2. Results and Discussion
Compound
1 was isolated as white amorphous powder. The molecular formula was deduced to be C
20H
20O
7 (corresponding to eleven degrees of unsaturation) on the basis of its HREIMS molecular ion cluster at 372.1209 [M]
+ (calcd. for C
20H
20O
7, 372.1204). The IR spectrum showed absorption bands at 3394 and 1643 cm
−1, which revealed the presence of hydroxyl and carbonyl groups. In the
1H NMR spectrum, the signals for two
meta-coupled aromatic protons at δ
H 6.03 (d,
J = 1.8 Hz, H-4′′) and 6.08 (d,
J = 1.8 Hz, H-6′′), two
E-configured olefinic protons at δ
H 6.28 (
J = 15.8 Hz, H-1′) and 6.58 (
J = 15.8 Hz, H-2′), one singlet olefinic proton (δ
H 6.16, H-3), one oxymethine (δ
H 5.11, H-5), one methylene (δ
H 3.07 and 2.70, H
2-4) and three methyls (δ
H 1.56, H
3-8; 2.39, H
3-8′′; 1.83, H
3-3′) were observed (
Table 1). The
1H–
1H COSY spectrum indicated two spin-systems of H-4/H-5 and H-1′/H-2′/H-3′, assigned to two fragments of –OCH–CH
2– and –CH=CH–CH
3 (
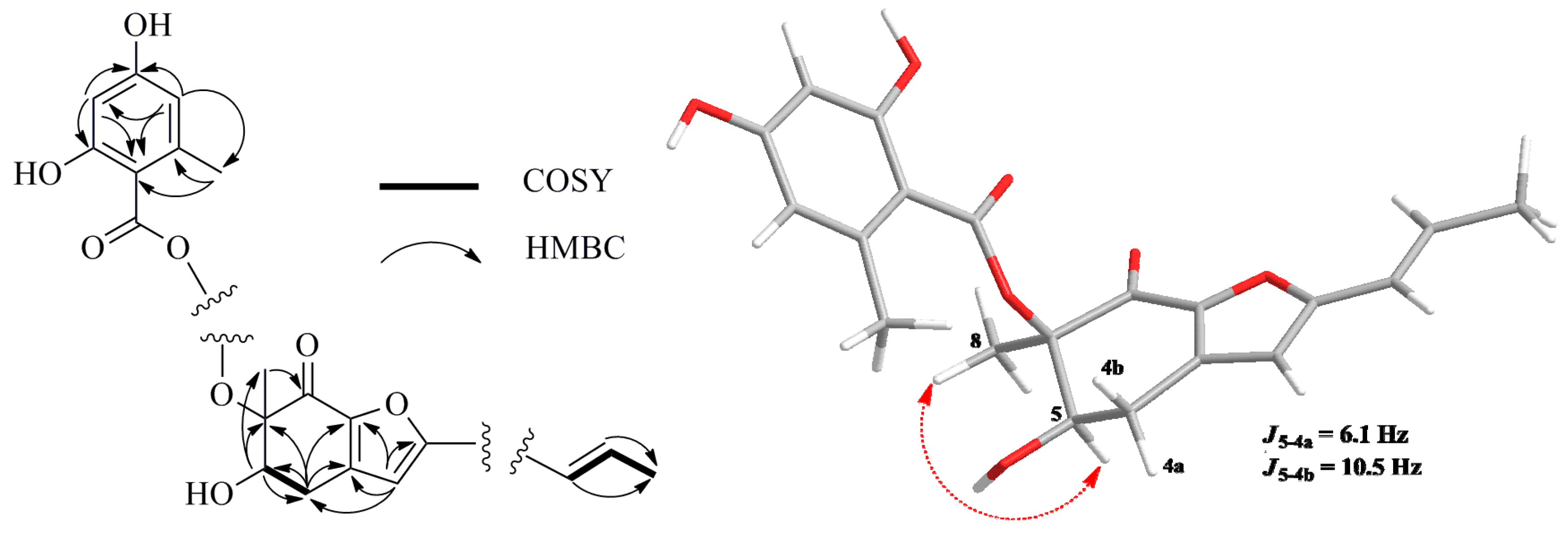
Figure 2), respectively. The HMBC correlations from H
3-8 to C-5, C-6 and C-7, from H-5 to C-4, C-6 and C-8, from H-4a to C-3, C-3a, C-5, C-6 and C-7a and from H-4b to C-5, C-3a and C-7a constructed a cyclohexenone fragment. The HMBC correlations from the singlet olefinic proton H-3 to C-1′, C-4, C-3a, C-7a and C-2 (δ
C 161.0) assembled a furan ring, which was connected with the cyclohexenone by sharing the bond of C-3a//C-7a. Judged by the following HMBC correlations of H-4′′ to C-3′′, C-5′′, C-6′′ and C-8′′; H-6′′ to C-2′′, C-4′′, C-5′′ and C-7′′ and H
3-8′′ to C-2′′, C-3′′ and C-4′′, the moiety of orsellinic acid was constructed, which was connected to C-6 (δc 87.3) on the basis of an HMBC correlation from H
3-8 to C-1′′. The HMBC correlations from H-1′ to C-2, C-3 and C-2′ determined that the fragment –CH=CH–CH
3 was connected to C-2. Thus, the planar structure of
1 was deduced.
Table 1.
13C NMR (125 MHz) and 1H NMR (500 MHz) data of 1 (CDCl3).
Table 1.
13C NMR (125 MHz) and 1H NMR (500 MHz) data of 1 (CDCl3).
| Position | δc, Type | δH, mult (J in Hz) |
|---|
| 2 | 161.0, C | |
| 3 | 107.8, CH | 6.16, s |
| 3a | 138.7, C | |
| 4 | 28.8, CH2 | 4a, 3.07, dd (16.6, 6.1) |
| | 4b, 2.70, dd (16.6, 10.5) |
| 5 | 69.6, CH | 5.11, dd (6.1, 10.5) |
| 6 | 87.3, C | |
| 7 | 181.9, C | |
| 7a | 144.1, C | |
| 8 | 16.4, CH3 | 1.56, s |
| 1′ | 119.0, CH | 6.28, dd (15.8, 1.4) |
| 2′ | 134.2, CH | 6.58, dd (15.8, 6.7) |
| 3′ | 18.9, CH3 | 1.83, d (6.7) |
| 1′′ | 170.3, C | |
| 2′′ | 104.8, C | |
| 3′′ | 144.2, C | |
| 4′′ | 112.2, CH | 6.03, d (1.8) |
| 5′′ | 161.6, C | |
| 6′′ | 101.2, CH | 6.08, d (1.8) |
| 7′′ | 164.9, C | |
| 8′′ | 24.4, CH3 | 2.39, s |
Figure 2.
Selected 1H–1H COSY (bold line), HMBC (arrow) and key NOESY (dashed lines) correlations of Compound 1.
Figure 2.
Selected 1H–1H COSY (bold line), HMBC (arrow) and key NOESY (dashed lines) correlations of Compound 1.
The relative configuration of
1 was established by interpretation of
1H NMR and NOESY data. In the
1H NMR spectrum, H-5 showed
ax/
eq coupling (
J = 6.1 Hz) to H-4a and
ax/
ax coupling (
J = 10.5 Hz) to H-4b. In the NOESY spectrum, the NOE correlation between H-5 and H
3-8 as shown in
Figure 2 supported the
syn relationship of H-5 and H
3-8. In order to establish the absolute configuration of
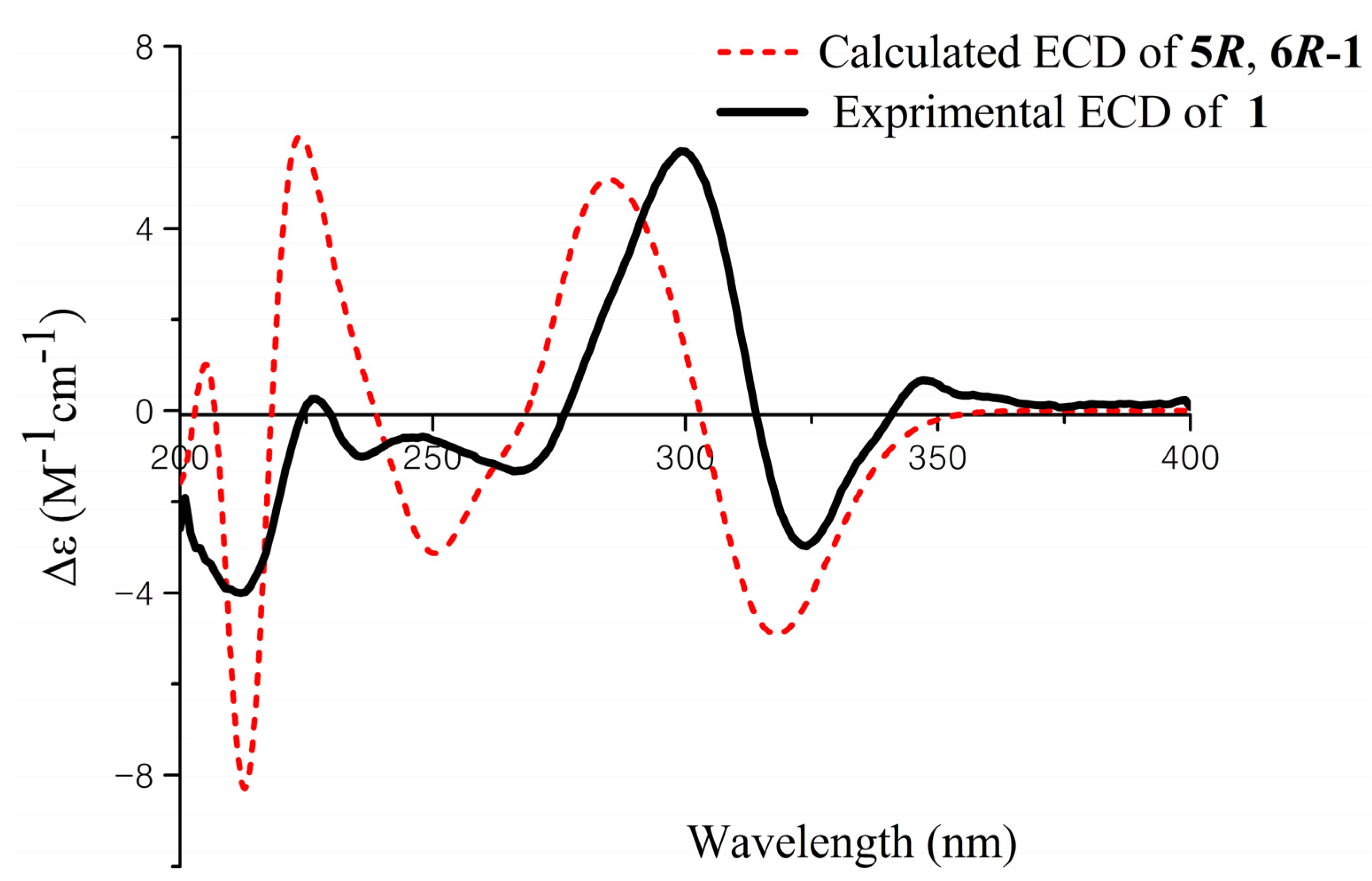
1, the ECD spectrum of
1 was recorded and compared with density functional theory (DFT)-calculated spectra for the 5
R, 6
R and 5
S, 6
S isomers of
1 (
Figure S25, Supplementary Information). The preliminary conformational distribution search was performed by Spartan 10 software using the MMFF94S force field. The corresponding minimum geometries were further fully optimized by DFT at the B3LYP/6-31G (d) level implemented in the Gaussian 03 program package. The obtained stable conformers were submitted to ECD calculation by the Time Dependent Density Functional Theory (TDDFT) cam-b3lyp/6-311+g (2d, p) method [
16,
17] (
Figure S26, Supplementary Information). The calculated ECD spectra for isomer 5
R, 6
R showed a good fit with the experimental CD of
1, which reproduced Cotton effects at 298 and 324 nm, respectively (
Figure 3). Thus, the configuration of
1 was assigned to be 5
R, 6
R, named aspergifuranone.
Figure 3.
Experimental and calculated ECD spectra of 1.
Figure 3.
Experimental and calculated ECD spectra of 1.
Compound
2 was obtained as a white amorphous powder. Its molecular formula was deduced to be C
12H
14O
6 on the basis of its HREIMS molecular ion cluster at
m/
z 254.0786 [M]
+ (calcd. for C
12H
14O
6, 254.0785). The IR spectrum displayed characteristic absorption bands at 3414 and 1661 cm
−1, suggesting the presence of hydroxyl and carbonyl groups. The
1H and
13C NMR spectra of
2 revealed the signals of a penta-substituted aryl ring (δ
H 6.62, 1H, s; δ
C 162.9, 162.4, 141.2, 111.4, 107.8 and 100.0), a oxygenated methine (δ
H 4.46, 1H, s; δ
C 69.8), a methoxyl (δ
H 3.38, 3H, s; δ
C 50.4) and two methyls (δ
H 1.63, 3H, s; δ
C 18.4; δ
H 2.08, 3H, s; δ
C 7.9) (
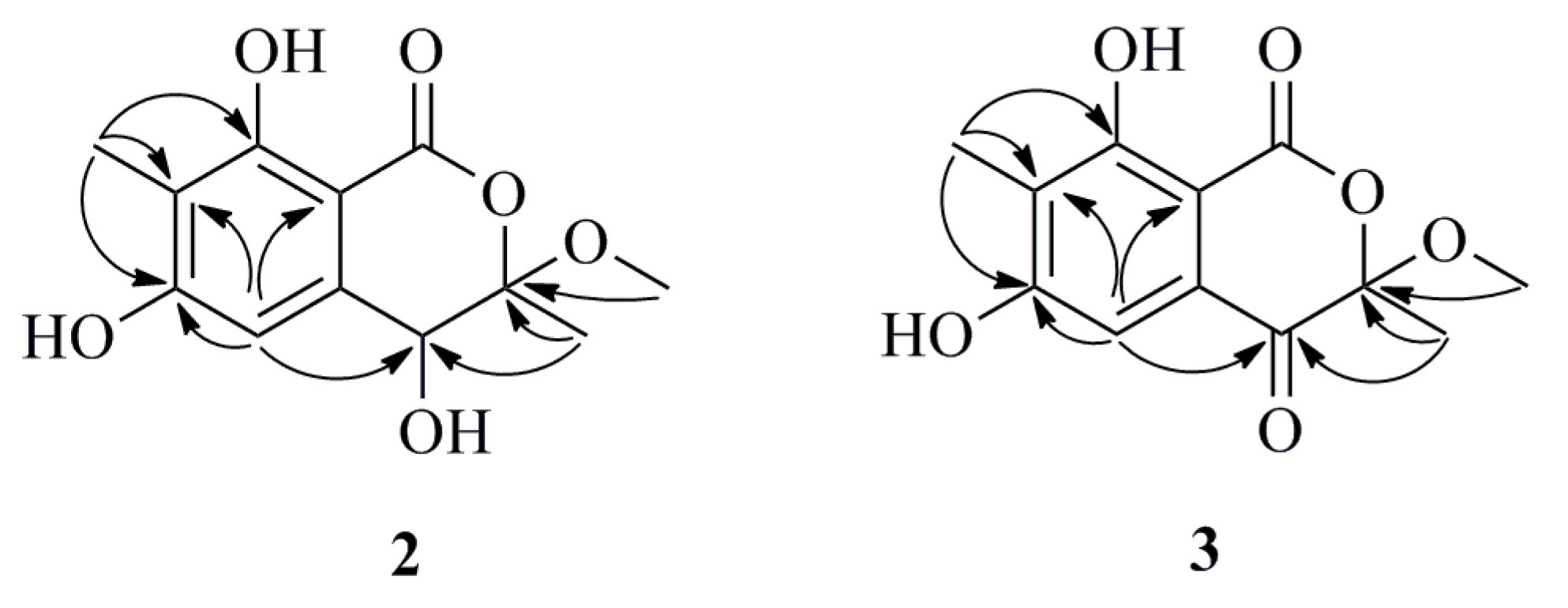
Table 2). In the HMBC spectrum, the correlations from 8-OH (δ
H 11.47) to C-7 and C-9, from H-5 to C-4, C-6, C-7 and C-9, from H
3-13 (δ
H 2.08) to C-6, C-7 and C-8 and from H
3-12 (δ
H 1.63) to C-3 and C-4 confirmed the existence of a 4,6,8-trihydroxy-3,7-dimethylisocoumarin unit (
Figure 4). Moreover, the methoxyl group was attached to the C-3 position due to the HMBC correlation between H
3-11 and C-3 (δ
C 108.3). Therefore, the planar structure of
2 was constructed as shown in
Figure 4. The absence of any CD signal of
2 and the value of optical rotation was zero, all indicating a racemic mixture of the possible enantiomer. Subsequently, (±) it was attempted to separate
2 by chiral HPLC using three types of chiral columns, but all failed due to only one symmetrical peak in the chromatogram, as detected by HPLC.
Compound
3 was obtained as a white amorphous powder. Its molecular formula was determined to be C
12H
12O
6 on the basis of its HREIMS molecular ion cluster at
m/
z 252.0630 [M]
+ (calcd. for C
12H
12O
6, 252.0628). Analysis of the
1H and
13C NMR data for
3 revealed the presence of nearly the same identical structural features as those found in
2, except that the carbon signal of C-4 (δ
C 69.8) in
2 was absent and replaced by a carboxyl group signal (δ
C 190.1), which indicated that Compound
3 was the oxidative product of Compound
2 (
Figure 4). However, the absence of any CD spectrum and zero optical rotation indicated that
3 was also a racemic mixture. Unfortunately, separation of
3’s enantiomers also failed.
Table 2.
1H and 13C NMR data of Compounds 2 a and 3 b.
Table 2.
1H and 13C NMR data of Compounds 2 a and 3 b.
| Position | 2 | 3 |
|---|
| δc, Type | δH, mult (J in Hz) | δc, Type | δH, mult (J in Hz) |
|---|
| 1 | 169.3, C | | 168.6, C | |
| 2 | | | | |
| 3 | 108.3, C | | 106.2, C | |
| 4 | 69.8, CH | 4.46, s | 190.1, C | |
| 5 | 107.8, CH | 6.62, s | 106.3, CH | 7.01, s |
| 6 | 162.9, C | | 164.1, C | |
| 7 | 111.4, C | | 120.9, C | |
| 8 | 162.4, C | | 162.6, C | |
| 9 | 100.0, C | | 103.2, C | |
| 10 | 141.2, C | | 130.6, C | |
| 11 | 50.4, CH3 | 3.38, s | 52.0, CH3 | 3.41, s |
| 12 | 18.4, CH3 | 1.63, s | 21.1, CH3 | 1.68, s |
| 13 | 7.9, CH3 | 2.08, s | 8.6, CH3 | 2.15, s |
| 8-OH | | 11.47, s | | |
Figure 4.
Selected HMBC (arrow) correlations of Compounds 2 and 3.
Figure 4.
Selected HMBC (arrow) correlations of Compounds 2 and 3.
Compound
4 was obtained as a white amorphous powder. Its molecular formula was assigned to be C
12H
14O
5 on the basis of its HREIMS molecular ion cluster at
m/
z 238.0835 [M]
+ (calcd. for C
12H
14O
5, 238.0836). The IR spectrum showed the presence of a hydroxyl group (3138 cm
−1) and a conjugated carbonyl group (1741 cm
−1). Overall inspections of the
1H and
13C NMR spectroscopic data revealed that Compound
4 also possessed a penta-substituted aryl ring (δ
H 6.59, 1H, s; δ
C 159.7, 152.2, 127.0, 125.7, 116.6 and 109.0) (
Table 3). By analysis of the
1H–
1H COSY spectrum, the correlation H
2-1′/H
2-2′ and H
2-2′/H
3-3′ indicated the presence of a propyl moiety. The HMBC correlations from H-4 to C-3 (δ
C 107.8), C-5 (δ
C 152.2), C-6 (δ
C 159.7), C-8 (δ
C 116.6) and C-9 (δ
C 127.0) and from H
3-10 to C-6 (δ
C 159.7), C-7 (δ
C 125.7) and C-8 (δ
C 116.6) indicated the position of two hydroxyl group were ortho. The correlations from H
3-10 to C-7 (δ
C 125.7) and from H-1′ to C-3 (δ
C 107.8) revealed that the methyl and propyl groups were attached to the C-7 and C-3 positions, respectively (
Figure 5). Thus, the planar structure of Compound
4 was constructed. The absolute configuration of
4 was established by comparing its optical rotation data and CD with those of the literature [
18]. The opposite CD Cotton effects to purpurester A at 286 nm (∆ɛ
max + 0.8) and 232 nm (∆ɛ
max − 1.5) and the opposite optical direction of
+30 (
c 0.1, MeOH) clearly revealed the 3
R-configuration
, named (
R)-3-demethylpurpurester A.
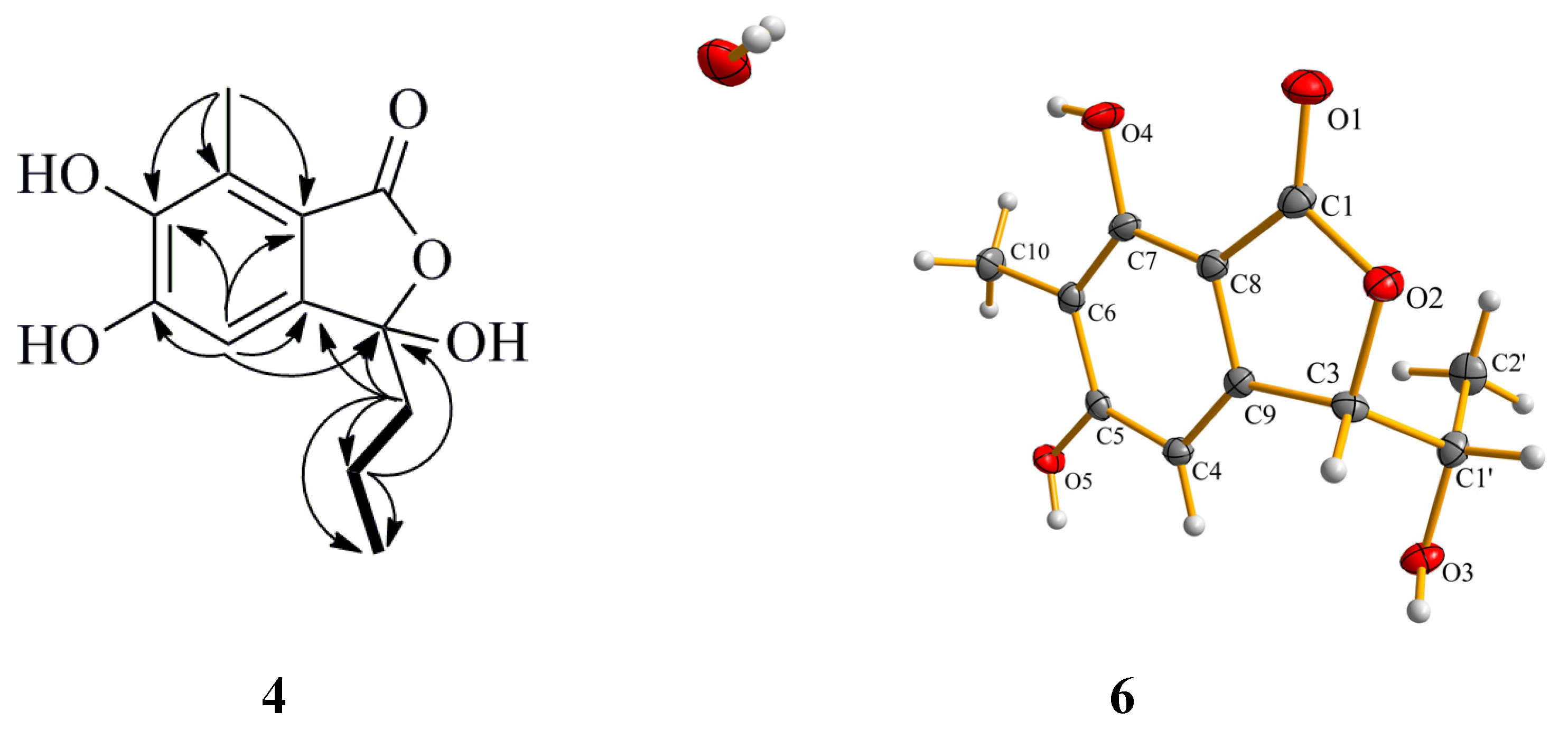
Figure 5.
Selected HMBC (arrow) correlations of Compound 4 and perspective Oak Ridge Thermal Ellipsoid Plot (ORTEP) drawing for Compound 6.
Figure 5.
Selected HMBC (arrow) correlations of Compound 4 and perspective Oak Ridge Thermal Ellipsoid Plot (ORTEP) drawing for Compound 6.
Table 3.
13C (125 MHz) and 1H (500 MHz) NMR data of Compounds 4 and 5 (methanol-d4).
Table 3.
13C (125 MHz) and 1H (500 MHz) NMR data of Compounds 4 and 5 (methanol-d4).
| Position | 4 | 5 |
|---|
| δc, Type | δH, mult (J in Hz) | δc, Type | δH, mult (J in Hz) |
|---|
| 1 | 171.6, C | | 167.5, C | |
| 2 | | | | |
| 3 | 107.8, C | | 143.7, C | |
| 4 | 109.0, CH | 6.59, s | 108.6, CH | 6.64, s |
| 5 | 152.2, C | | 151.6, C | |
| 6 | 159.7, C | | 158.1, C | |
| 7 | 125.7, C | | 123.7, C | |
| 8 | 116.6, C | | 114.9, C | |
| 9 | 127.0, C | | 117.3, C | |
| 10 | 9.4, CH3 | 2.35, s | 9.8, CH3 | 2.37, s |
| 1′ | 40.3, CH2 | 2.18, m | 109.9, CH | 5.69, t (7.7) |
| 2′ | 18.1, CH2 | 1.26, m | 19.5, CH2 | 2.40, m |
| | 1.06, m | | |
| 3′ | 14.3, CH3 | 0.88, t (7.4) | 14.8, CH3 | 1.11, t (7.7) |
The structures of the known compounds were identified as purpuresters B (
5) [
18] (
Table 3) and pestaphthalides A (
6) [
19] by comparison of their spectroscopic and optical rotation data with those reported in the literature. The structure of Compound
6 was further confirmed by single-crystal X-ray diffraction experiment using CuKα radiation [
20] (
Figure 5) (Cambridge Crystallographic Data Centre (CCDC) Number 1053019).
All compounds were tested for their
in vitro inhibitory activities against α-glucosidase along with the clinical α-glucosidase inhibitor acarbose (positive control) [
21]. As a result (
Table 4), Compound
1 was the most active and showed better inhibitory potential (IC
50 = 9.05 ± 0.60 μM) than acarbose (IC
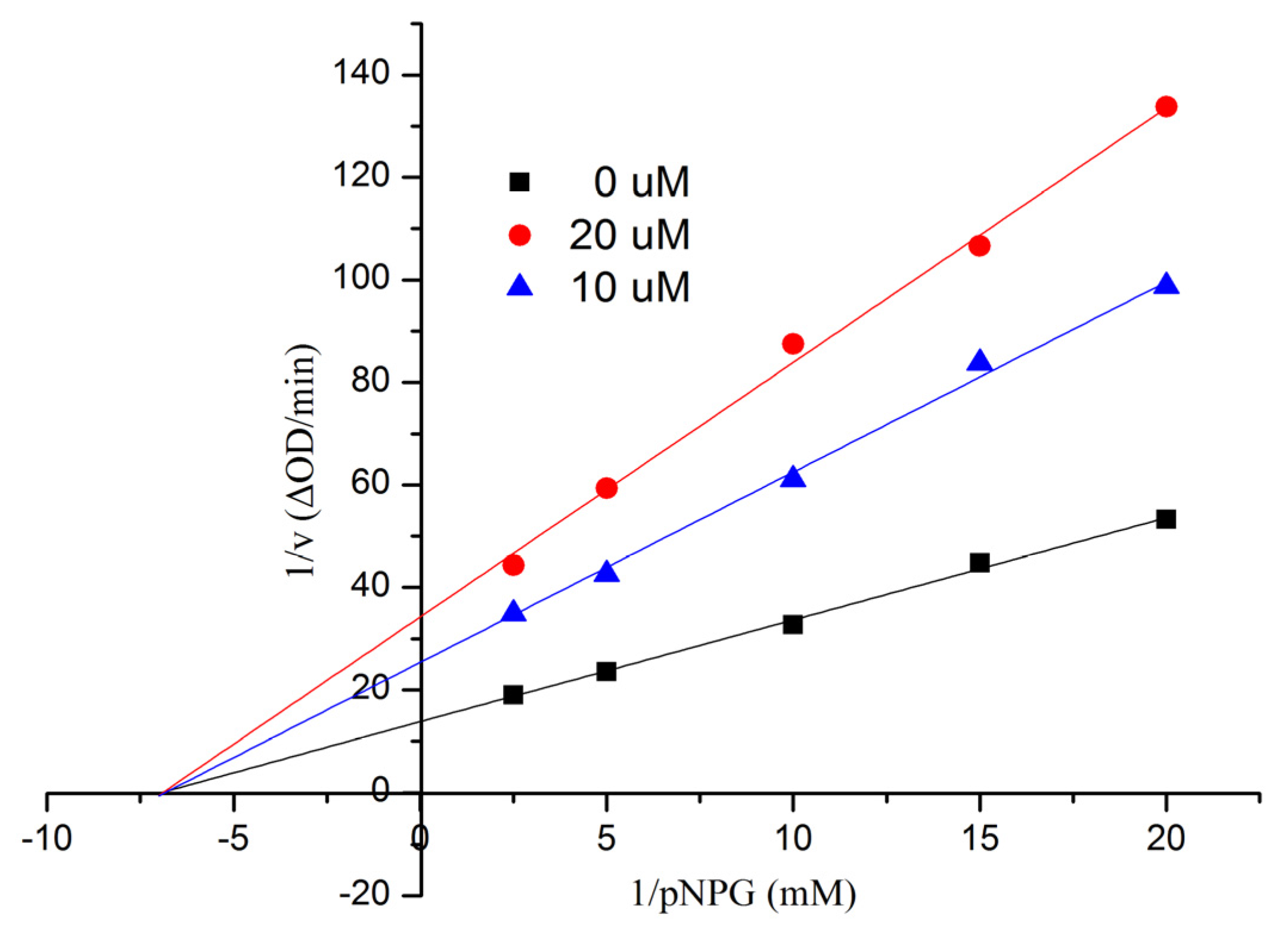
50 = 553.7 ± 6.8 μM). In order to examine the inhibition type of Compound
1, further kinetic studies were carried out by the Lineweaver-Burk plot method. The results are shown in
Figure 6, indicating that
1 was a noncompetitive inhibitor of α-glucosidase. Compounds
2 and
6 exhibited more efficacy than that of the positive control, with IC
50 values of 90.4 ± 2.9 and 69.6 ± 3.5 μM, respectively. More details are given in the
Supplementary Information.
Table 4.
Inhibitory effects of the isolates against α-glucosidase a.
Table 4.
Inhibitory effects of the isolates against α-glucosidase a.
| Compound | 1 | 2 | 3 | 4 | 5 | 6 | Acarbose b |
|---|
| IC50 (μM) | 9.05 ± 0.60 | 90.4 ± 2.9 | >200 | >200 | >200 | 69.6 ± 3.5 | 553.7 ± 6.8 |
Figure 6.
Kinetic analysis of the inhibition of α-glucosidase of 1.
Figure 6.
Kinetic analysis of the inhibition of α-glucosidase of 1.
3. Experimental Section
3.1. General
Melting points were measured on an X-4 micro-melting-point apparatus (Cany Precision Instruments Co., Ltd., Shanghai, China) and are uncorrected. Optical rotations were recorded with an MCP 300 (Anton Paar, Shanghai, China) polarimeter at 25 °C. UV data were measured on a UV-240 spectrophotometer (Shimadzu, Beijing, China). IR spectra were recorded on a Nicolet Nexus 670 spectrophotometer (Thermo Fisher Scientific, Inc., Hudson, NH, USA) in KBr discs. CD data were measured on a Chirascan™ CD spectrometer (Applied Photophysics, London, UK). 1H and 13C NMR spectra were recorded on a Bruker AVANCE 500 spectrometer (Bruker BioSpin Corporation, Bellerica, MA, USA) in CDCl3, acetone-d6 or methanol-d4. Chemical shifts were reported in δ (ppm), using Tetramethylsilane (TMS) as the internal standard, and coupling constants (J) were reported in Hertz (Hz). EIMS spectra were measured on a Thermo DSQ EIMS spectrometer and HREIMS on a Thermo MAT95XP high-resolution mass spectrometer. Single-crystal data were measured on an Agilent Gemini Ultra diffractometer (CuKα radiation). Column chromatography (CC) was performed using silica gel (200–300 mesh, Qingdao Huang Hai Chemical Group Co., Qingdao, China; G60, F-254) and Sephadex LH-20 (GE Healthcare, Buckinghamshire, UK) stationery phases.
3.2. Fungal Material
The mangrove endophytic fungal strain 16-5B used in this study was isolated from the leaves of Sonneratia apetala, which was collected from Dongzhaigang Mangrove National Nature Reserve in Hainan Island, China, in April, 2009. The strain was identified by Yayue Liu as Aspergillus sp., according to morphologic traits and molecular identification. Its 444 base pair internal transcribed spacer (ITS) sequence had 99% sequence identity to that of Aspergillus sp. (No. JF312217). The sequence data had been submitted to GenBank with Accession Number KP059102. A voucher specimen (Registration Number 16-5B) has been deposited at the School of Chemistry and Chemical Engineering, Sun Yat-Sen University, Guangzhou, China.
3.3. Extraction and Isolation
The fungus Aspergillus sp. was cultivated in Czapek’s medium (3 g NaNO3, 1 g K2HPO4, 0.5 g MgSO4, 1 g KCl, 0.01 g FeSO4, 20 g sucrose and 20 g agar in 1 L water) in 1-L Erlenmeyer flasks, each containing 300 mL of culture broth at 25 °C without shaking for 28 days. The culture (40 L) was filtered to separate the mycelia from culture broth, and then, the mycelia were extracted with MeOH three times. The organic solvent was filtered and concentrated under reduced pressure to yield 6.9 g of organic extract, which was subjected to silica gel CC using gradient elution with petroleum ether-EtOAc from 90:10 to 0:100 (v/v) to give nine fractions (Fractions 1–9). Fraction 3 was further purified by silica gel CC using 40% EtOAc-petroleum ether to afford seven subfractions (Fractions 3.1–3.7). Fraction 3.3 was applied to Sephadex LH-20 CC, eluted with MeOH to obtain Compound 1 (3.2 mg). Fraction 6 was applied to Sephadex LH-20 CC, eluted with CHCl3/MeOH (1:1) to afford five subfractions (Fractions 6.1–6.5). Fraction 6.3 was further purified with Sephadex LH-20 CC, eluted with MeOH to obtain Compound 2 (1.5 mg). Fraction 2 was further purified by silica gel CC using 1% MeOH/CHCl3 to obtain Compound 3 (2.1 mg). Fraction 4 was further purified by silica gel CC using gradient elution with MeOH/CHCl3 from 1:99–1:9 (v/v) to afford five subfractions (Fractions 4.1–4.5). Fraction 4.2 was further purified using 3% MeOH/CHCl3 to obtain Compound 5 (5.8 mg). Fraction 4.4 was further purified by Sephadex LH-20 CC eluted with CHCl3/MeOH (1:1) to obtain Compound 4 (5.8 mg). Compound 6 (20.5 mg) was obtained by recrystallization with acetone from Fraction 5.
Compound
1: white amorphous powder; mp 121.1–121.6 °C;
–110 (
c 0.1, MeOH); UV (MeOH) λ
max: 319 (3.33), 268 (3.11) and 217 (3.55) nm; CD (CH
3OH) λ
max (∆ɛ) 324 (–3.1), 298 (+3.6), 267 (–1.3), 221 (+0.3), 211 (–4.0) nm; IR (KBr)
υmax 3394, 2933, 2851, 1643, 1505, 1450, 1380, 1321, 1266, 1171, 1079, 998 and 847 cm
−1;
1H NMR (CDCl
3, 500 MHz) and
13C NMR (CDCl
3, 125 MHz), see
Table 1; EIMS
m/
z 372 [M]
+; HREIMS
m/
z 372.1209 [M]
+ (calcd. for C
20H
20O
7, 372.1204)
Compound
2: white amorphous powder; mp 174.4–174.9 °C,
0 (
c 0.1, MeOH); UV (MeOH) λ
max: 309 (2.81), 277 (3.21) and 218 (3.50) nm; IR (KBr)
υmax 3365, 2922, 1653, 1510, 1450, 1330, 1206, 1101, 1041 and 836 cm
−1;
1H NMR (acetone-
d6, 500 MHz) and
13C NMR (acetone-
d6, 125 MHz), see
Table 2; EIMS 254 [M]
+; HREIMS
m/
z 254.0786 [M]
+ (calcd. for C
12H
14O
6, 254.0785).
Compound
3: white amorphous powder; mp 134.2–134.8 °C;
0 (
c 0.1, MeOH); UV(MeOH) λ
max: 342 (2.72), 319 (2.86) and 251 (3.47) nm; IR (KBr)
υmax 3239, 2922, 1721, 1627, 1309, 1189, 1085, 1037 and 870 cm
−1;
1H NMR (methanol-
d4, 500 MHz) and
13C NMR (methanol-
d4, 125 MHz), see
Table 2; EIMS
m/
z 252 [M]
+; HREIMS
m/
z 252.0630 [M]
+ (calcd. for C
12H
12O
6, 252.0628).
Compound
4: white amorphous powder; mp 201.2–201.9 °C
+30 (
c 0.1, MeOH); UV(MeOH) λ
max: 326 (2.68), 258 (2.76) and 209 (3.59) nm; CD (CH
3OH) λ
max (∆ɛ) 329 (+0.2), 286 (+0.8), 232 (–1.5) nm; IR (KBr)
υmax 3601, 3311, 3151, 2961, 1734, 1623, 1521, 1460, 1394, 1343, 1279, 1208, 1098 and 864 cm
−1;
1H NMR (methanol-
d4, 500 MHz) and
13C NMR (methanol-
d4, 125 MHz), see
Table 3; EIMS
m/
z 238 [M]
+; HREIMS
m/
z 238.0835 [M]
+ (calcd. for C
12H
14O
5, 238.0836).
3.4. Assay for α-Glucosidase Inhibitory Activity
All of the tests for α-glucosidase inhibitory activity were according to a previously described method [
22,
23]. The reaction mixture (final volume, 1 mL) consisted of the enzyme solution (20 μL, Sigma 9001-42-7, E.C 3.2.1.20), substrate (10 mM
p-nitrophenyl-α-glucopyranoside, 20 μL, Fluka, BioChemika, Buchs, Switzerland) in 50 mM potassium phosphate buffer (pH 7.0) and 20 μL DMSO or inhibitor (test sample dissolved in DMSO (10 μmol/mL)). The inhibitors were pre-incubated with the enzyme at 37 °C for 20 min, and the substrate was then added. The reaction was monitored spectrophotometrically by measuring the absorbance at 400 nm for 1-min intervals. Calculations were performed according to the equation: inhibition rates (%) = ((OD
control − OD
control blank) − (OD
test − OD
test blank))/(OD
control − OD
control blank) × 100%. The IC
50 values of the compounds were calculated by nonlinear regression analysis and expressed as the mean ± SD of two distinct experiments. Kinetic parameters were determined using the Lineweaver-Burk double-reciprocal plot method at increasing concentrations of substrates and inhibitors.
3.5. Quantum Mechanical Calculation
In theoretical calculations, the preliminary conformational distribution search was performed by Spartan’10 software (Wavefunction, Inc., Irvine, CA, USA) using the MMFF94S force field. The corresponding minimum geometries were further fully optimized with the Gaussian 03 (Gaussian, Wallingford, CT, USA) program package at the B3LYP/6-31G (d) computational level as frequency calculations. The obtained stable conformers were submitted to CD calculation by the TDDFT cam-b3lyp/6-311+g (2d, p) method. ECD spectra were generated using the program SpecDis 1.6 (University of Würzburg, Würzburg, Germany) and OriginPro 8.5 (OriginLab, Ltd., Northampton, MA, USA) from dipole-length rotational strengths by applying Gaussian band shapes with sigma = 0.19 electron volt (ev). All calculations were performed with the High-Performance Grid Computing Platform of Sun Yat-Sen University.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




