
Involvement of JNK and Caspase Activation in Hoiamide A-Induced Neurotoxicity in Neocortical Neurons

 and
and 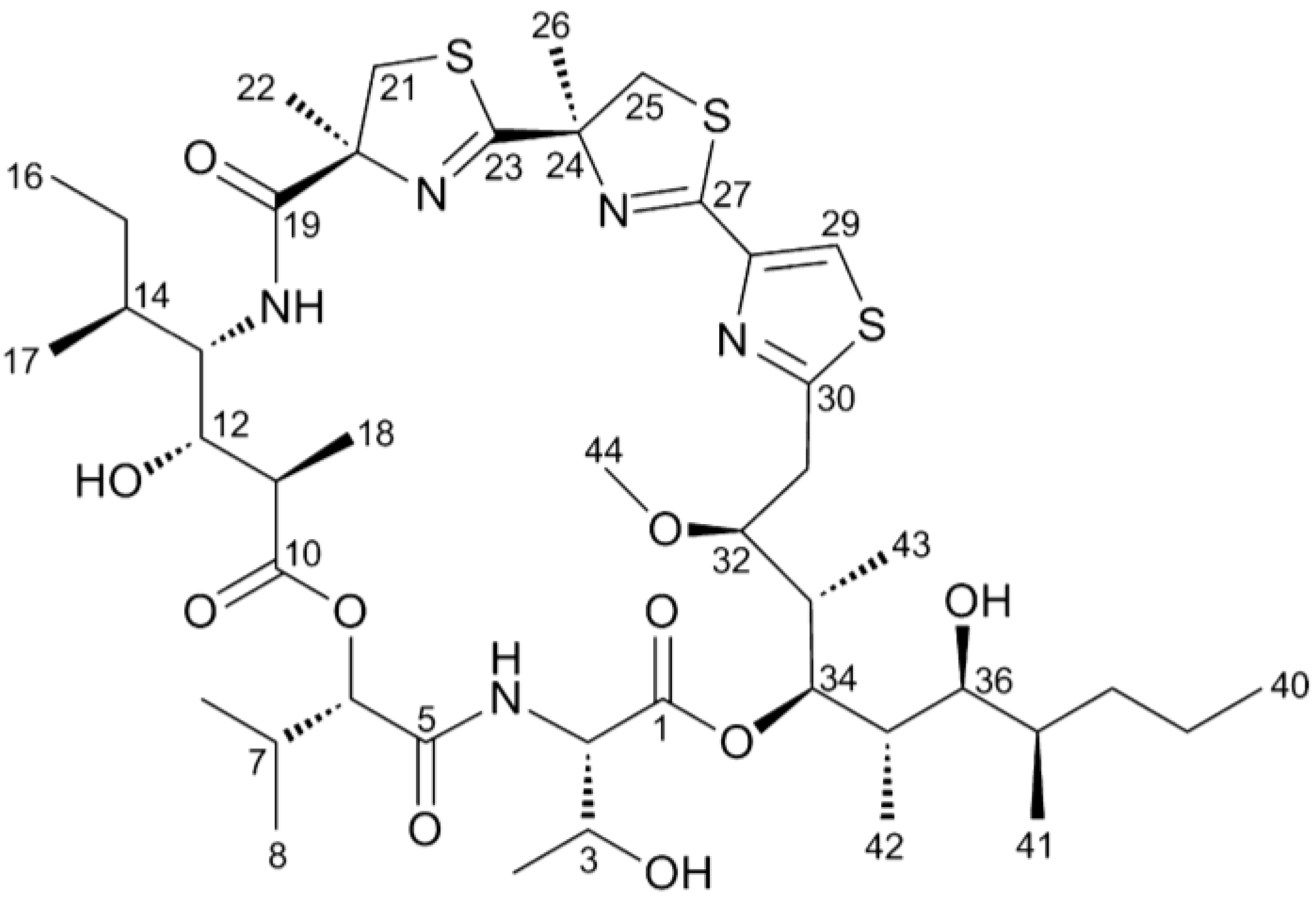{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
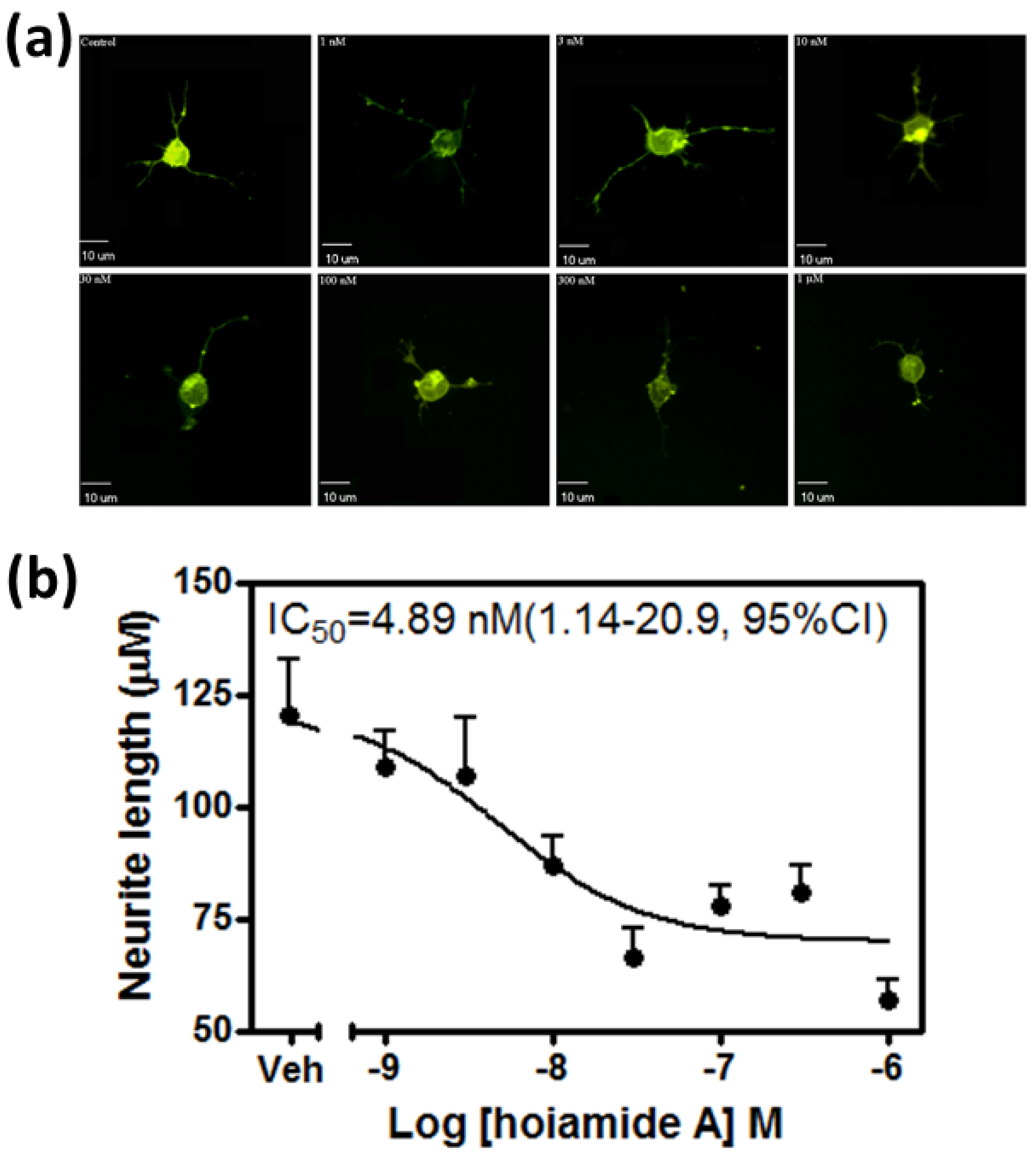
2.1. Hoiamide A Produces Neurite Retraction in Neocortical Neurons


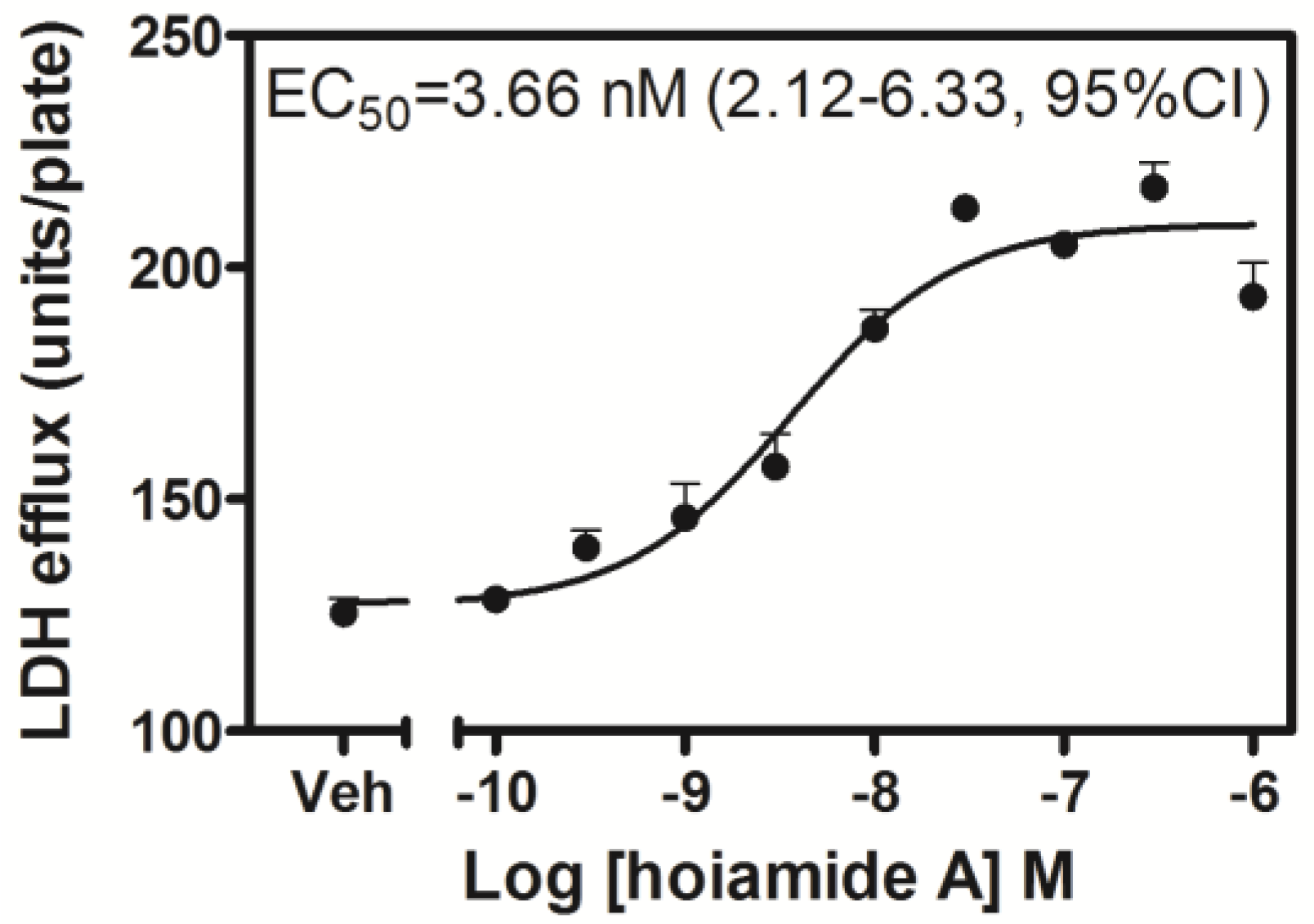
2.2. Hoiamide A Produces LDH Efflux in Neocortical Neurons


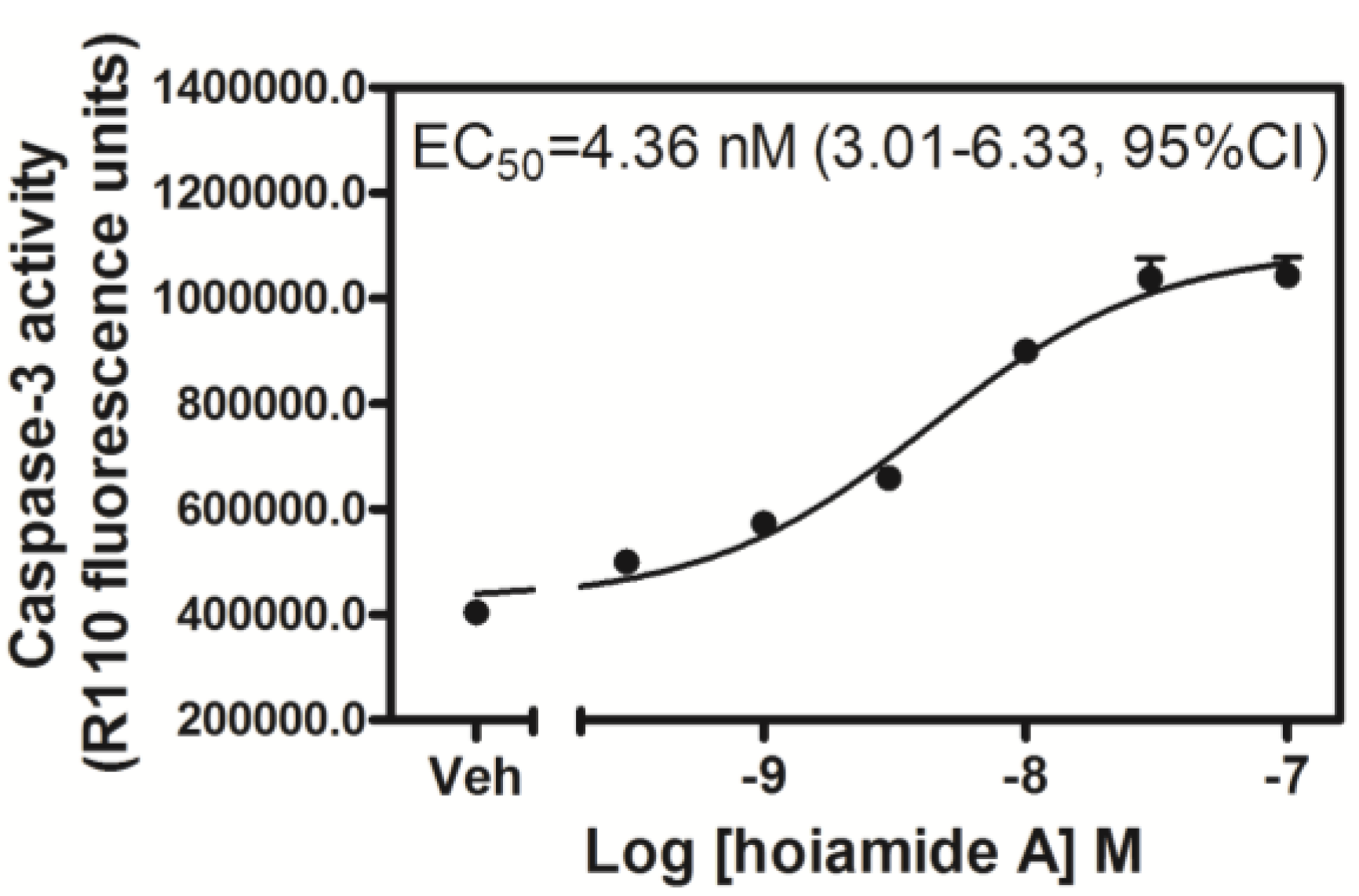
2.3. Hoiamide A Stimulates Caspase-3 Activation in Neocortical Neurons

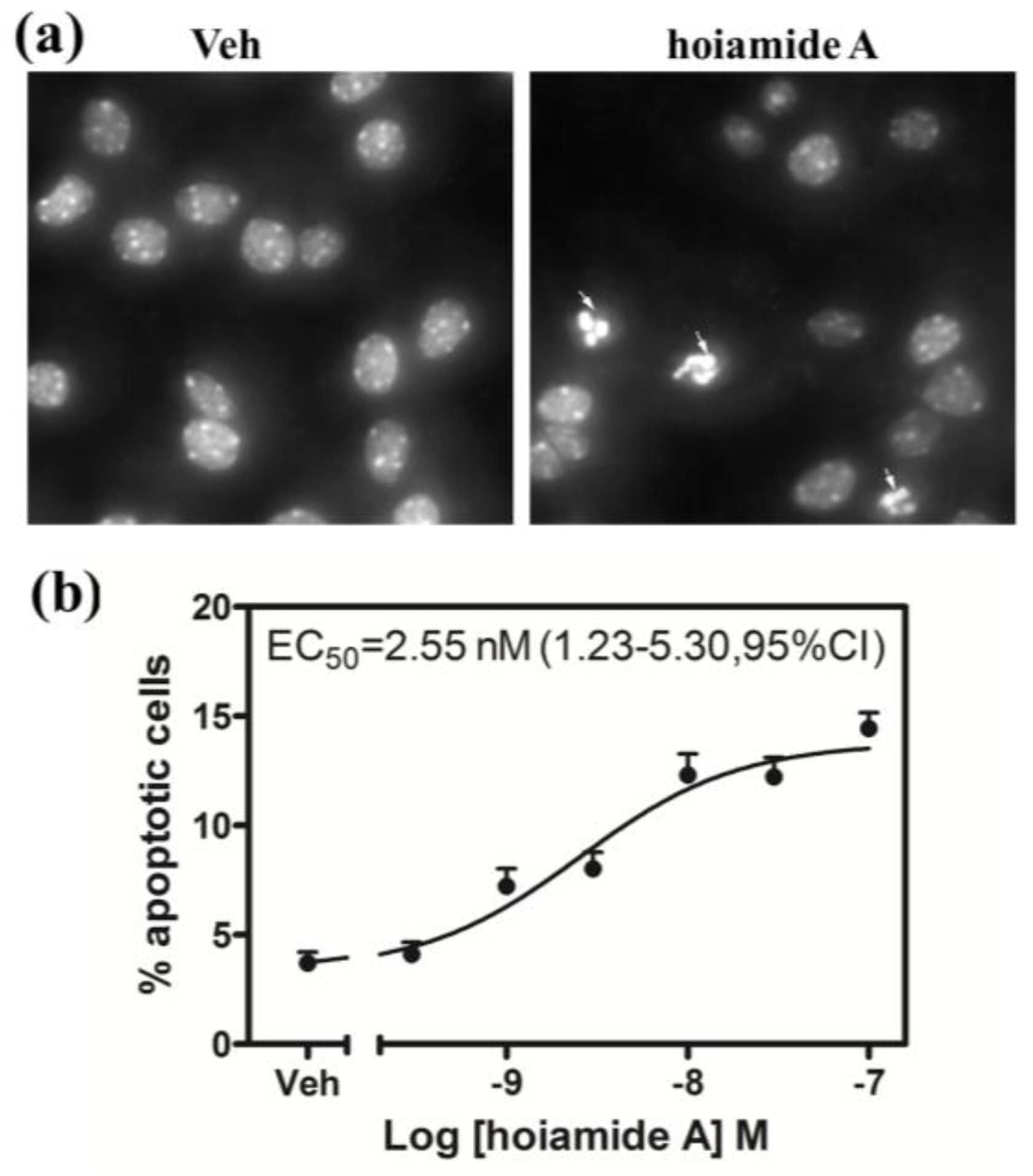
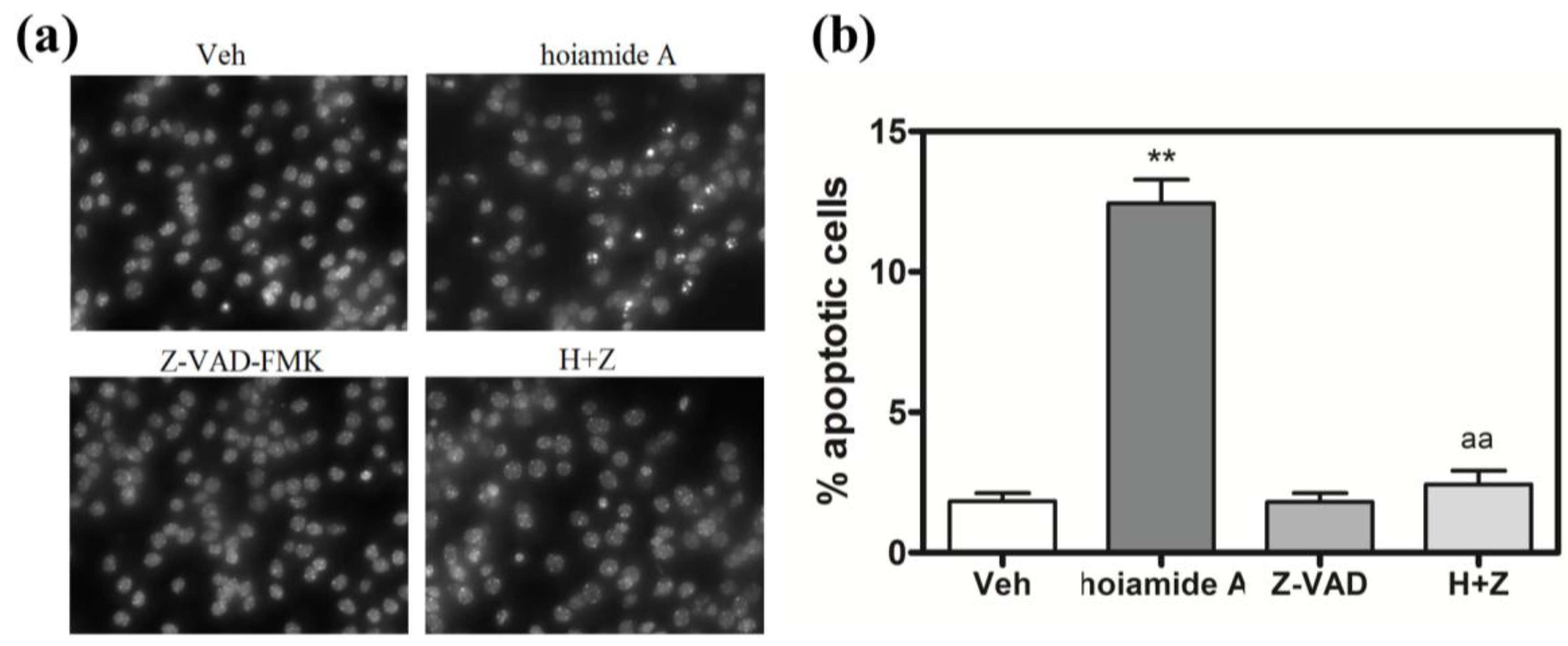
2.4. Hoiamide A Produces Nuclear Condensation in Neocortical Neurons

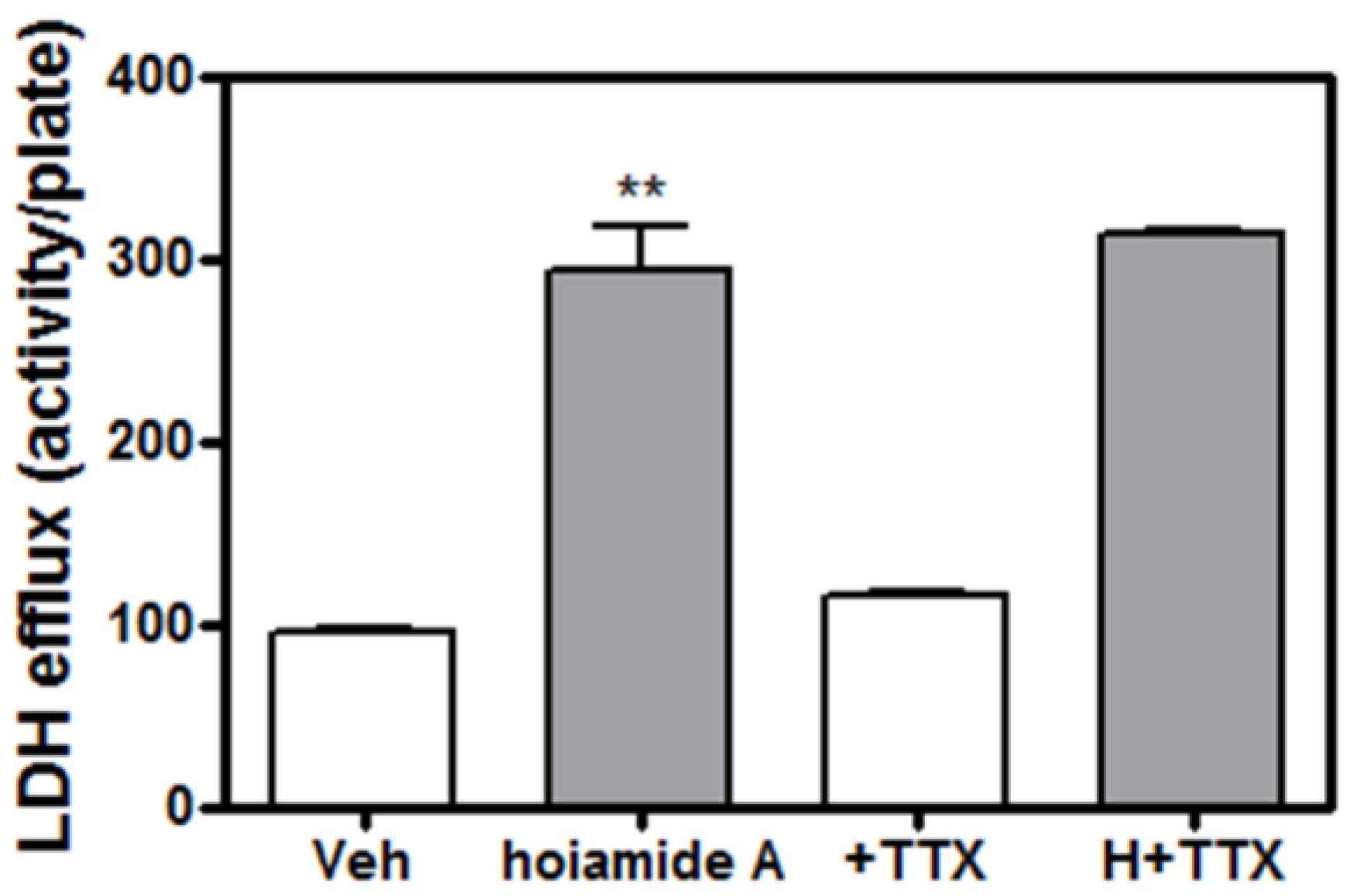
2.5. Tetrodotoxin (TTX) is without Effect on Hoiamide A-Induced Neurotoxicity

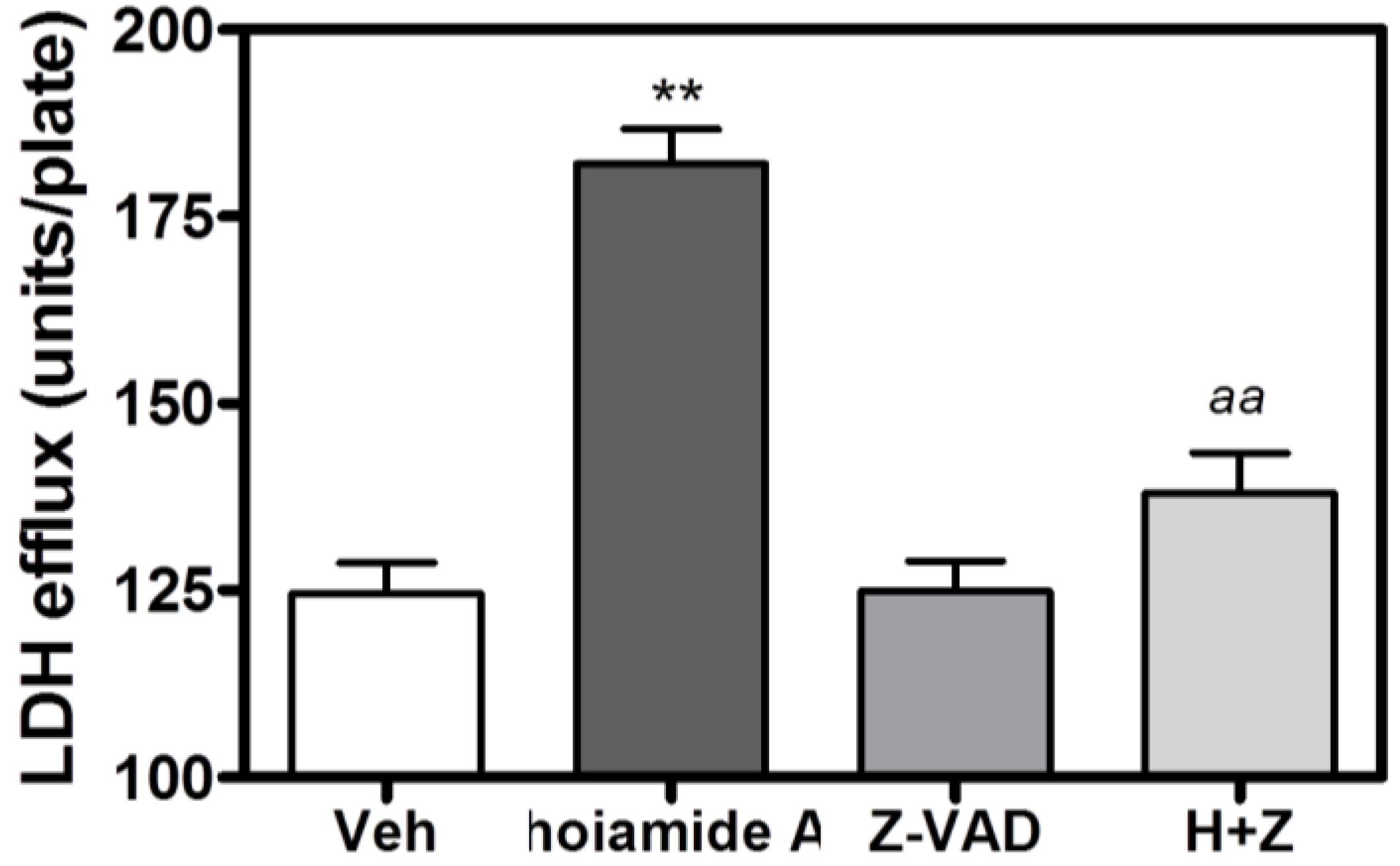
2.6. Caspase Inhibitor, Z-VAD-FMK, Antagonizes Hoiamide A-Induced Neurotoxicity


2.7. Lack of Effect of MK-801, Nifedipine, NBQX and KBR-7943 on Hoiamide A-Induced Neurotoxicity in Neocortical Neurons
2.8. The JNK Inhibitor, SP 600125, Protects Neocortical Neurons from Hoiamide A-Induced Neurotoxicity

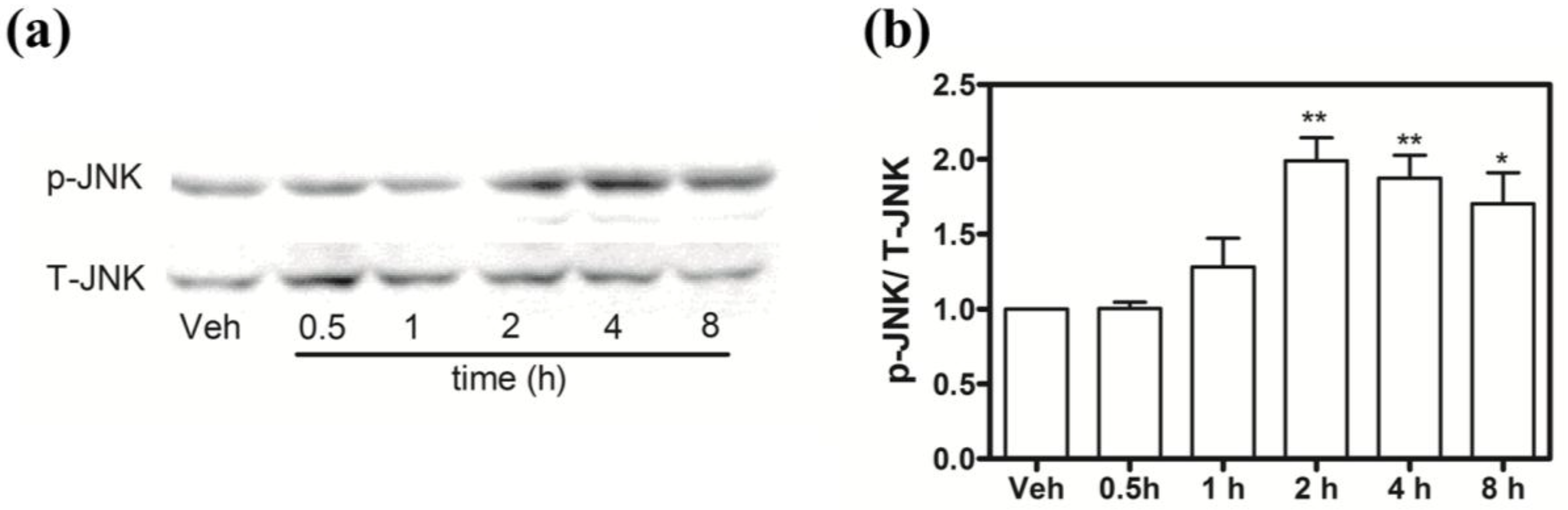
2.9. Hoiamide A Stimulates JNK Phosphorylation on Neocortical Neurons

3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Neocortical Neuron Culture
4.3. Lactate Dehydrogenase (LDH) Activity Assay
4.4. Hoechst 33342 Staining
4.5. Caspase-3 Activity Assay
4.6. Western Blotting
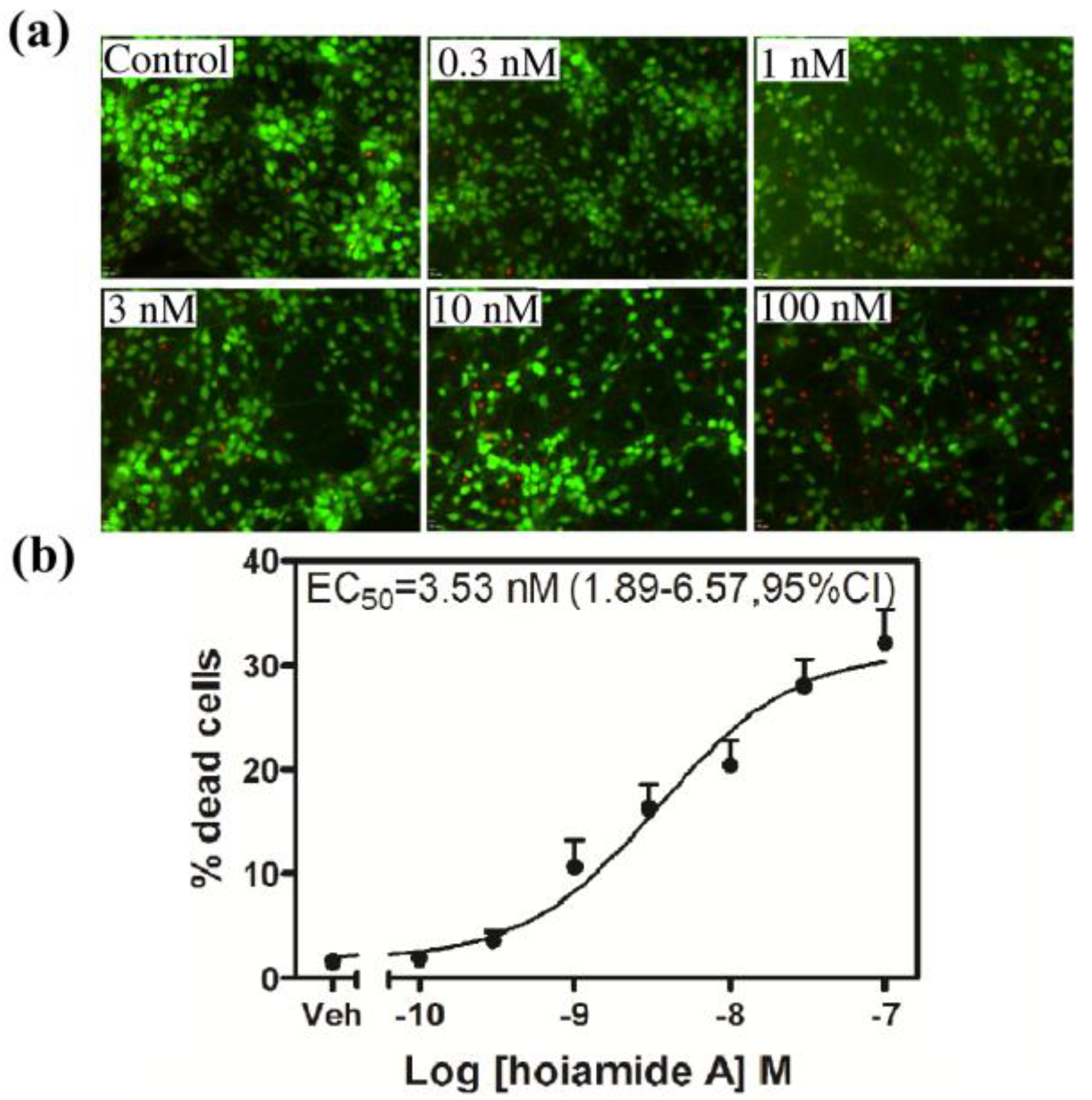
4.7. Fluorescein Diacetate (FDA) and Propidium Iodide (PI) Staining
4.8. Diolistic Labeling
4.9. Data Analysis
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gerwick, W.H.; Tan, L.T.; Sitachitta, N. Nitrogen-containing metabolites from marine cyanobacteria. Alkaloids Chem. Biol. 2001, 57, 75–184. [Google Scholar] [PubMed]
- Nogle, L.M.; Williamson, R.T.; Gerwick, W.H. Somamides a and b, two new depsipeptide analogues of dolastatin 13 from a Fijian cyanobacterial assemblage of Lyngbya majuscula and schizothrix species. J. Nat. Prod. 2001, 64, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Bayne, C.J.; Gerwick, L. The acute phase response and innate immunity of fish. Dev. Comp. Immunol. 2001, 25, 725–743. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.; Friedman, A.J.; Choi, H.; Hogan, J.; McCammon, J.A.; Hook, V.; Gerwick, W.H. The marine cyanobacterial metabolite gallinamide A is a potent and selective inhibitor of human cathepsin l. J. Nat. Prod. 2014, 77, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.J.; Shaw, G.R.; Webb, P.M. Health effects of recreational exposure to Moreton bay, Australia waters during a Lyngbya majuscula bloom. Environ. Int. 2007, 33, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.; O’Neil, J.M.; Udy, J.W.; Ahern, K.S.; O’Sullivan, C.M.; Dennison, W.C. Blooms of the cyanobacterium Lyngbya majuscula in coastal Queensland, Australia: Disparate sites, common factors. Mar. Pollut. Bull. 2005, 51, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.J.T.; Webb, P.M.; Shaw, G.R. The toxins of lyngbya majuscula and their human and ecological health effects. Environ. Int. 2001, 27, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Izumi, A.K.; Moore, R.E. Seaweed (Lyngbya majuscula) dermatitis. Clin. Dermatol. 1987, 5, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.J. Occupational dermatitis caused by Lyngbya majuscula in Australia. Int. J. Dermatol. 2012, 51, 122–123. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Lee, P.P.; Tan, L.T. Hantupeptins b and c, cytotoxic cyclodepsipeptides from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 2010, 71, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Lee, P.P.; Tan, L.T. Hantupeptin a, a cytotoxic cyclic depsipeptide from a Singapore collection of Lyngbya majuscula. J. Nat. Prod. 2009, 72, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Chan, K.P.; Chen, D.Y.; Tan, L.T. Lagunamide c, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 2011, 72, 2369–2375. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Nunnery, J.K.; Engene, N.; Esquenazi, E.; Byrum, T.; Dorrestein, P.C.; Gerwick, W.H. Palmyramide A, a cyclic depsipeptide from a Palmyra Atoll collection of the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2010, 73, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.T.; Okino, T.; Gerwick, W.H. Hermitamides a and b, toxic malyngamide-type natural products from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Han, B.N.; Gross, H.; Goeger, D.E.; Mooberry, S.L.; Gerwick, W.H. Aurilides B and C, cancer cell toxins from a papua new guinea collection of the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2006, 69, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.T.; Chang, Y.Y.; Ashootosh, T. Besarhanamides A and B from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 2008, 69, 2067–2069. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.; Andrianasolo, E.H.; Shin, W.K.; Goeger, D.E.; Yokochi, A.; Schemies, J.; Jung, M.; France, D.; Cornell-Kennon, S.; Lee, E.; et al. Structural and synthetic investigations of tanikolide dimer, a SIRT2 selective inhibitor, and tanikolide seco acid from the Madagascar marine cyanobacterium Lyngbya majuscula. J. Org. Chem. 2009, 74, 5267–5275. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.; Suyama, T.L.; Engene, N.; Wingerd, J.S.; Matainaho, T.; Gerwick, W.H. Apratoxin D, a potent cytotoxic cyclodepsipeptide from papua new guinea collections of the marine cyanobacteria Lyngbya majuscula and Lyngbya sordida. J. Nat. Prod. 2008, 71, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.; Tidgewell, K.; Capson, T.L.; Engene, N.; Almanza, A.; Schemies, J.; Jung, M.; Gerwick, W.H. Malyngolide dimer, a bioactive symmetric cyclodepside from the Panamanian marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2010, 73, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Li, W.I.; Berman, F.W.; Okino, T.; Yokokawa, F.; Shioiri, T.; Gerwick, W.H.; Murray, T.F. Antillatoxin is a marine cyanobacterial toxin that potently activates voltage-gated sodium channels. Proc. Natl. Acad. Sci. USA 2001, 98, 7599–7604. [Google Scholar] [CrossRef] [PubMed]
- Berman, F.W.; Gerwick, W.H.; Murray, T.F. Antillatoxin and kalkitoxin, ichthyotoxins from the tropical cyanobacterium Lyngbya majuscula, induce distinct temporal patterns of NMDA receptor-mediated neurotoxicity. Toxicon 1999, 37, 1645–1648. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.; Cao, Z.Y.; Murray, T.F.; Gerwick, W.H. Hoiamide a, a sodium channel activator of unusual architecture from a consortium of two Papua New Guinea cyanobacteria. Chem. Biol. 2009, 16, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Pereira, A.R.; Cao, Z.; Shuman, C.F.; Engene, N.; Byrum, T.; Matainaho, T.; Murray, T.F.; Mangoni, A.; Gerwick, W.H. The hoiamides, structurally intriguing neurotoxic lipopeptides from Papua New Guinea marine cyanobacteria. J. Nat. Prod. 2010, 73, 1411–1421. [Google Scholar] [CrossRef] [PubMed]
- Malloy, K.L.; Choi, H.; Fiorilla, C.; Valeriote, F.A.; Matainaho, T.; Gerwick, W.H. Hoiamide D, a marine cyanobacteria-derived inhibitor of p53/MDM2 interaction. Bioorg. Med. Chem. Lett. 2012, 22, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, Z.; Ye, T. Total synthesis of hoiamide C. Org. Lett. 2011, 13, 2506–2509. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Dravid, S.M.; Prakash, A.; Xie, J.; Peterson, J.; Jabba, S.V.; Baden, D.G.; Murray, T.F. Sodium channel activation augments NMDA receptor function and promotes neurite outgrowth in immature cerebrocortical neurons. J. Neurosci. 2009, 29, 3288–3301. [Google Scholar] [CrossRef] [PubMed]
- Jabba, S.V.; Prakash, A.; Dravid, S. M.; Gerwick, W.H.; Murray, T.F. I Antillatoxin, a novel lipopeptide, enhances neurite outgrowth in immature cerebrocortical neurons through activation of voltage-gated sodium channels. J. Pharmacol. Exp. Ther. 2010, 332, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.M.; Gunasekar, P.G.; Li, L.; Borowitz, J.L.; Isom, G.E. Differential susceptibility of brain areas to cyanide involves different modes of cell death. Toxicol. Appl. Pharmacol. 1999, 156, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Mills, L.D.; Zhang, L.; Marler, R.; Svingen, P.; Fernandez-Barrena, M.G.; Dave, M.; Bamlet, W.; McWilliams, R.R.; Petersen, G.M.; Faubion, W.; et al. Inactivation of the transcription factor GLI1 accelerates pancreatic cancer progression. J. Biol. Chem. 2014, 289, 16516–16525. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Maravei, D.V.; Bergeron, L.; Wang, S.; Perez, G.I.; Tsutsumi, O.; Taketani, Y.; Asano, M.; Horai, R.; Korsmeyer, S.J.; et al. Caspase-2 deficiency prevents programmed germ cell death resulting from cytokine insufficiency but not meiotic defects caused by loss of ataxia telangiectasia-mutated (Atm) gene function. Cell Death Differ. 2001, 8, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Maravei, D.V.; Trbovich, A.M.; Perez, G.I.; Tilly, K.I.; Banach, D.; Talanian, R.V.; Wong, W.W.; Tilly, J.L. Cleavage of cytoskeletal proteins by caspases during ovarian cell death: Evidence that cell-free systems do not always mimic apoptotic events in intact cells. Cell Death Differ. 1997, 4, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.Y.; LePage, K.T.; Frederick, M.O.; Nicolaou, K.C.; Murray, T.F. Involvement of caspase activation in azaspiracid-induced neurotoxicity in neocortical neurons. Toxicol. Sci. 2010, 114, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Xiao, A.Y.; Wang, X.Q.; Yang, A.Z.; Yu, S.P. Slight impairment of na+,k+-atpase synergistically aggravates ceramide- and β-amyloid-induced apoptosis in cortical neurons. Brain Res. 2002, 955, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Xiao, A.Y.; Wei, L.; Xia, S.L.; Rothman, S.; Yu, S.P. Ionic mechanism of ouabain-induced concurrent apoptosis and necrosis in individual cultured cortical neurons. J. Neurosci. 2002, 22, 1350–1362. [Google Scholar] [PubMed]
- Yu, S.P. Na+, k+-ATPase: The new face of an old player in pathogenesis and apoptotic/hybrid cell death. Biochem. Pharmacol. 2003, 66, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Ying, D.J.; Cui, L.; Langsdorf, J.; Yu, S.P. Necrosis, apoptosis and hybrid death in the cortex and thalamus after barrel cortex ischemia in rats. Brain Res. 2004, 1022, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [PubMed]
- Davis, R.J. Signal transduction by the jnk group of map kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Cui, Y.; Busse, E.; Mehrotra, S.; Rainier, J.D.; Murray, T.F. Gambierol inhibition of voltage-gated potassium channels augments spontaneous Ca2+ oscillations in cerebrocortical neurons. J. Pharmacol. Exp. Ther. 2014, 350, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.Y.; Choi, D.W. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J. Neurosci. Methods 1987, 20, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.H.; Senft, J. A. An improved method to determine cell viability by simultaneous staining with fluorescein diacetate-propidium iodide. J. Histochem. Cytochem. 1985, 33, 77–79. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Z.; Li, X.; Zou, X.; Greenwood, M.; Gerwick, W.H.; Murray, T.F. Involvement of JNK and Caspase Activation in Hoiamide A-Induced Neurotoxicity in Neocortical Neurons. Mar. Drugs 2015, 13, 903-919. https://doi.org/10.3390/md13020903
Cao Z, Li X, Zou X, Greenwood M, Gerwick WH, Murray TF. Involvement of JNK and Caspase Activation in Hoiamide A-Induced Neurotoxicity in Neocortical Neurons. Marine Drugs. 2015; 13(2):903-919. https://doi.org/10.3390/md13020903
Chicago/Turabian StyleCao, Zhengyu, Xichun Li, Xiaohan Zou, Michael Greenwood, William H. Gerwick, and Thomas F. Murray. 2015. "Involvement of JNK and Caspase Activation in Hoiamide A-Induced Neurotoxicity in Neocortical Neurons" Marine Drugs 13, no. 2: 903-919. https://doi.org/10.3390/md13020903
APA StyleCao, Z., Li, X., Zou, X., Greenwood, M., Gerwick, W. H., & Murray, T. F. (2015). Involvement of JNK and Caspase Activation in Hoiamide A-Induced Neurotoxicity in Neocortical Neurons. Marine Drugs, 13(2), 903-919. https://doi.org/10.3390/md13020903