]
- Mareš, J.; Hájek, J.; Urajová, P.; Kopecký, J.; Hrouzek, P. A hybrid non-ribosomal peptide/polyketide synthetase containing fatty-acyl ligase (FAAL) synthesizes the β-amino fatty acid lipopeptides Puwainaphycins in the cyanobacterium Cylindrospermum alatosporum. PLoS ONE 2014, 9, e111904. [Google Scholar] [CrossRef] [PubMed]
- Hoefler, B.C.; Gorzelnik, K.V.; Yang, J.Y.; Hendricks, N.; Dorrestein, P.C.; Straight, P.D. Enzymatic resistance to the lipopeptide surfactin as identified through imaging mass spectrometry of bacterial competition. Proc. Natl. Acad. Sci. USA 2012, 109, 13082–13087. [Google Scholar] [CrossRef] [PubMed]
- Di Blasio, B.; del Duca, V.; Lombardi, A.; Pedone, C.; Lorenzi, G.P.; Benedetti, E. Conformation and structure of linear peptides with regularly alternating l- and d-residues: Structure of the blocked hexapeptide Tert-butyloxycarbonyl-(d-alloisoleucyl-l-isoleucyl)3 methyl ester monohydrate. Int. J. Pept. Protein Res. 1995, 45, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.D.; Buriak, J.M.; Kobayashi, K.; Isler, M.P.; McRee, D.E.; Ghadiri, M.R. Cylindrical β-sheet peptide assemblies. J. Am. Chem. Soc. 1998, 120, 8949–8962. [Google Scholar] [CrossRef]
- Jensen, K.J.; Alsina, J.; Songster, M.F.; Vágner, J.; Albericio, F.; Barany, G. Backbone amide linker (BAL) strategy for solid-phase synthesis of C-terminal-modified and cyclic peptides. J. Am. Chem. Soc. 1998, 120, 5441–5452. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bornancin, L.; Boyaud, F.; Mahiout, Z.; Bonnard, I.; Mills, S.C.; Banaigs, B.; Inguimbert, N. Isolation and Synthesis of Laxaphycin B-Type Peptides: A Case Study and Clues to Their Biosynthesis. Mar. Drugs 2015, 13, 7285-7300. https://doi.org/10.3390/md13127065
Bornancin L, Boyaud F, Mahiout Z, Bonnard I, Mills SC, Banaigs B, Inguimbert N. Isolation and Synthesis of Laxaphycin B-Type Peptides: A Case Study and Clues to Their Biosynthesis. Marine Drugs. 2015; 13(12):7285-7300. https://doi.org/10.3390/md13127065
Chicago/Turabian StyleBornancin, Louis, France Boyaud, Zahia Mahiout, Isabelle Bonnard, Suzanne C. Mills, Bernard Banaigs, and Nicolas Inguimbert. 2015. "Isolation and Synthesis of Laxaphycin B-Type Peptides: A Case Study and Clues to Their Biosynthesis" Marine Drugs 13, no. 12: 7285-7300. https://doi.org/10.3390/md13127065
APA StyleBornancin, L., Boyaud, F., Mahiout, Z., Bonnard, I., Mills, S. C., Banaigs, B., & Inguimbert, N. (2015). Isolation and Synthesis of Laxaphycin B-Type Peptides: A Case Study and Clues to Their Biosynthesis. Marine Drugs, 13(12), 7285-7300. https://doi.org/10.3390/md13127065