
Can Some Marine-Derived Fungal Metabolites Become Actual Anticancer Agents?





Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Cancer Epidemiology
1.2. The Role of Natural Products in Cancer Therapy
2. Marine Fungal-Derived Metabolites versus Cancer
2.1. Pro-Apoptotic Metabolites











2.2. Metabolites That Kill Cancer Cells without Direct Pro-Apoptotic Effects










2.3. Metabolites with in Vivo Antitumor Activity or Those Entering Clinical Trials







3. Which Compounds Are the Most Promising Marine-Derived Fungal Metabolites for Use as Potential Anticancer Agents?
4. How Could We Increase the Rate of Discovery of Marine-Derived Fungal Metabolites as Potential Anticancer Agents, at Least in Vitro?
4.1. Are Pro-Apoptotic Compounds Still Valuable Weapons for Combating Cancer?
4.2. Pharmacological and Toxicological Strategies
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gulland, A. Global cancer prevalence is growing at “alarming pace,” says WHO. BMJ 2014, 348, g1338. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Giddings, L.-A. Natural products as leads to antitumor drugs. Phytochem. Rev. 2014, 13, 123–137. [Google Scholar] [CrossRef]
- Schiff, P.B.; Horwitz, S.B. Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 1980, 77, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Yared, J.A.; Tkaczuk, K.H. Update on taxane development: New analogues and new formulations. Drug Des. Dev. Ther. 2012, 6, 371–384. [Google Scholar]
- Duflos, A.; Kruczynski, A.; Barret, J.M. Novel aspects of natural and modified vinca alkaloids. Curr. Med. Chem. Anticancer Agents 2002, 2, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Mathijseen, R.H.; Loos, W.J.; Verweij, J.; Sparreboom, A. Pharmacology of topoisomerase I inhibitors irinotecan (CPT-11) and topotecan. Curr. Cancer Drug Targets 2002, 2, 103–123. [Google Scholar] [CrossRef]
- Basili, S.; Moro, S. Novel camptothecin derivatives as topoisomerase I inhibitors. Expert. Opin. Ther. Pat. 2009, 19, 555–574. [Google Scholar] [CrossRef] [PubMed]
- Gordaliza, M.; Castro, M.A.; del Corral, J.M.; Feliciano, A.S. Antitumor properties of podophyllotoxin and related compounds. Curr. Pharm. Des. 2000, 6, 1811–1839. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Xu, H. Recent advances in semisynthesis, biosynthesis, biological activities, mode of action, and structure-activity relationship of podophyllotoxins: An update (2008–2010). Mini Rev. Med. Chem. 2011, 11, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Demain, A.L. Importance of microbial natural products and the need to revitalize their discovery. J. Ind. Microbiol. Biotechnol. 2014, 41, 185–201. [Google Scholar] [CrossRef] [PubMed]
- Indumathy, S.; Dass, C.R. Finding chemo: The search for marine-based pharmaceutical drugs active against cancer. J. Pharm. Pharmacol. 2013, 65, 1280–1301. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed marine natural products in the pharmaceutical and cosmeceutical industries: Tips for success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Marine-sourced anti-cancer and cancer pain control agents in clinical and late preclinical development. Mar. Drugs 2014, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Dicato, M.; Diederich, M. Epigenetic modulators from “The Big Blue”: A treasure to fight against cancer. Cancer Lett. 2014, 351, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, W.; Feeney, R.J. Contributions to the study of marine products. XXXII. The nucleosides of sponges I. J. Org. Chem. 1951, 16, 981–987. [Google Scholar] [CrossRef]
- Cancer Treatment: Cytarabine. National Cancer Institute. Available online: http://www.cancer.gov/cancertopics/druginfo/cytarabine (accessed on 17 February 2015).
- Rinehart, K.L.; Holt, T.G.; Fregeau, N.L.; Stroh, J.G.; Keifer, P.A.; Sun, F.; Li, L.H.; Martin, D.G. Ecteinascidins 729, 743, 745, 759A, 759B, and 770: Potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4512–4515. [Google Scholar] [CrossRef]
- Wright, A.E.; Forleo, D.A.; Gunawardana, G.P.; Gunasekera, S.P.; Koehn, F.E.; McConnell, O.J. Antitumor tetrahydroisoquinoline alkaloids from the colonial ascidian Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4508–4512. [Google Scholar] [CrossRef]
- European Medicines Agency. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/000773/WC500059175.pdf (accessed on 17 February 2015).
- Cuevas, C.; Pérez, M.; Martín, M.J.; Chicharro, J.L.; Fernández-Rivas, C.; Flores, M.; Francesch, A.; Gallego, P.; Zarzuelo, M.; de La Calle, F.; et al. Synthesis of ecteinascidin ET-743 and phthalascidin Pt-650 from cyanosafracin B. Org. Lett. 2000, 2, 2545–2548. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Uemura, D. Halichondrins—Antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef]
- FDA Approval for Eribulin Mesylate. National Cancer Institute. Available online: http://www.cancer.gov/cancertopics/druginfo/fda-eribulinmesylate (accessed on 17 February 2015).
- FDA Approval for Brentuximab Vedotin. National Cancer Institute. Available online: http://www.cancer.gov/cancertopics/druginfo/fda-brentuximabvedotin (accessed on 17 February 2015).
- Pettit, G.R.; Kamano, Y.; Herald, C.L.; Tuinman, A.A.; Boettner, F.E.; Kizu, H.; Schmidt, J.M.; Baczynskyj, L.; Tomer, K.B.; Bontems, R.J. The isolation and structure of a remarkable marine animal antineoplastic constituent: Dolastatin 10. J. Am. Chem. Soc. 1987, 109, 6883–6885. [Google Scholar] [CrossRef]
- Williamson, R.T.; Chapin, E.L.; Carr, A.W.; Gilbert, J.R.; Graupner, P.R.; Lewer, P.; McKamey, P.; Carney, J.R.; Gerwick, W.H. New diffusion-edited NMR experiments to expedite the dereplication of known compounds from natural product mixtures. Org. Lett. 2000, 2, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Moore, R.E.; Paul, V.J.; Mooberry, S.L.; Corbett, T.H. Isolation of dolastatin 10 from the marine cyanobacterium Symploca species VP642 and total stereochemistry and biological evaluation of its analogue symplostatin 1. J. Nat. Prod. 2001, 64, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Petit, K.; Biard, J.-F. Marine natural products and related compounds as anticancer agents: An overview of their clinical status. Anticancer Agents Med. Chem. 2013, 13, 603–631. [Google Scholar] [CrossRef] [PubMed]
- Aly, A.H.; Debbab, A.; Proksch, P. Fifty years of drug discovery from fungi. Fungal Divers. 2011, 50, 3–19. [Google Scholar] [CrossRef]
- Kück, U.; Bloemendal, S.; Teichert, I. Putting fungi to work: Harvesting a cornucopia of drugs, toxins, and antibiotics. PLoS Pathog. 2014, 10, e1003950. [Google Scholar] [CrossRef] [PubMed]
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef] [PubMed]
- Bugni, T.S.; Ireland, C.M. Marine-derived fungi: A chemically and biologically diverse group of microorganisms. Nat. Prod. Rep. 2004, 21, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.; Shaiq Ali, M.; Hussain, S.; Jabbar, A.; Ashraf, M.; Lee, Y.S. Marine natural products of fungal origin. Nat. Prod. Rep. 2007, 24, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.P. A glimpse of the early history of the cephalosporins. Rev. Infect. Dis. 1979, 1, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Evidente, A.; Kornienko, A.; Cimmino, A.; Andolfi, A.; Lefranc, F.; Mathieu, V.; Kiss, R. Fungal metabolites with anticancer activity. Nat. Prod. Rev. 2014, 31, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Cotter, T.G. Apoptosis and cancer: The genesis of a research field. Nat. Rev. Cancer 2009, 9, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Giansanti, V.; Tillhon, M.; Mazzini, G.; Prosperi, E.; Lombardi, P. Scovassi, A.I. Killing of tumor cells: A drama in two acts. Biochem. Pharmacol. 2011, 82, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Indran, I.R.; Tufo, G.; Pervaiz, S.; Brenner, C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim. Biophys. Acta 2011, 1807, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Nagaprashantha, L.; Vartak, N.; Awasthi, S.; Awasthi, S.; Singhal, S.S. Novel anti-cancer compounds for developing combinatorial therapies to target anoikis-resistant tumors. Pharm. Res. 2012, 29, 621–636. [Google Scholar] [CrossRef] [PubMed]
- Taddei, M.L.; Giannoni, E.; Fiaschi, T.; Chiarugi, P. Anoikis: An emerging hallmark in health and diseases. J. Pathol. 2012, 226, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Diff. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Kornienko, A.; Mathieu, V.; Rastogi, S.K.; Lefranc, F.; Kiss, R. Therapeutic agents triggering nonapoptotic cancer cell death. J. Med. Chem. 2013, 56, 4823–4839. [Google Scholar] [CrossRef] [PubMed]
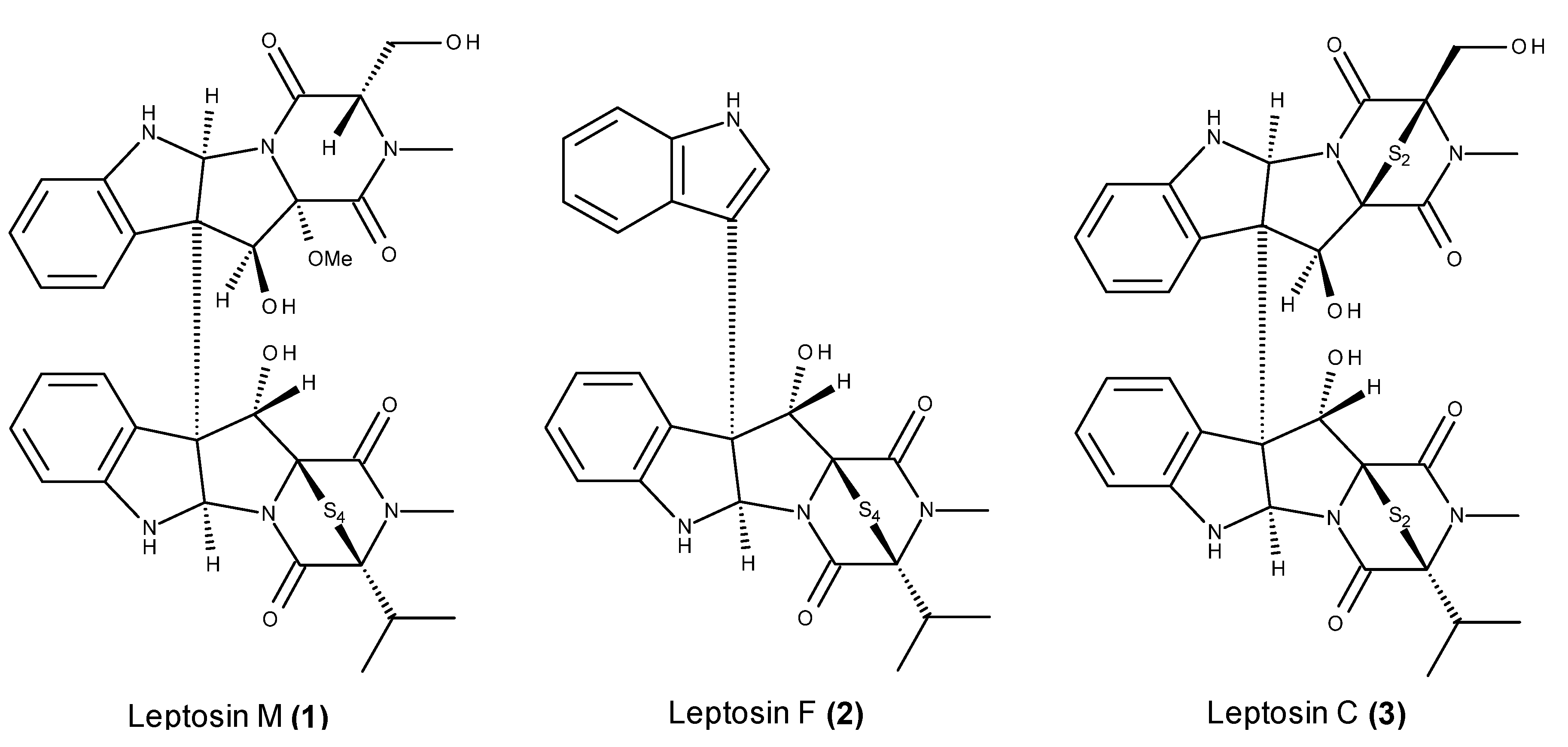
- Takahashi, C.; Numata, A.; Ito, Y.; Matsumura, E.; Araki, H.; Iwaki, H.; Kushida, K. Leptosins, antitumour metabolites of a fungus isolated from a marine alga. J. Chem. Soc. Perkin Trans. 1994, 1, 1859–1864. [Google Scholar] [CrossRef]
- Takahashi, C.; Numata, A.; Matsumura, E.; Minoura, K.; Eto, H.; Shingu, T.; Ito, T.; Hasegawa, T. Leptosins I and J, cytotoxic substances produced by a Leptosphaeria sp. Physico-chemical properties and structures. J. Antibiot. (Tokyo) 1994, 47, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, C.; Takai, Y.; Kimura, Y.; Numata, A.; Shigematsu, N.; Tanaka, H. Cytotoxic metabolites from a fungal adherent of a marine alga. Phytochemistry 1995, 38, 155–158. [Google Scholar] [CrossRef]
- Takahashi, C.; Minoura, K.; Yamada, T.; Numata, A.; Kushida, K.; Shingu, T.; Hagishita, S.; Nakai, H.; Sato, T.; Harada, H. Potent cytotoxic metabolites from a Leptosphaeria species. Structure determination and conformational analysis. Tetrahedron 1995, 51, 3483–3498. [Google Scholar] [CrossRef]
- Yamada, T.; Iwamoto, C.; Yamagaki, N.; Yamanouchi, T.; Minoura, K.; Yamori, T.; Uehara, Y.; Andoh, T.; Umemura, K.; Numata, A. Leptosins M-N1, cytotoxic metabolites from a Leptosphaeria species separated from a marine alga. Structure determination and biological activities. Tetrahedron 2002, 58, 479–487. [Google Scholar] [CrossRef]
- Yamada, T.; Iwamoto, C.; Yamagaki, N.; Yamanouchi, T.; Minoura, K.; Hagishita, S.; Numata, A. Leptosins O-S, cytotoxic metabolites from a strain of Leptosphaeria sp. isolated from a marine alga. Heterocycles 2004, 63, 641–653. [Google Scholar] [CrossRef]
- Yanagihara, M.; Sasaki-Takahashi, N.; Sugahara, T.; Yamamoto, S.; Shinomi, M.; Yamashita, I.; Hayashida, M.; Yamanoha, B.; Numata, A.; Yamori, T.; et al. Leptosins isolated from marine fungus Leptoshaeria species inhibit DNA topoisomerases I and/or II and induce apoptosis by inactivation of Akt/protein kinase B. Cancer Sci. 2005, 96, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Kalimuthu, S.; Se-Kwon, K. Cell survival and apoptosis signaling as therapeutic target for cancer: Marine bioactive compounds. Int. J. Mol. Sci. 2013, 14, 2334–2354. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Park, S.J.; Kim, Y.J.; Jung, J.H.; Lee, J.K.; Kwon, H.C.; Yang, H.O. Apoptosis-inducing effect of diketopiperazine disulfides produced by Aspergillus sp. KMD901 isolated from marine sediment on HCT116 colon cancer cell lines. J. Appl. Microbiol. 2011, 110, 304–313. [Google Scholar] [CrossRef] [PubMed]
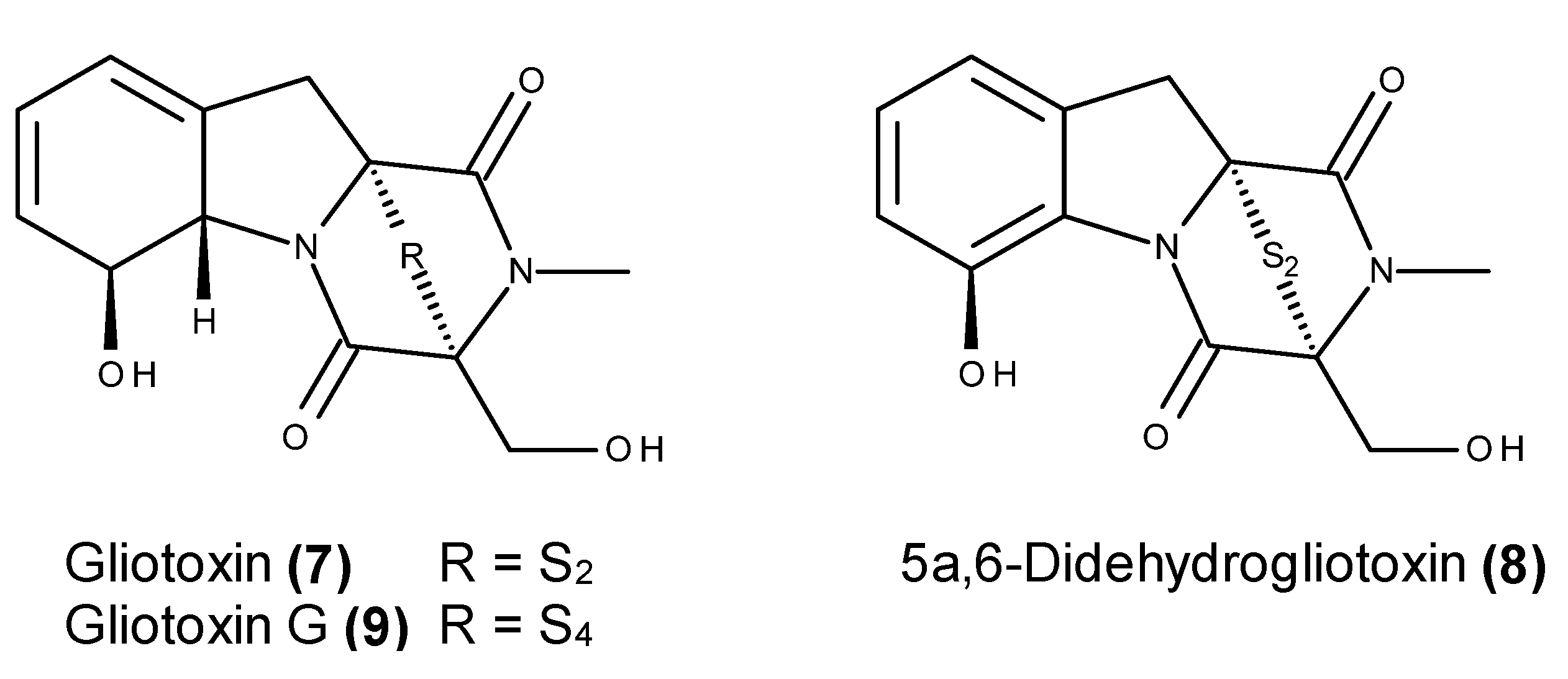
- Li, X.; Kim, S.K.; Nam, K.W.; Kang, J.S.; Choi, H.D.; Son, B.W. A new bacterial dioxopiperazine alkaloid related to gliotoxin from a marine isolate of the fungus Pseudallescheria. J. Antibiot. 2006, 59, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Takada, K.; Takemoto, Y.; Yoshida, M.; Nogi, Y.; Okada, S.; Matsunaga, S. Gliotoxin analogues from a marine-derived fungus, Penicillium sp., and their cytotoxic and histone methyltransferase inhibitory activities. J. Nat. Prod. 2012, 75, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.L.; le, X.; Li, H.J.; Yang, X.L.; Chen, J.X.; Xu, J.; Liu, H.L.; Wang, L.Y.; Wang, K.T.; Hu, K.C.; et al. Exploring the chemodiversity and biological activities of the secondary metabolites from the marine fungus Neosartorya pseudofischeri. Mar. Drugs 2014, 12, 5657–5676. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Lee, J.S.; Qian, Z.J.; Li, Y.X.; Kim, K.N.; Heo, S.J.; Jeon, Y.J.; Park, W.S.; Choi, I.W.; Je, J.Y.; et al. Gliotoxin isolated from marine fungus Aspergillus sp. induces apoptosis of human cervical cancer and chondrosarcoma cells. Mar. Drugs 2014, 12, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.L.; Casey, P.J. Protein prenylation: Molecular mechanisms and functional consequences. Annu. Rev. Biochem. 1996, 65, 241–269. [Google Scholar] [CrossRef] [PubMed]
- Vigushin, D.M.; Mirsaidi, N.; Brooke, G.; Sun, C.; Pace, P.; Inman, L.; Moody, C.J.; Coombes, R.C. Gliotoxin is a dual inhibitor of farnesyltransferase and geranylgeranyltransferase I with antitumor activity against breast cancer in vivo. Med. Oncol. 2004, 21, 21–30. [Google Scholar] [CrossRef]
- Albert, M.; Helin, K. Histone methyltransferases in cancer. Semin. Cell. Dev. Biol. 2010, 21, 209–220. [Google Scholar] [CrossRef] [PubMed]
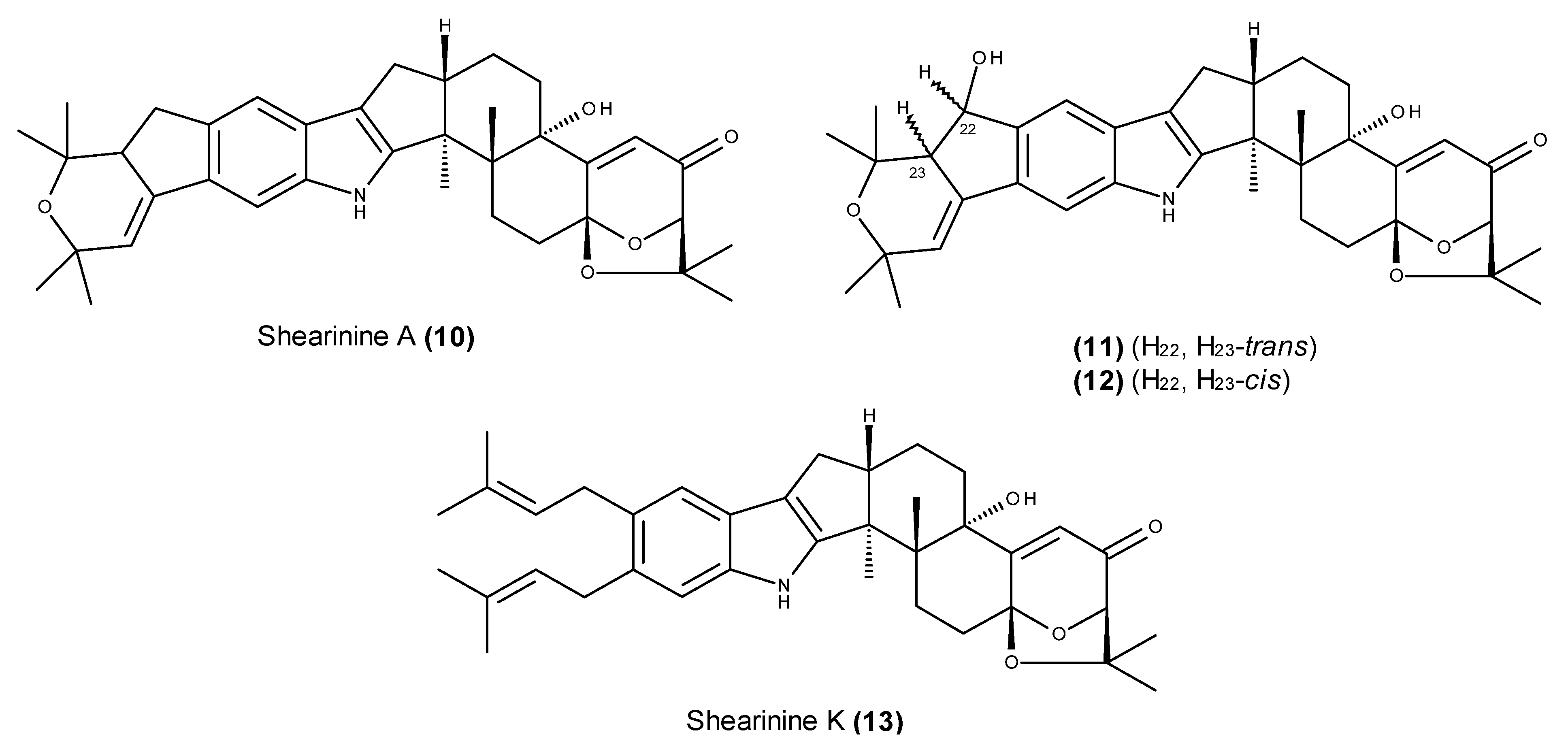
- Xu, M.; Gessner, G.; Groth, I.; Lange, C.; Christner, A.; Bruhn, T.; Deng, Z.; Li, X.; Heinemann, S.H.; Grabley, S.; et al. Shearinines D–K, new indole triterpenoids from an endophytic Penicillium sp. (strain HKI0459) with blocking activity on large-conductance calcium-activated potassium channels. Tetrahedron 2007, 63, 435–444. [Google Scholar] [CrossRef]
- Smetanina, O.F.; Kalinovsky, A.I.; Khudyakova, Y.V.; Pivkin, M.V.; Dmitrenok, P.S.; Fedorov, S.N.; Ji, H.; Kwak, J.-Y.; Kuznetsova, T.A. Indole alkaloids produced by a marine fungus isolate of Penicillium janthinellum Biourge. J. Nat. Prod. 2007, 70, 906–909. [Google Scholar] [CrossRef] [PubMed]
- Smetanina, O.F.; Kalinovsky, A.I.; Khudyakova, Y.V.; Pivkin, M.V.; Dmitrenok, P.S.; Fedorov, S.N.; Ji, H.; Kwak, J.-Y.; Kuznetsova, T.A. Erratum [Indole alkaloids produced by a marine fungus isolate of Penicillium janthinellum Biourge.]. J. Nat. Prod. 2007, 70, 2054. [Google Scholar] [CrossRef]
- Du, L.; Feng, T.; Zhao, B.; Li, D.; Cai, S.; Zhu, T.; Wang, F.; Xiao, X.; Gu, Q. Alkaloids from a deep ocean sediment-derived fungus Penicillium sp. and their antitumor activities. J. Antibiot. 2010, 63, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Li, D.; Zhu, T.; Cai, S.; Wang, F.; Xiao, X.; Gu, Q. New alkaloids and diterpenes from a deep ocean sediment derived fungus Penicillium sp. Tetrahedron 2009, 65, 1033–1039. [Google Scholar] [CrossRef]
- Shang, Z.; Li, X.; Meng, L.; Li, C.; Gao, S.; Huang, C.; Wang, B. Chemical profile of the secondary metabolites produced by a deep-sea sediment-derived fungus Penicillium commune SD-118. Chin. J. Oceanol. Limnol. 2012, 30, 305–314. [Google Scholar] [CrossRef]
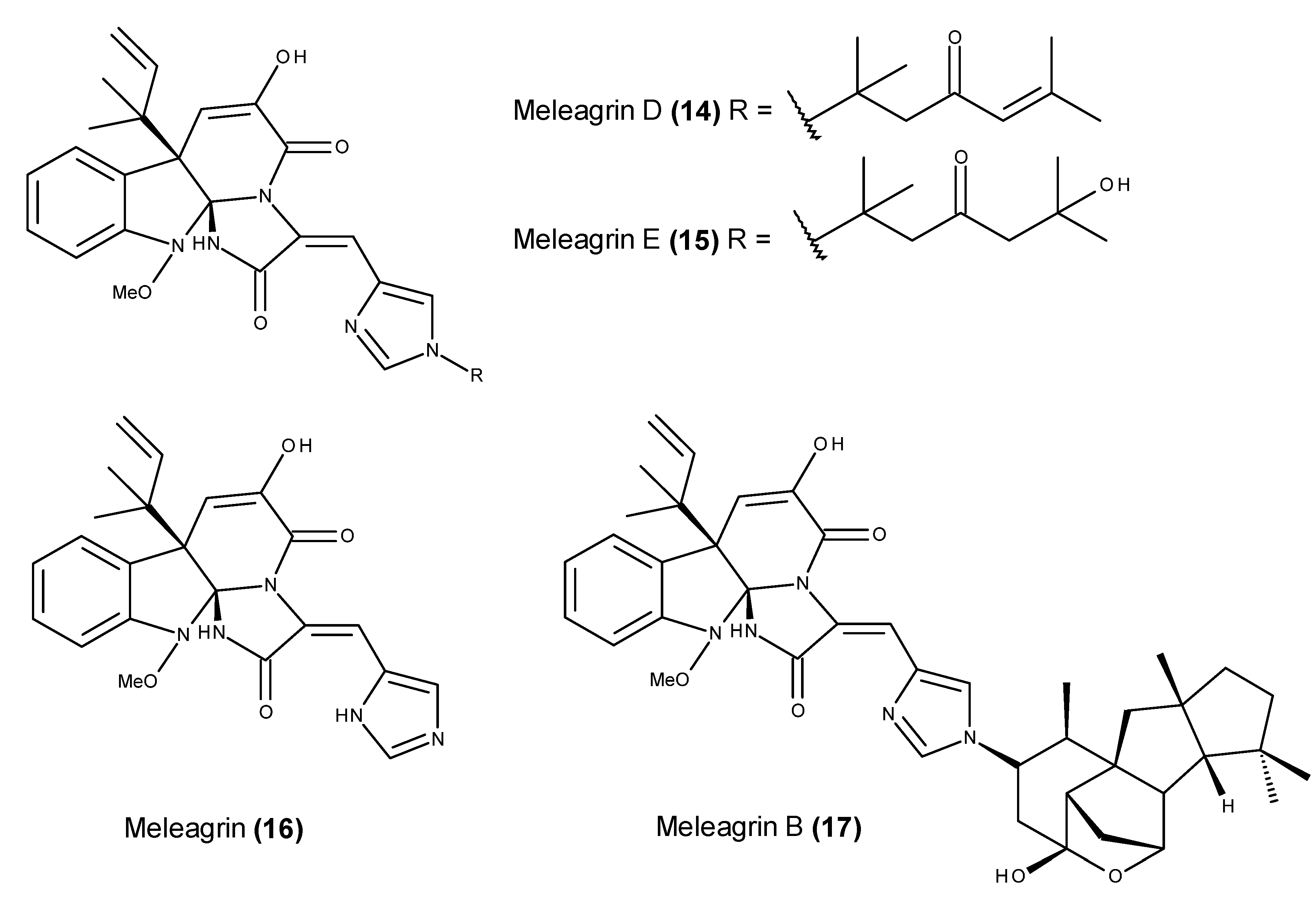
- Zheng, C.J.; Sohn, M.-J.; Lee, S.; Kim, W.-G. Meleagrin, a new FabI inhibitor from Penicillium chryosogenum with at least one additional mode of action. PLoS ONE 2013, 8, e78922. [Google Scholar] [CrossRef] [PubMed]
- Li, D.L.; Li, X.M.; Li, T.G.; Dang, H.Y.; Wang, B.G. Dioxopiperazine alkaloids produced by the marine mangrove derived endophytic fungus Eurotium rubrum. Helv. Chim. Acta 2008, 91, 1888–1893. [Google Scholar] [CrossRef]
- Gomes, N.M.; Dethoup, T.; Singburaudom, N.; Gales, L.; Silva, A.M.S.; Kijjoa, A. Eurocristatine, a new diketopiperazine dimer from the marine sponge-associated fungus Eurotium cristatum. Phytochem. Lett. 2012, 5, 717–720. [Google Scholar] [CrossRef]
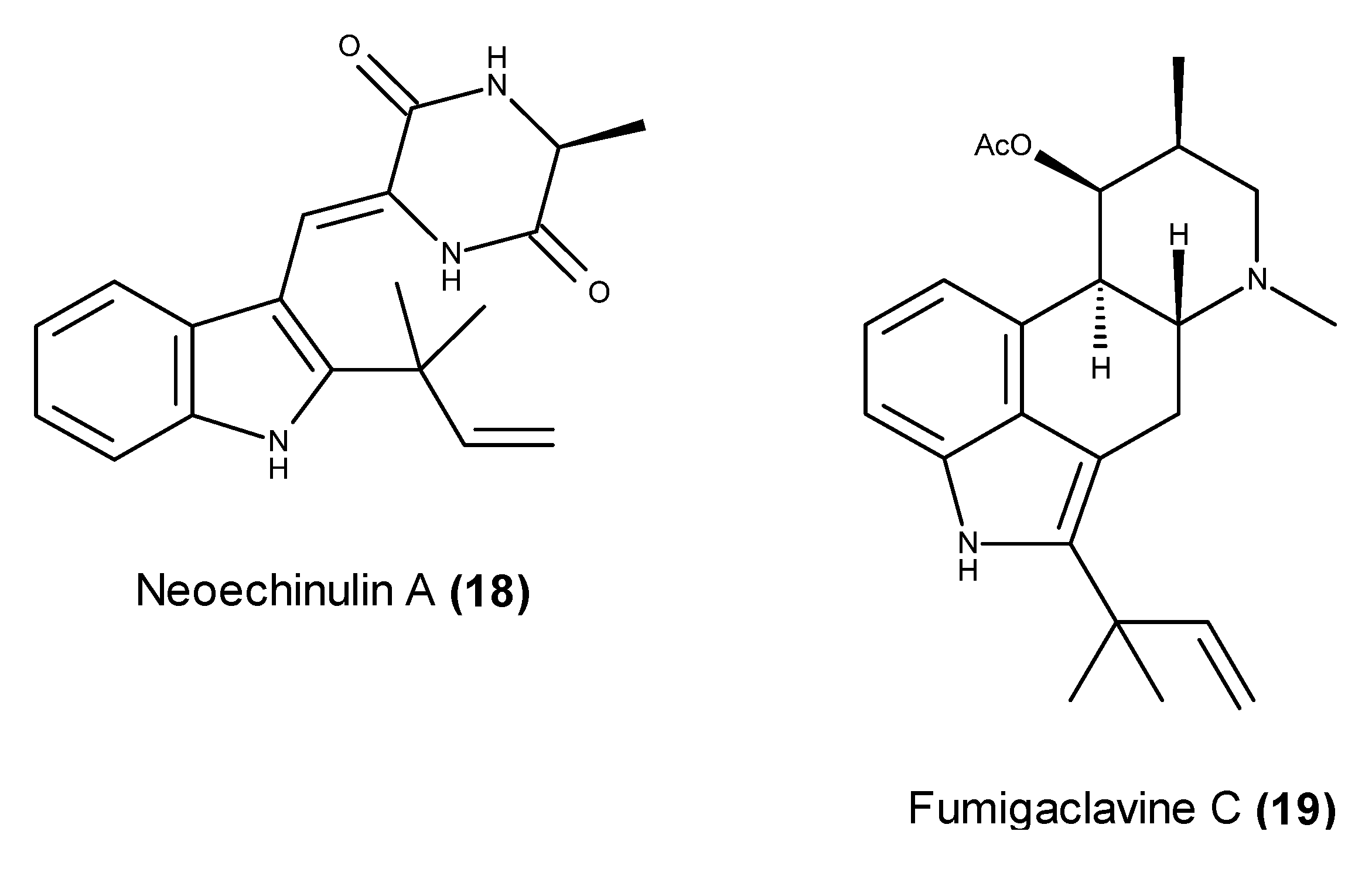
- Kim, K.S.; Cui, X.; Lee, D.S.; Sohn, J.H.; Yim, J.H.; Kim, Y.C.; Oh, H. Anti-inflammatory effect of neoechinulin A from the marine fungus Eurotium sp. SF-5989 through the suppression of NF-κB and p38 MAPK pathways in lipopolysaccharide-stimulated RAW264.7 macrophages. Molecules 2013, 18, 13245–13259. [Google Scholar] [CrossRef] [PubMed]
- Wijesekara, I.; Li, X.Y.; Uo, T.S.; van Ta, Q.; Ngo, D.H.; Kim, S.K. Induction of apoptosis in human cervical carcinoma HeLa cells by neoechinulin A from marine-derived fungus Microsporum sp. Process Biochem. 2013, 48, 68–72. [Google Scholar] [CrossRef]
- Li, Y.X.; Himaya, S.W.A.; Dewapriya, P.; Zhang, C.; Kim, S.K. Fumigaclavine C from a marine-derived fungus Aspergillus fumigatus induces apoptosis in MCF-7 breast cancer cells. Mar. Drugs 2013, 11, 5063–5086. [Google Scholar] [CrossRef] [PubMed]
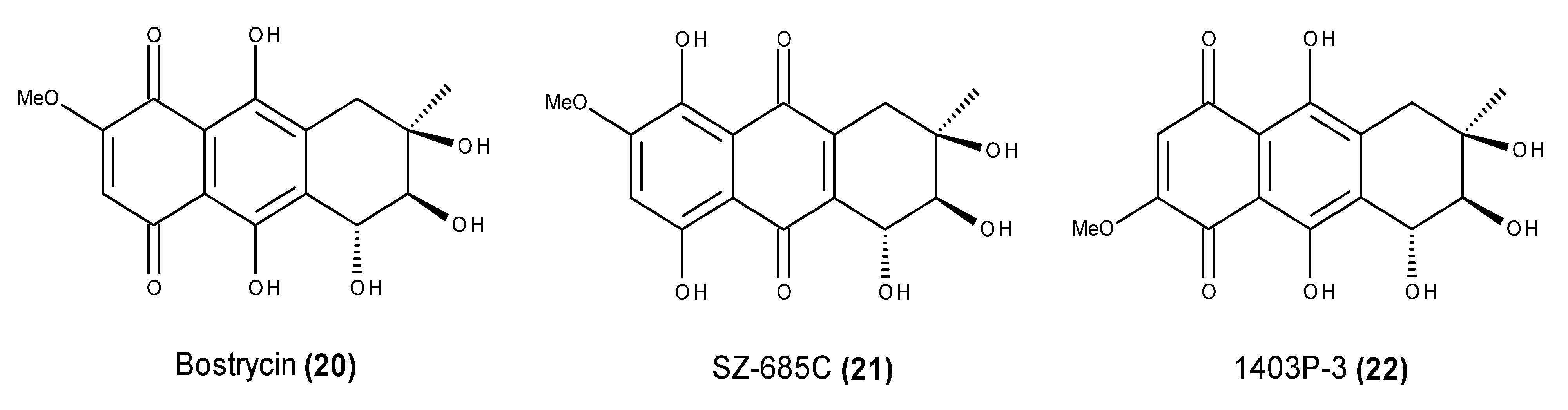
- Jiang, G.C.; Lin, Y.C.; Zhou, S.N.; Vrumoed, L.L.P.; Jones, E.B.G. Studies of the secondary metabolites of mangrove fungus No. 1403 from the South China Sea. Acta Sci. Nat. Univ. Sunyatseni 2000, 39, 68–72. [Google Scholar]
- Xu, J.; Nakazawa, T.; Ukai, K.; Kobayashi, H.; Mangindaan, R.E.P.; Wewengkang, D.S.; Rotinsulu, H.; Namikoshi, M. Tetrahydrobostrycin and 1-deoxytetrahydrobostrycin, two new hexahydroanthrone derivatives, from a marine-derived fungus Aspergillus sp. J. Antibiot. 2008, 61, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Trisuwan, K.; Khamthong, N.; Rukachaisirikul, V.; Phongpaichit, S.; Preedanon, S.; Sakayroj, J. Anthraquinone, cyclopentanone, and naphthoquinone derivatives from the sea fan-derived fungi Fusarium spp. PSU-F14 and PSU-F135. J. Nat. Prod. 2010, 73, 1507–1511. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Sun, X.; Lin, S.; Xiao, Z.; Li, H.; Bo, D.; She, Z. Xylanthraquinone, a new anthraquinone from the fungus Xylaria sp. 2508 from the South China Sea. Nat. Prod. Res. 2014, 28, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.S.; Hou, J.N.; Guo, Y.B.; Yang, H.L.; Xie, C.M.; Lin, Y.C.; She, Z.G. Bostrycin inhibits proliferation of human lung carcinoma A549 cells via downregulation of the PI3K/Akt pathway. J. Exp. Clin. Cancer Res. 2011, 30, 17. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, J.; Gao, Y.; Lin, H.; Du, L.; Yang, S.; Long, S.; She, Z.; Cai, X.; Zhou, S.; et al. The anthracenedione compound bostrycin induces mitochondria-mediated apoptosis in the yeast Saccharomyces cerevisiae. FEMS Yeast Res. 2010, 10, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhong, L.; Long, Y.; Li, J.; Wu, J.; Liu, L.; Chen, S.; Lin, Y.; Li, M.; Zhu, X.; et al. Studies on the synthesis of derivatives of marine-derived bostrycin and their structure-activity relationship against tumor cells. Mar. Drugs 2012, 10, 932–952. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Zhu, X.; Li, Q.; Gu, M.; He, Z.; Wu, J.; Li, J.; Lin, Y.; Li, M.; She, Z.; et al. SZ-685C, a marine anthraquinone, is a potent inducer of apoptosis with anticancer activity by suppression of the Akt/FOXO pathway. Br. J. Pharmacol. 2010, 159, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, S.; Liu, Q.; Wang, M.; Wang, C.; Yang, H. SZ-685C exhibits potent anticancer activity in both radiosensitive and radioresistant NPC cells through the miR-205-PTEN-Akt pathway. Oncol. Rep. 2013, 29, 2341–2347. [Google Scholar] [PubMed]
- Zhu, X.; He, Z.; Wu, J.; Yuan, J.; Wen, W.; Hu, Y.; Jiang, Y.; Lin, C.; Zhang, Q.; Lin, M.; et al. A marine anthraquinone SZ-685C overrides adriamycin-resistance in breast cancer cells through suppressing Akt signaling. Mar. Drugs 2012, 10, 694–711. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; He, Z.; Wu, J.; Lin, Y.; Zhu, X. A novel adriamycin analogue derived from marine microbes induces apoptosis by blocking Akt activation in human breast cancer cells. Mol. Med. Rep. 2011, 4, 261–265. [Google Scholar] [PubMed]
- Zhang, J.Y.; Wu, H.Y.; Xia, X.K.; Liang, Y.J.; Yan, Y.Y.; She, Z.G.; Lin, Y.C.; Fu, L.W. Anthracenedione derivative 1403P-3 induces apoptosis in KB and KBv200 cells via reactive oxygen species-independent mitochondrial pathway and death receptor pathway. Cancer Biol. Ther. 2007, 6, 1409–1417. [Google Scholar] [CrossRef]
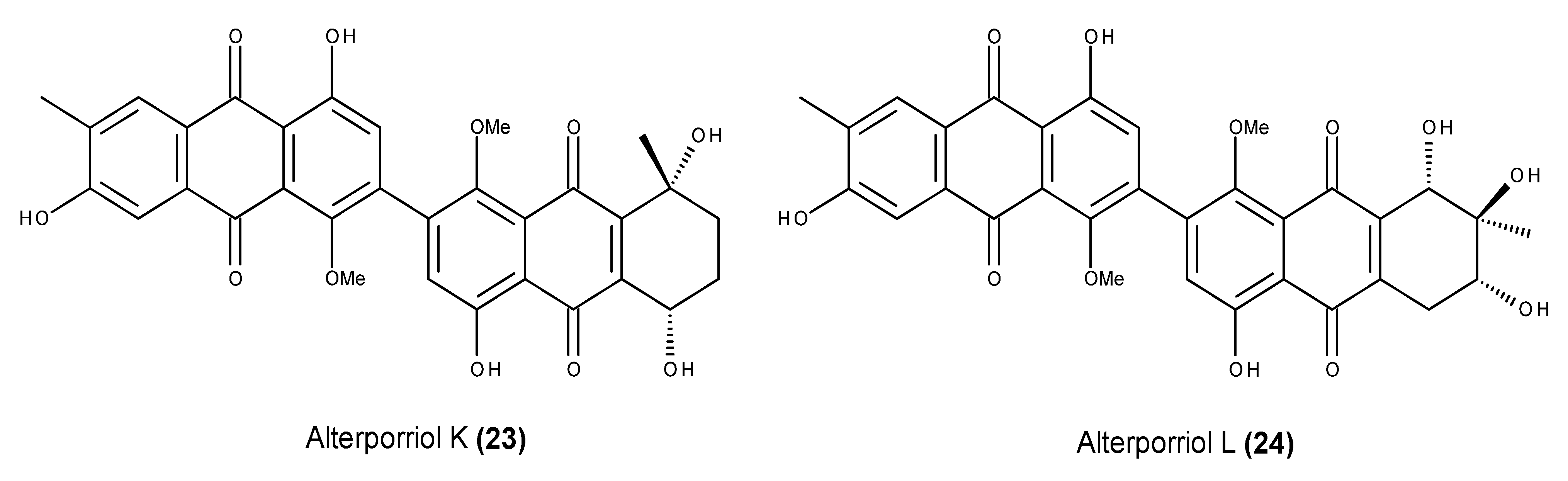
- Huang, C.-H.; Pan, J.-H.; Chen, B.; Yu, M.; Huang, H.-B.; Zhu, X.; Lu, Y.-J.; She, Z.-G.; Lin, Y.-C. Three bianthraquinone derivatives from the mangrove endophytic fungus Alternaria sp. ZJ9–6B from the South China Sea. Mar. Drugs 2011, 9, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Jin, H.; Song, B.; Zhu, X.; Zhao, H.; Cai, J.; Lu, Y.; Chen, B.; Lin, Y. The cytotoxicity and anticancer mechanisms of alterporriol L, a marine bianthraquinone, against MCF-7 human breast cancer cells. Appl. Microbiol. Biotechnol. 2012, 93, 777–785. [Google Scholar] [CrossRef] [PubMed]
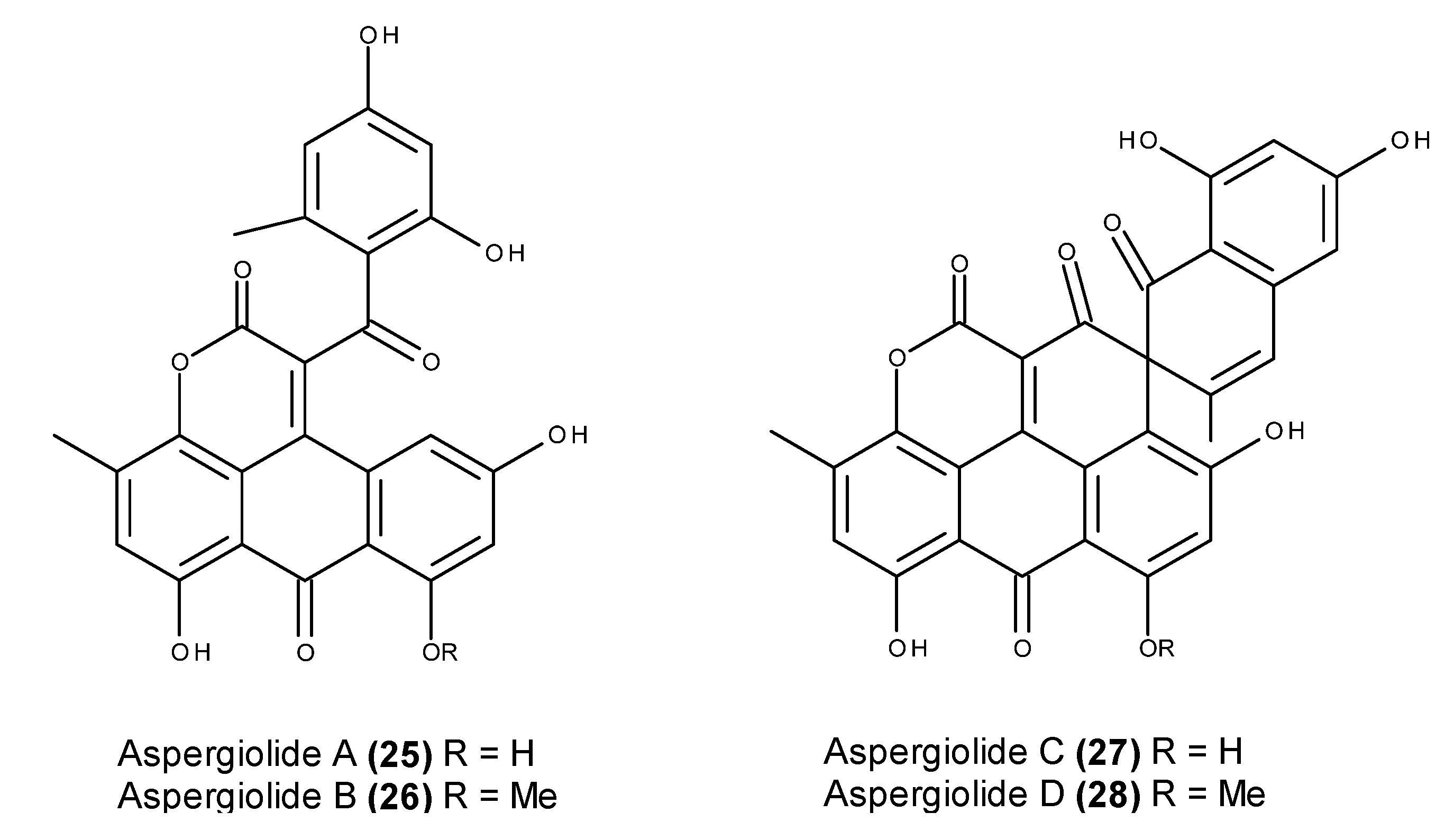
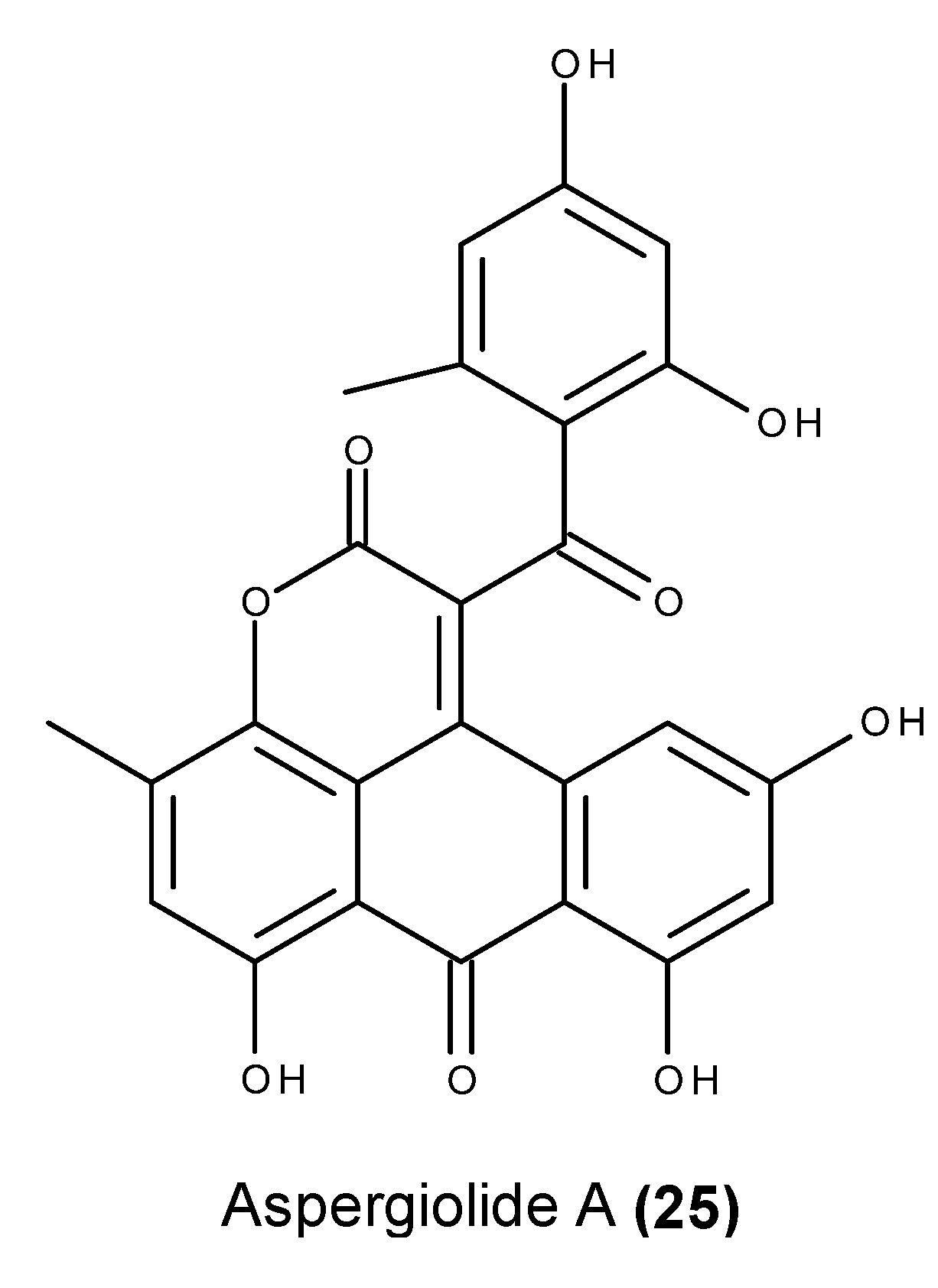
- Du, L.; Zhu, T.; Fang, Y.; Liu, H.; Gu, Q.; Zhu, W. Aspergiolide A, a novel anthraquinones derivative with naphtho[1,2,3-de]chromene-2,7-dione skeleton isolated from a marine-derived fungus Aspergillus glaucus. Tetrahedron 2007, 63, 1085–1088. [Google Scholar] [CrossRef]
- Du, L.; Zhu, T.; Fang, Y.; Liu, H.; Gu, Q.; Zhu, W. Corrigendum to “Aspergiolide A, a novel anthraquinones derivative with naphtho[1,2,3-de]chromene-2,7-dione skeleton isolated from a marine-derived fungus Aspergillus glaucus” [Tetrahedron 63 (2007) 1085]. Tetrahedron 2008, 64, 4657. [Google Scholar] [CrossRef]
- Du, L.; Zhu, T.; Liu, H.; Fang, Y.; Zhu, W.; Gu, Q. Cytotoxic polyketides from a marine-derived fungus Aspergillus glaucus. J. Nat. Prod. 2008, 71, 1837–1842. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Du, L.; Ai, J.; Li, D.; Zhu, T.; Wang, Y.; Knauer, M.; Bruhn, T.; Liu, H.; Geng, M.; et al. Aspergiolides C and D: Spirocyclic aromatic polyketides with potent protein kinase c-Met inhibitory effects. Chem. Eur. J. 2011, 17, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qi, X.; Li, D.; Zhu, T.; Mo, X.; Li, J. Anticancer efficacy and absorption, distribution, metabolism and toxicity studies of Aspergiolide A in early drug development. Drug Design Dev. Ther. 2014, 8, 1965–1977. [Google Scholar]
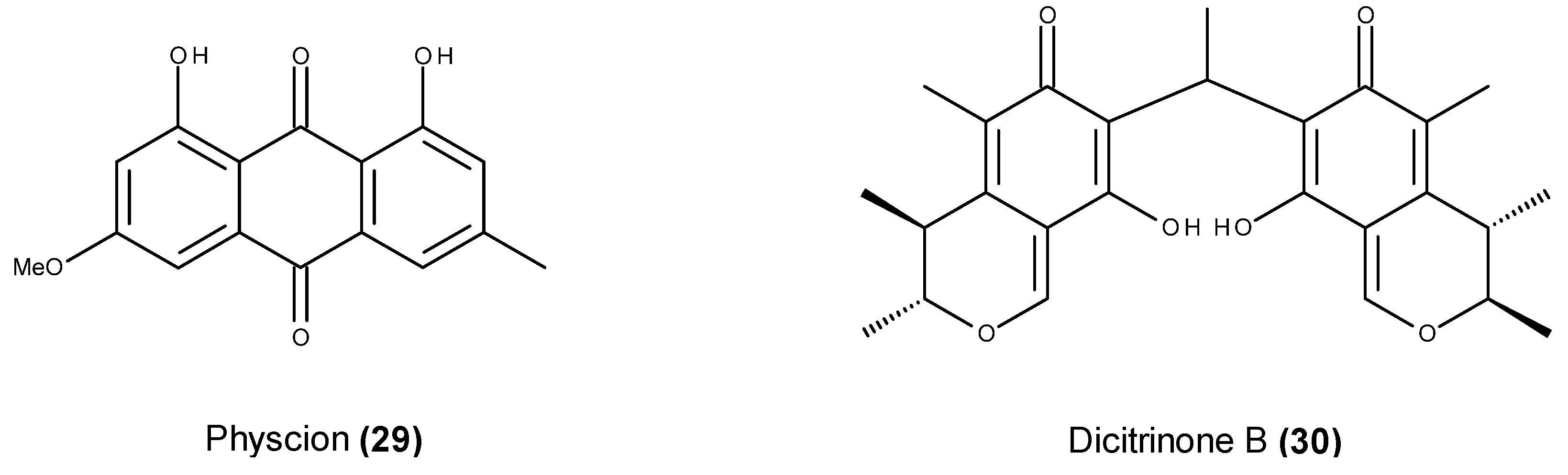
- Wijesekara, I.; Zhang, C.; Ta, Q.V.; Vo, T.S.; Li, Y.X.; Kim, S.K. Physcion from marine-derived fungus Microsporum sp. induces apoptosis in human cervical carcinoma HeLa cells. Microbiol. Res. 2014, 169, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Li, D.; Zhang, G.; Zhu, T.; Ai, J.; Gu, Q. Novel carbon-bridged citrinin dimers from a volcano-ash derived fungus Penicillium citrinum and their cytotoxic and cell cycle arrest activities. Tetrahedron 2010, 66, 9286–9290. [Google Scholar] [CrossRef]
- Chen, L.; Gong, M.W.; Peng, Z.F.; Zhou, T.; Ying, M.G.; Zheng, Q.H.; Liu, Q.Y.; Zhang, Q.Q. The marine fungal metabolite, dicitrinone B, induces A375 cell apoptosis through the ROS-related caspase pathway. Mar. Drugs 2014, 12, 1939–1958. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Y.; Zhu, Q.; Gu, Q. Studies on chemical constituents of secondary metabolites of marine-derived Aspergillus fumigatus. J. Qingdao Univ. Sci. Technol. (Nat. Sci. Ed.) 2007, 3, 199–201. [Google Scholar]
- Klaiklay, S.; Rukachaisirikul, V.; Sukpondma, Y.; Phongpaichit, S.; Buatong, J.; Bussaban, B. Metabolites from the mangrove-derived fungus Xylaria cubensis PSU-MA34. Arch. Pharm. Res. 2012, 35, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.Q.; Lin, X.P.; Liu, J.; Kaliyaperumal, K.; Ai, W.; Ju, Z.R.; Yang, B.; Wang, J.; Yang, X.W.; Liu, Y. Ascomycotin A, a new citromycetin analogue produced by Ascomycota sp. Ind19F07 isolated from deep sea sediment. Nat. Prod. Res. 2015, 29, 820–826. [Google Scholar] [CrossRef] [PubMed]
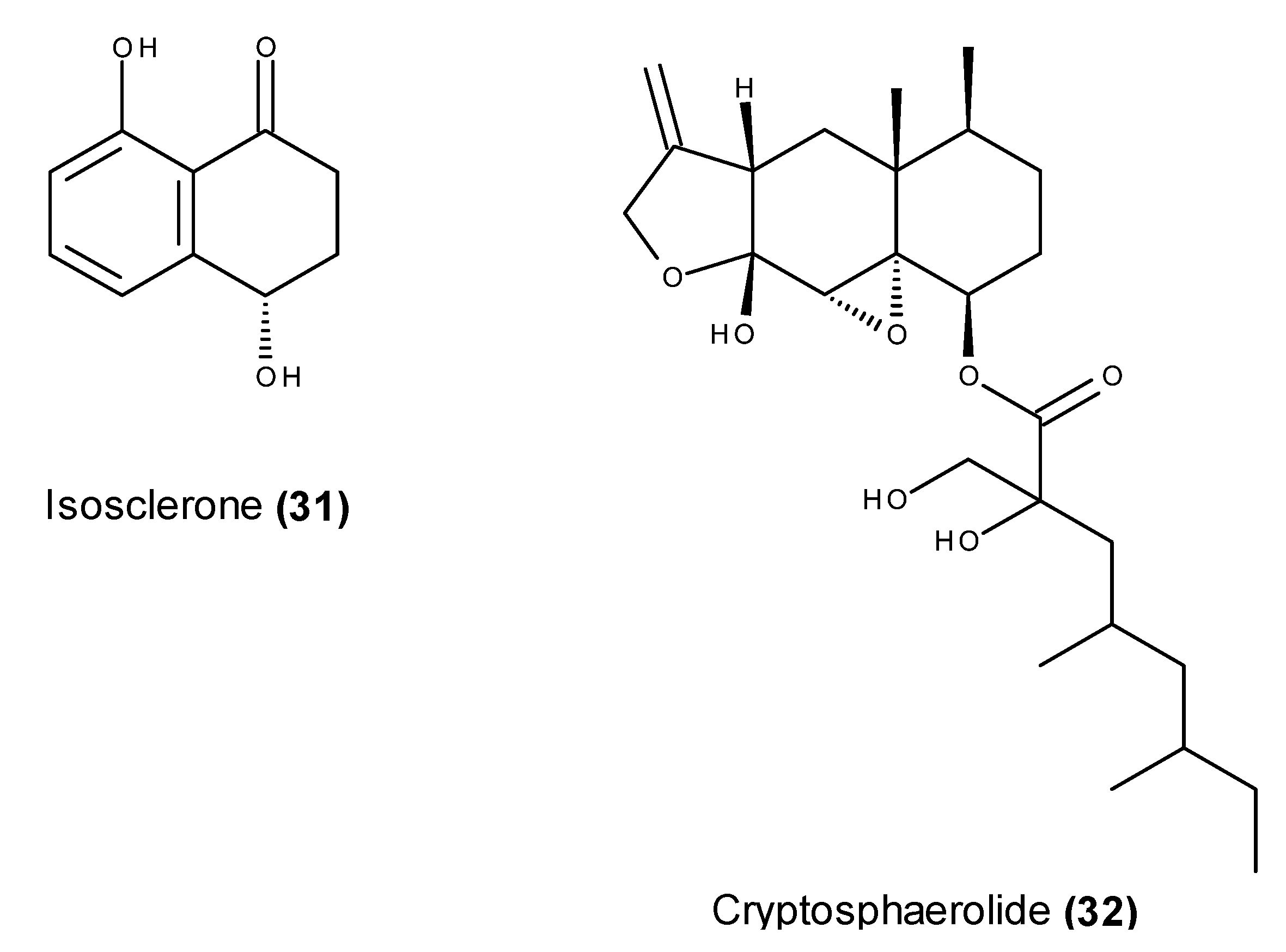
- Li, Y.X.; Himaya, S.W.A.; Dewapriya, P.; Kim, H.J.; Kim, S.K. Anti-proliferative effects of isosclerone isolated from marine fungus Aspergillus fumigatus in MCF-7 human breast cancer cells. Process Biochem. 2014, 49, 2292–2298. [Google Scholar] [CrossRef]
- Li, Y.X.; Kang, K.H.; Kim, H.J.; Kim, S.K. In vitro induction of apoptosis by isosclerone from marine-derived fungus Aspergillus fumigatus. Bioorg. Med. Chem. Lett. 2014, 24, 3923–3927. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Jensen, P.R.; Murphy, B.T.; Fiorilla, C.; Sullivan, J.F.; Ramsey, T.; Fenical, W. Cryptosphaerolide: A Cytotoxic Mcl-1 Inhibitor from a Marine-Derived Ascomycete Related to the Genus Cryptosphaeria. J. Nat. Prod. 2010, 73, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Belmar, J.; Fesik, S.W. Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol. Ther. 2015, 145C, 76–84. [Google Scholar] [CrossRef] [PubMed]
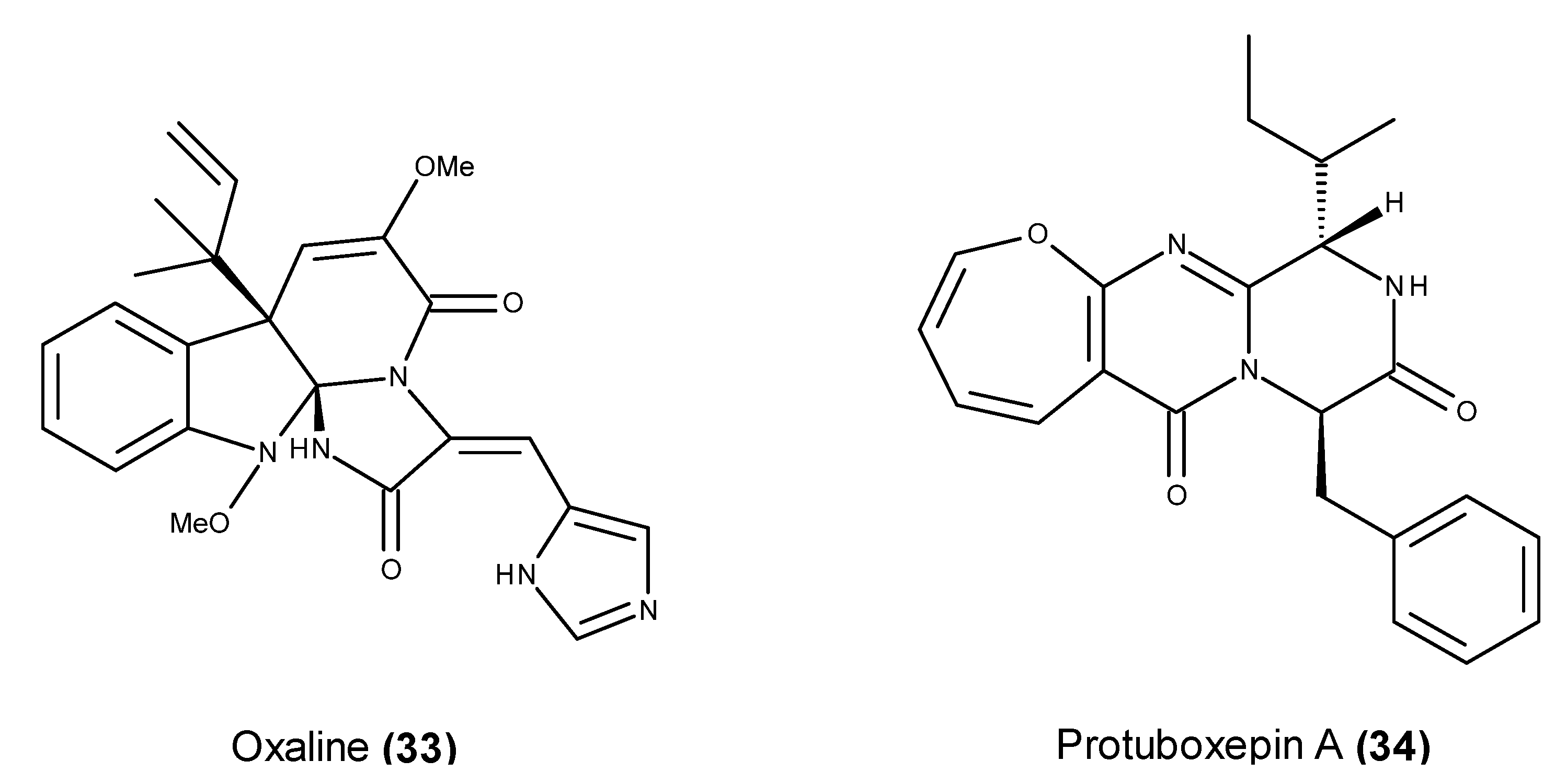
- Li, Y.; Li, X.F.; Kim, D.S.; Choi, H.D.; Son, B.W. Indolyl alkaloid derivatives, Nb-acetyltryptamine and oxaline from a marine-derived fungus. Arch. Pharm. Res. 2003, 26, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Jadulco, R.; Edrada, R.A.; Ebel, R.; Berg, A.; Schaumann, K.; Wray, V.; Steube, K.; Proksch, P. New communesin derivatives from the fungus Penicillium sp. derived from the Mediterranean sponge Axinella verrucosa. J. Nat. Prod. 2004, 67, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, Y.; Arai, M.; Tomoda, H.; Omura, S. Oxaline, a fungal alkaloid, arrests the cell cycle in M phase by inhibition of tubulin polymerization. Biochim. Biophys. Acta 2004, 1693, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.U.; Asami, Y.; Lee, D.; Jang, J.H.; Ahn, J.S.; Oh, H. Protuboxepins A and B and protubonines A and B from the marine-derived fungus Aspergillus sp. SF-5044. J. Nat. Prod. 2011, 74, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Ebada, S.S.; Fischer, T.; Hamacher, A.; Du, F.Y.; Roth, Y.O.; Kassack, M.U.; Wang, B.G.; Roth, E.H. Psychrophilin E, a new cyclotripeptide, from co-fermentation of two marine alga-derived fungi of the genus Aspergillus. Nat. Prod. Res. 2014, 28, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Asami, Y.; Jang, J.-H.; Soung, N.-K.; He, L.; Moon, D.O.; Kim, J.W.; Oh, H.; Muroi, M.; Osada, H.; Kim, B.Y.; et al. Protuboxepin A, a marine fungal metabolite, inducing metaphase arrest and chromosomal misalignment in tumor cells. Bioorg. Med. Chem. 2012, 20, 3799–3806. [Google Scholar] [CrossRef] [PubMed]
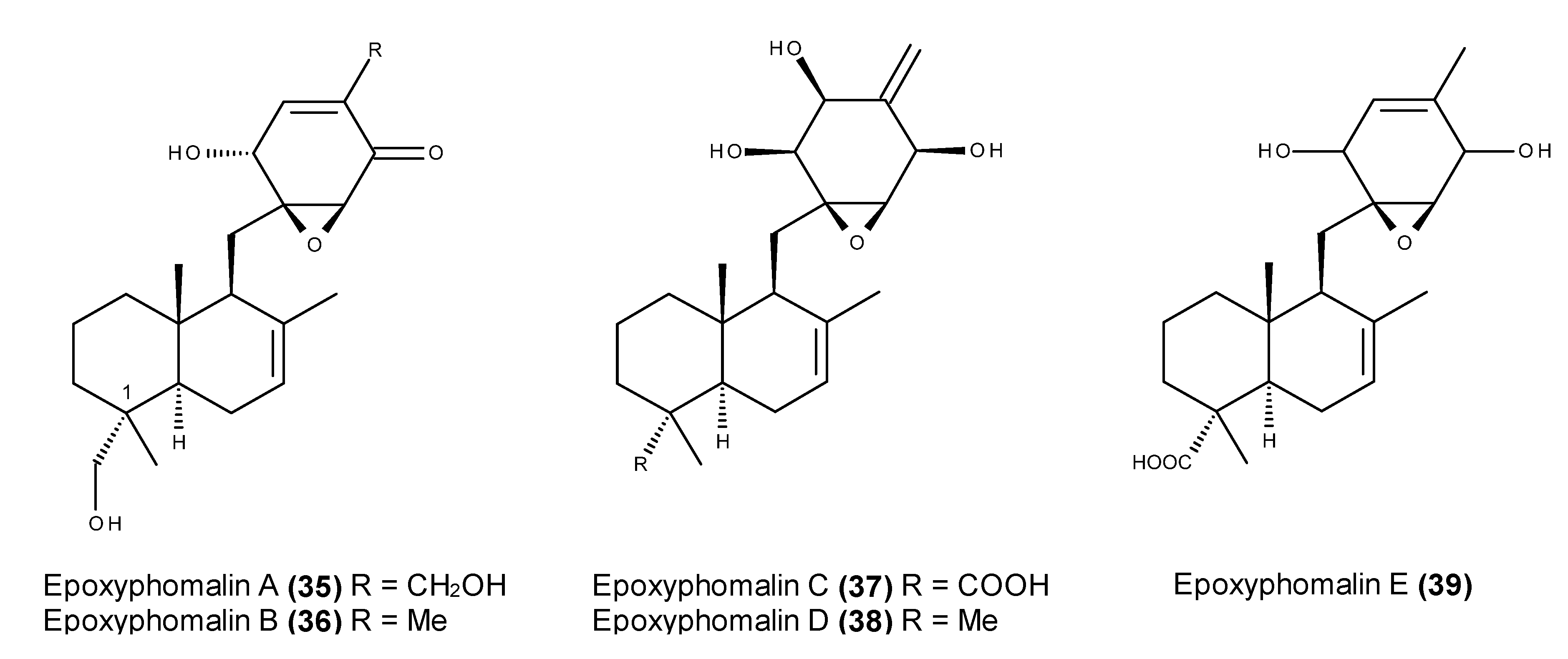
- Mohamed, I.E.; Gross, H.; Pontius, A.; Kehraus, S.; Krick, A.; Kelter, G.; Maier, A.; Fiebig, H.H.; König, G.M. Epoxyphomalin A and B, prenylated polyketides with potent cytotoxicity from the marine-derived fungus Phoma sp. Org. Lett. 2009, 11, 5014–5017. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, I.E.; Kehraus, S.; Krick, A.; König, G.M.; Kelter, G.; Maier, A.; Fiebig, H.-H.; Kalesse, M.; Malek, N.P.; Gross, H. Mode of action of epoxyphomalins a and b and characterization of related metabolites from the marine-derived fungus Paraconiothyrium sp. J. Nat. Prod. 2010, 73, 2053–2056. [Google Scholar] [CrossRef] [PubMed]
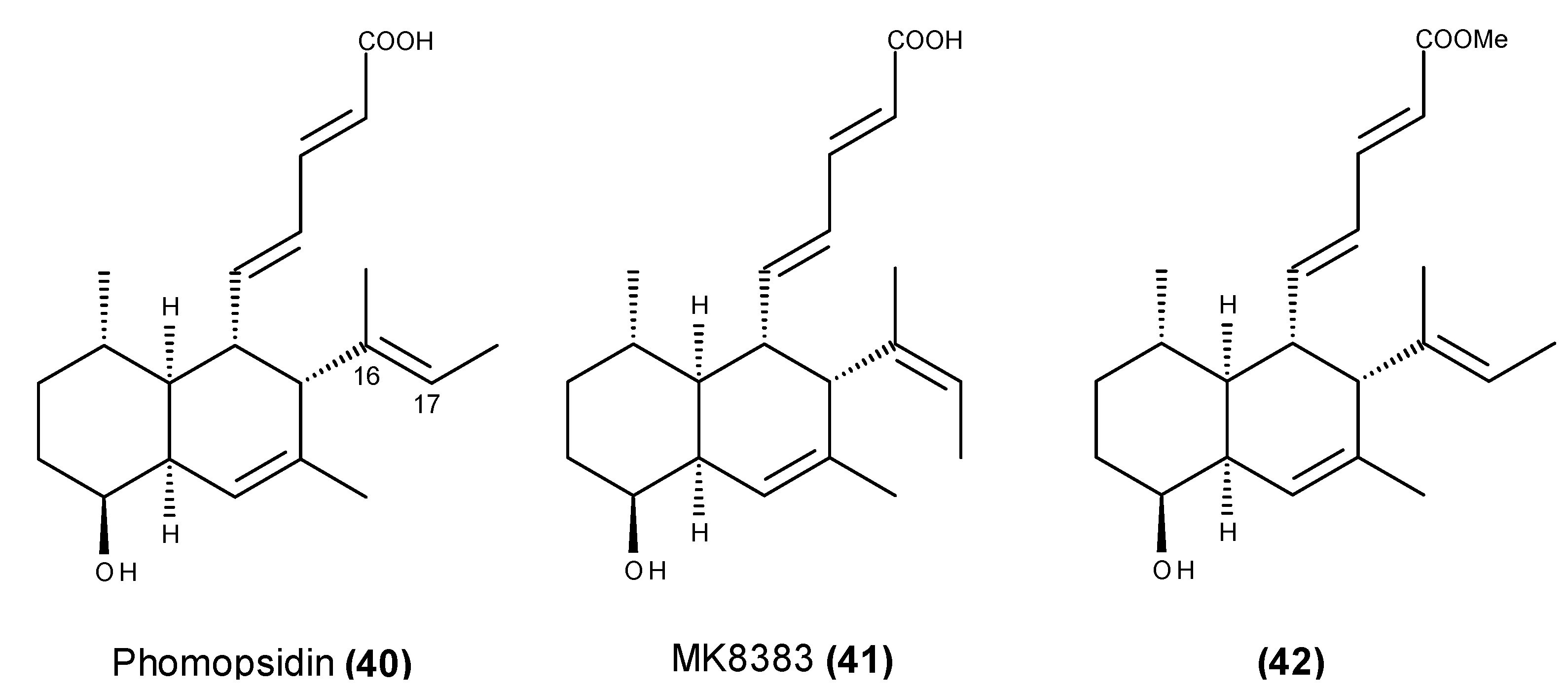
- Namikoshi, M.; Kobayashi, H.; Yoshimoto, T.; Hosoya, T. Phomopsidin, a new inhibitor of microtubule assembly produced by Phomopsis sp. isolated from coral reef in Pohnpei. J. Antibiot. (Tokyo) 1997, 50, 890–892. [Google Scholar] [CrossRef]
- Namikoshi, M.; Kobayashi, H.; Yoshimoto, T.; Meguro, S.; Akano, K. Isolation and characterization of bioactive metabolites from marine-derived filamentous fungi collected from tropical and sub-tropical coral reefs. Chem. Pharm. Bull. 2000, 48, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Sunaga, R.; Furihata, K.; Morisaki, N.; Iwasaki, S. Isolation and structures of an antifungal antibiotic, fusarielin A, and related compounds produced by a Fusarium sp. J. Antibiot. (Tokyo) 1995, 48, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Meguro, S.; Yoshimoto, T.; Namikoshi, M. Absolute structure, biosynthesis, and anti-microtubule activity of phomopsidin, isolated from a marine-derived Phomopsis sp. Tetrahedron 2003, 59, 455–459. [Google Scholar] [CrossRef]
- Kobayashi, H.; Meguro, S.; Yoshimoto, T.; Namikoshi, M. Corrigendum to “Absolute structure, biosynthesis, and anti-microtubule activity of phomopsidin, isolated from a marine-derived Phomopsis sp.” [Tetrahedron 59 (2003) 455–459]. Tetrahedron 2004, 60, 1255. [Google Scholar]
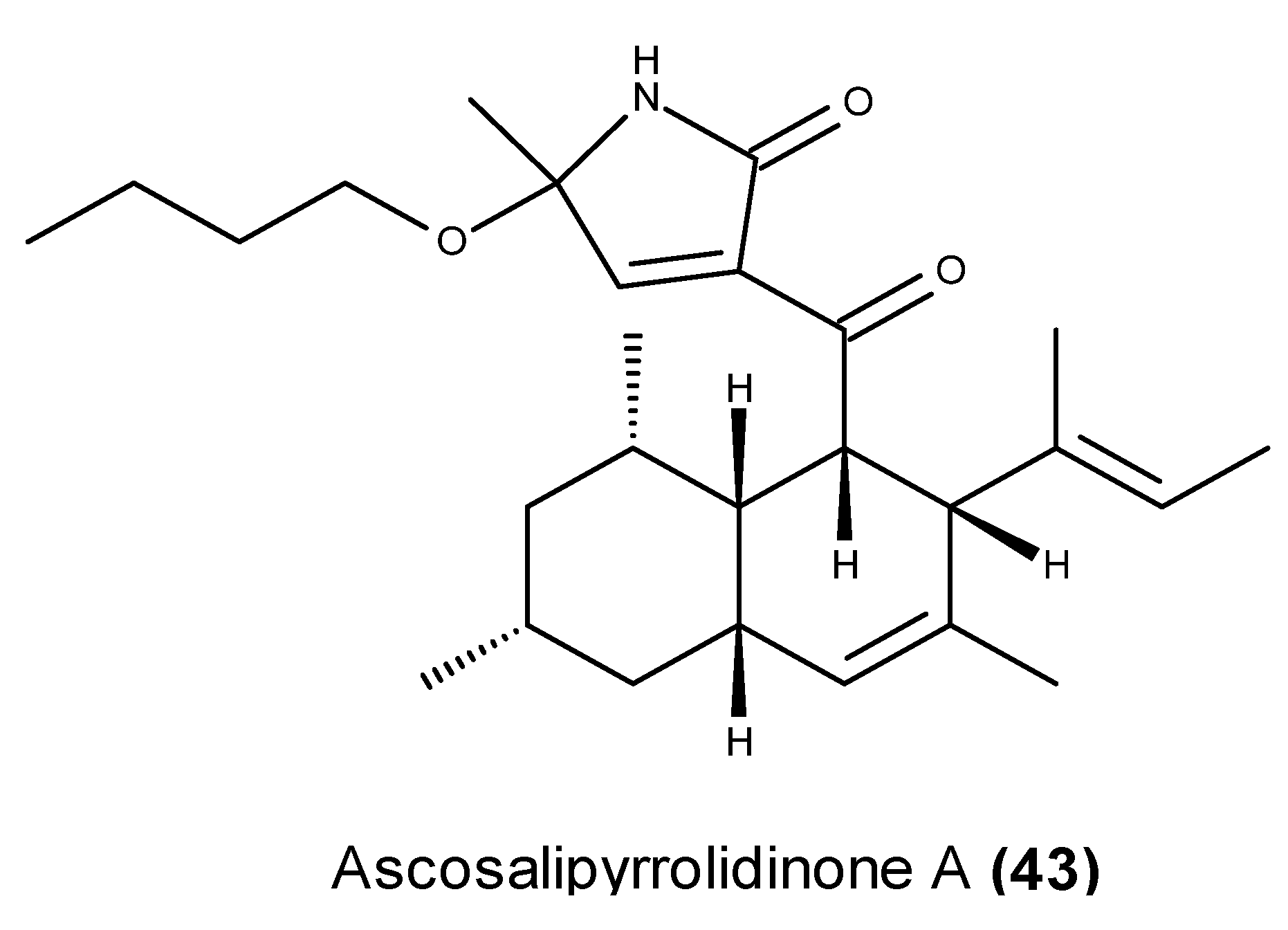
- Osterhage, C.; Kaminsky, R.; Kӧnig, G.M.; Wright, A.D. Ascosalipyrrolidinone A, an antimicrobial alkaloid, from the obligate marine fungus Ascochyta salicorniae. J. Org. Chem. 2000, 65, 6412–6417. [Google Scholar] [CrossRef] [PubMed]
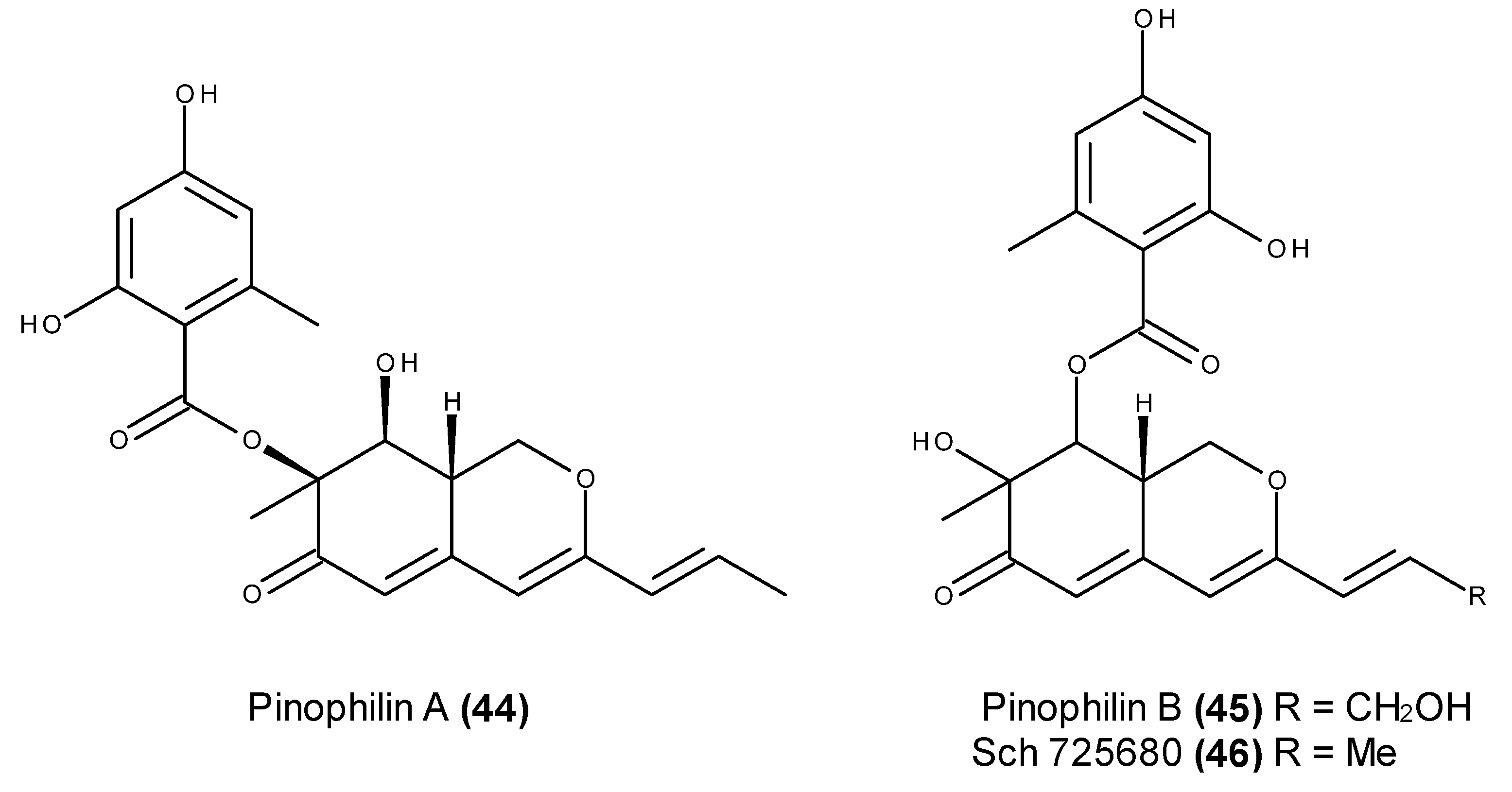
- Myobatake, Y.; Takeuchi, T.; Kuramochi, K.; Kuriyama, I.; Ishido, T.; Hirano, K.; Sugawara, F.; Yoshida, H.; Mizushina, Y. Pinophilins A and B, inhibitors of mammalian A-, B-, and Y-family DNA polymerases and human cancer cell proliferation. J. Nat. Prod. 2012, 75, 135–141. [Google Scholar] [CrossRef] [PubMed]
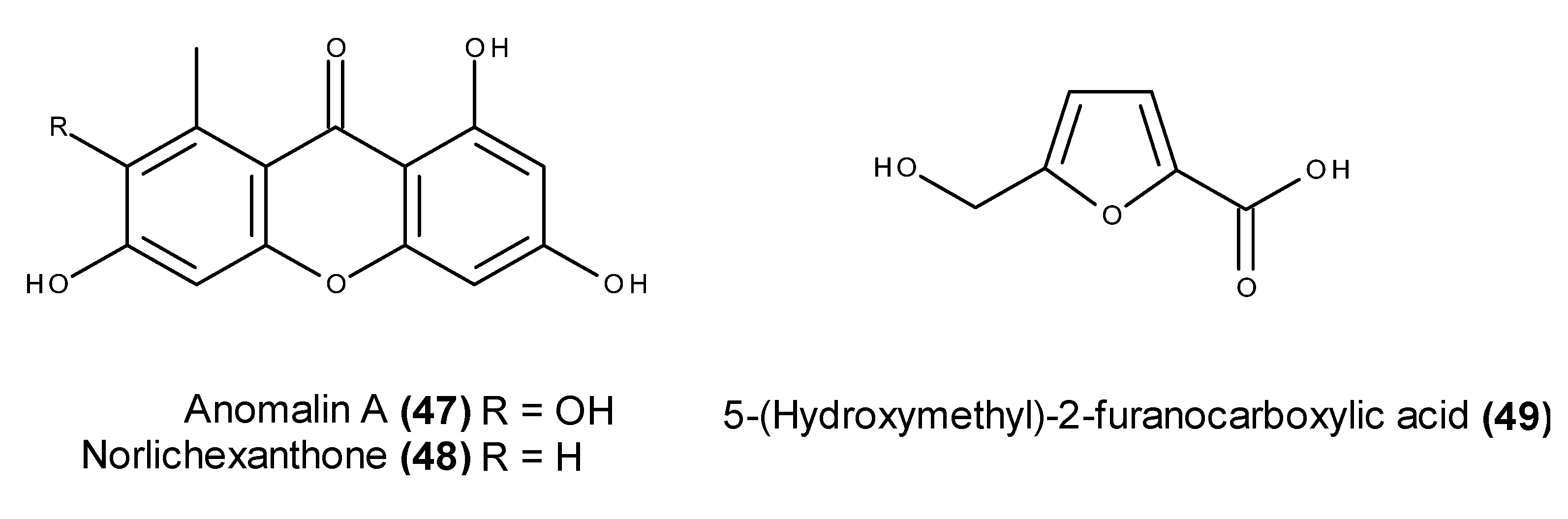
- Abdel-Lateff, A.; Klemke, C.; Kӧnig, G.M.; Wright, A.D. Two new xanthone derivatives from the algicolous marine fungus Wardomyces anomalus. J. Nat. Prod. 2003, 66, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Ebada, S.S.; Schulz, B.; Wray, V.; Totzke, F.; Kubbutat, M.H.G.; Muller, W.E.G.; Hamacher, A.; Kassack, M.U.; Lin, W.; Proksch, P. Arthrinins A–D: Novel diterpenoids and further constituents from the sponge derived fungus Arthrinium sp. Bioorg. Med. Chem. 2011, 19, 4644–4651. [Google Scholar] [CrossRef] [PubMed]
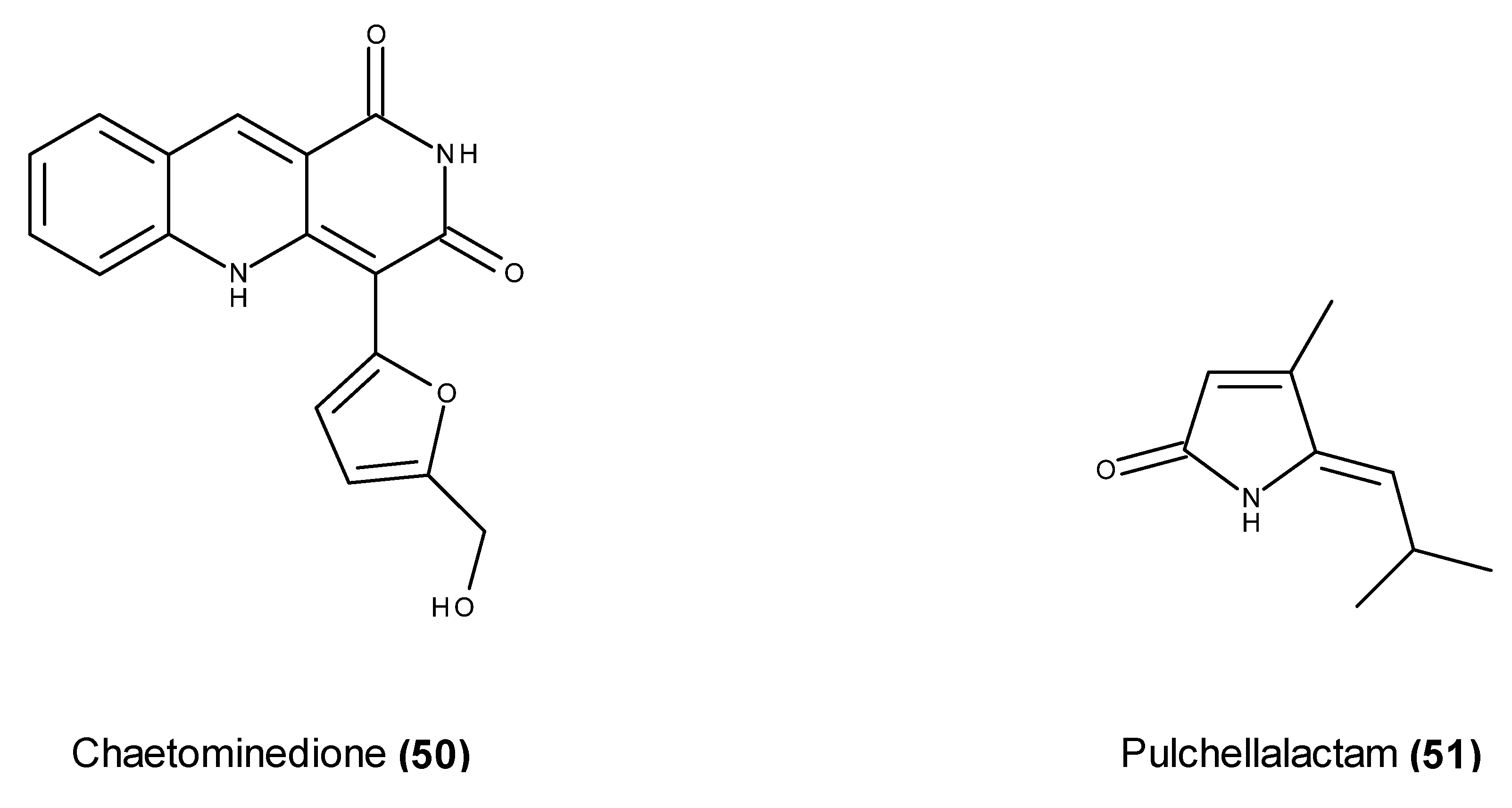
- Abdel-Lateff, A. Chaetominedione, a new tyrosine kinase inhibitor isolated from the algicolous marine fungus Chaetomium sp. Tetrahedron Lett. 2008, 49, 6398–6400. [Google Scholar] [CrossRef]
- Szigligeti, P.; Neumeier, L.; Duke, E.; Chougnet, C.; Takimoto, K.; Lee, S.M.; Filipovich, A.H.; Conforti, L. Signalling during hypoxia in human T lymphocytes—Critical role of the Src protein tyrosine kinase p56Lck in the O2 sensitivity of Kv1.3 channels. J. Physiol. 2006, 573, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Alvi, K.A.; Casey, A.; Nair, B.G. Pulchellalactam: A CD45 protein tyrosine phosphatase inhibitor from the marine fungus Corollospora pulchella. J. Antibiot. (Tokyo) 1998, 51, 515–517. [Google Scholar] [CrossRef] [PubMed]
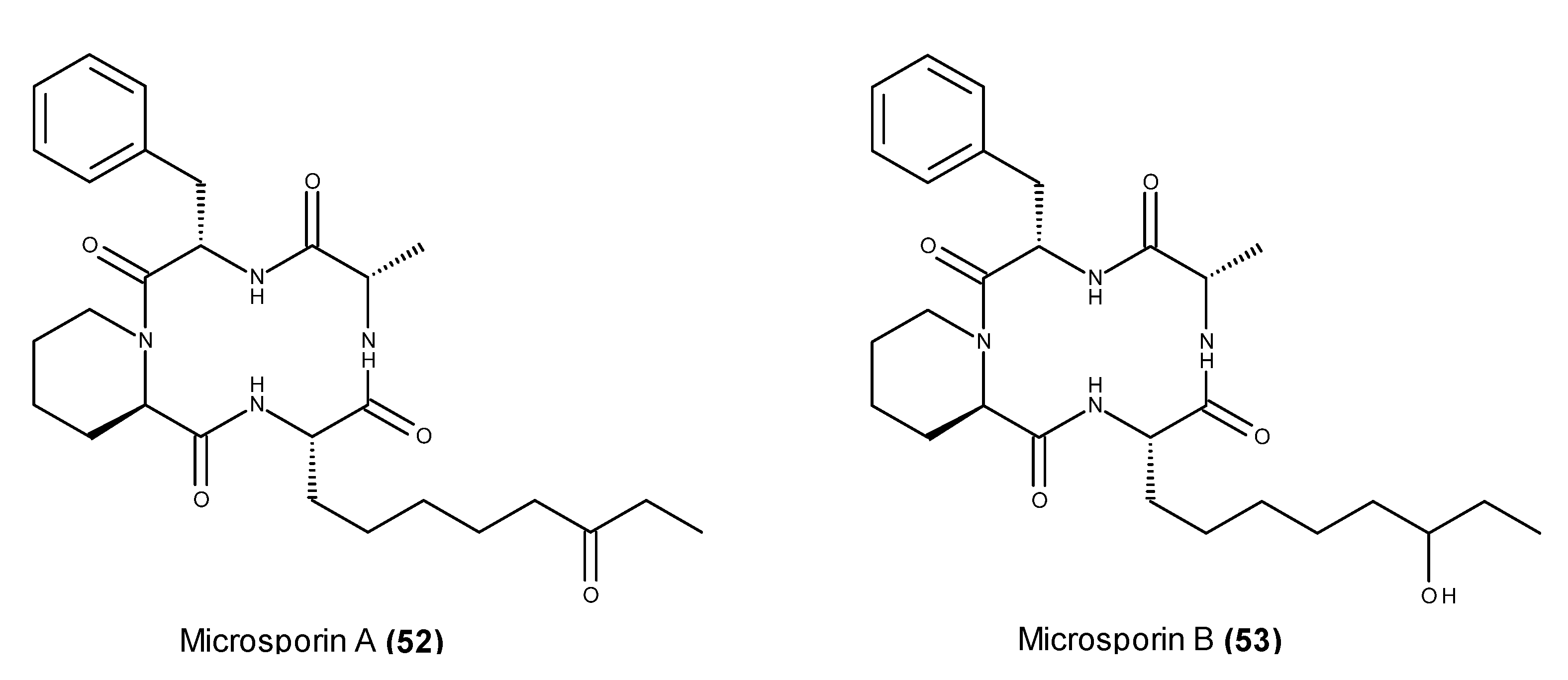
- Gu, W.; Cueto, M.; Jensen, P.R.; Fenical, W.; Silverman, R.B. Microsporins A and B: New histone deacetylase inhibitors from the marine-derived fungus Microsporum cf. gypseum and the solid-phase synthesis of microsporin A. Tetrahedron 2007, 63, 6535–6541. [Google Scholar]
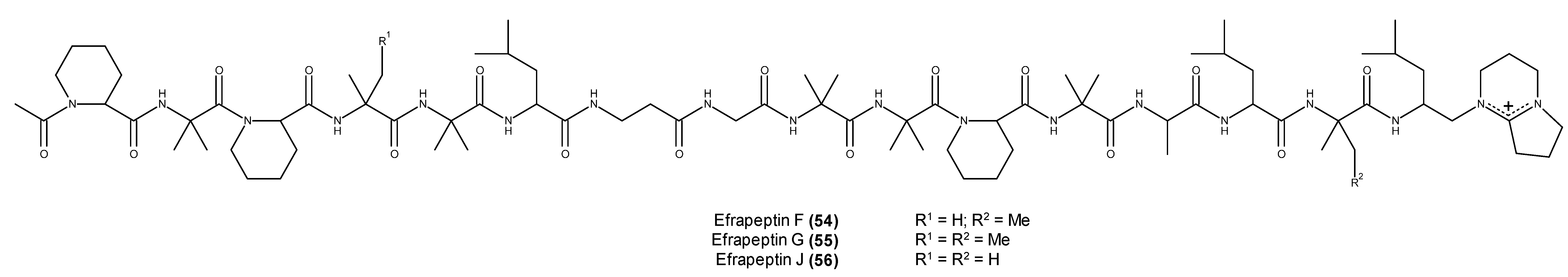
- Hayakawa, Y.; Hattori, Y.; Kawasaki, T.; Kanoh, K.; Adachi, K.; Shizuri, Y.; Shin-Ya, K. Efrapeptin J, a new down-regulator of the molecular chaperone GRP78 from a marine Tolypocladium sp. J. Antibiot. 2008, 61, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Roller, C.; Maddalo, D. The molecular chaperone GRP78/BiP in the development of chemoresistance: Mechanism and possible treatment. Front. Pharmacol. 2013, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Shenolikar, S. Dangerous liaisons: Flirtations between oncogenic BRAF and GRP78 in drug-resistant melanomas. J. Clin. Invest. 2014, 124, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Numata, A.; Iritani, M.; Yamada, T.; Minoura, K.; Matsumura, E.; Yamori, T.; Tsuruo, T. Novel antitumor metabolites produced by a fungal strain from a sea hare. Tetrahedron Lett. 1997, 38, 8215–8218. [Google Scholar] [CrossRef]
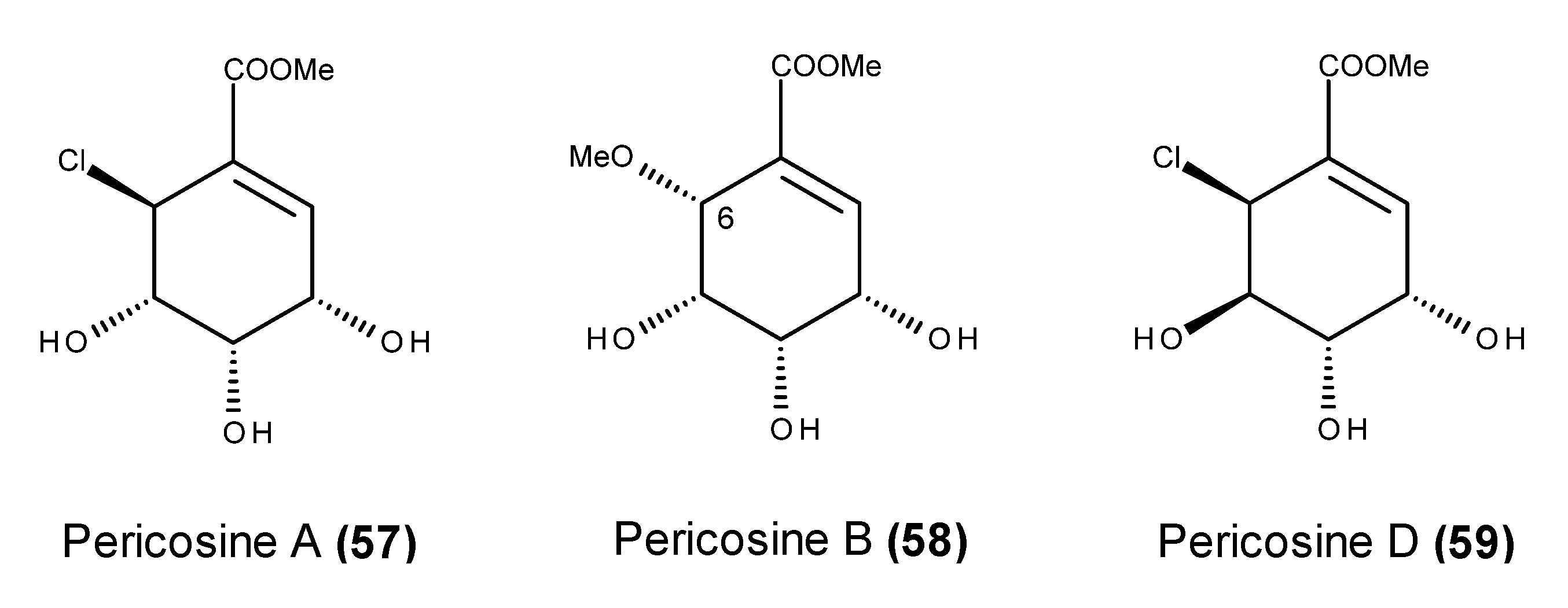
- Yamada, T.; Iritani, M.; Ohishi, H.; Tanaka, K.; Minoura, K.; Doi, M.; Numata, A. Pericosines, antitumour metabolites from the sea hare-derived fungus Periconia byssoides. Structures and biological activities. Org. Biomol. Chem. 2007, 5, 3979–3986. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Hatsuno, C.; Yamamoto, H.; Tanabe, M.; Numata, A. Synthesis of the epimer of pericosine B from (−)-quinic acid. Chem. Pharm. Bull. 2004, 52, 1130–1133. [Google Scholar] [CrossRef] [PubMed]
- Darro, F.; Decaestecker, C.; Gaussin, J.F.; Mortier, S.; van Ginckel, R.; Kiss, R. Are syngeneoic mouse tumor models still valuable experimental models in the field of anti-cancer drug discovery? Int. J. Oncol. 2005, 27, 607–616. [Google Scholar] [PubMed]
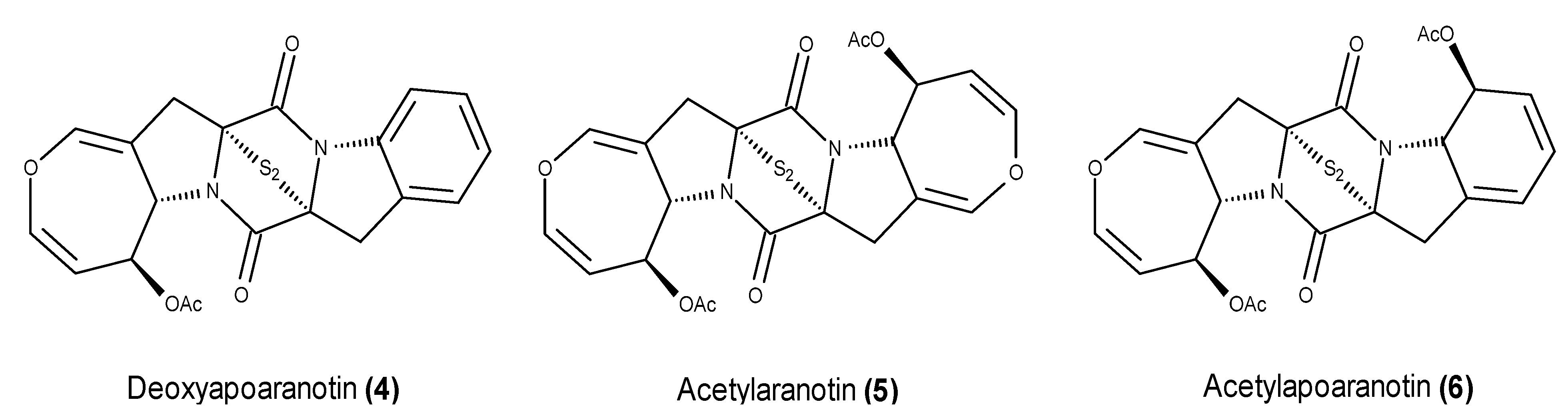
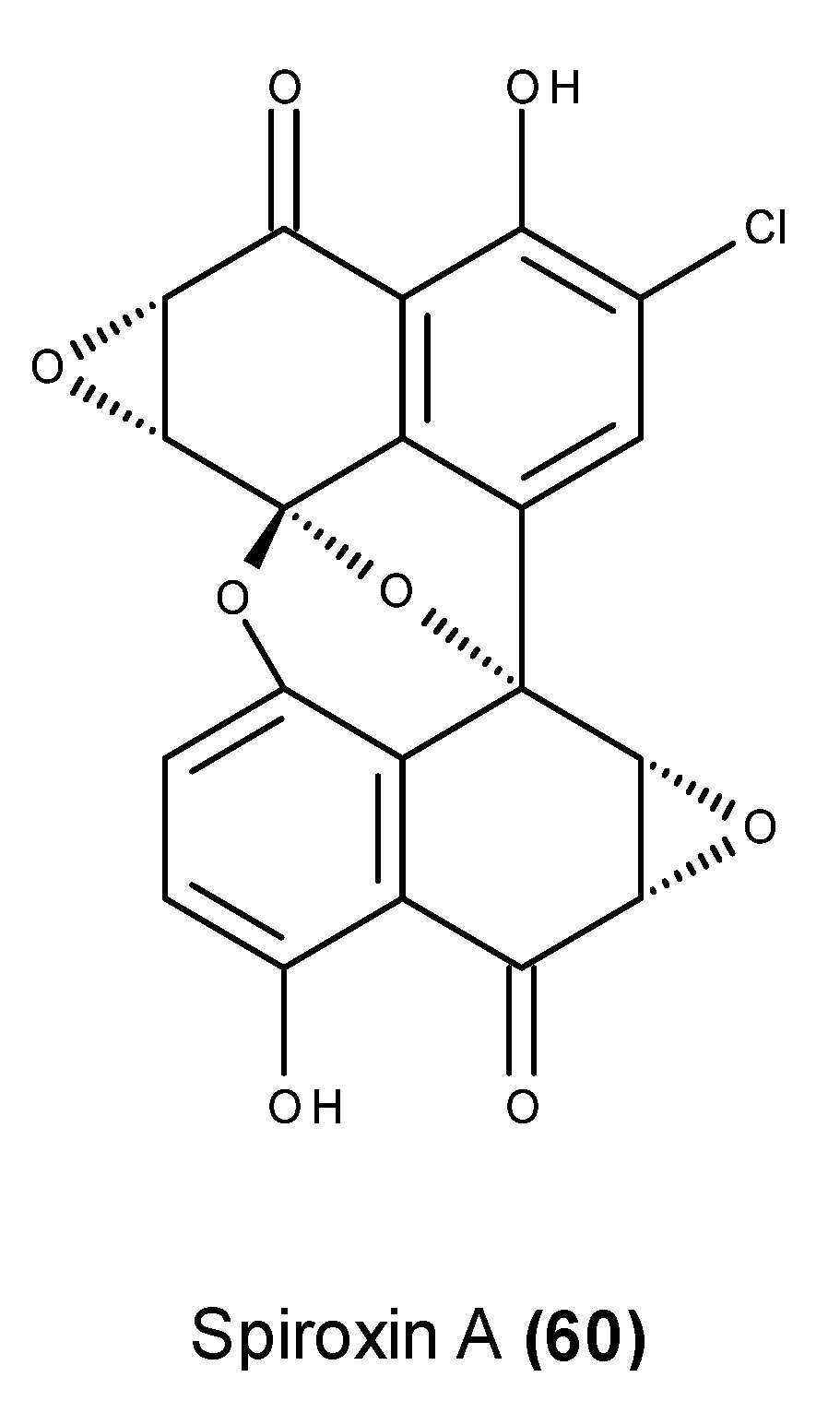
- McDonald, L.A.; Abbanat, D.R.; Barbieri, L.R.; Beruan, V.S.; Discafani, C.M.; Greenstein, M.; Janota, K.; Korshalla, J.D.; Lassota, P.; Tischler, M.; et al. Spiroxins, DNA cleaving antitumor antibiotics from a marine-derived fungus. Tetrahedron Lett. 1999, 40, 2489–2492. [Google Scholar] [CrossRef]
- Westin, S.N.; Herzog, T.J.; Coleman, R.L. Investigational agents in development for the treatment of ovarian cancer. Invest. New Drugs 2013, 31, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Lao, J.; Madani, J.; Puértolas, T.; Alvarez, M.; Hernández, A.; Pazo-Cid, R.; Artal, A.; Antón Torres, A. Liposomal Doxorubicin in the treatment of breast cancer patients: A review. J. Drug Deliv. 2013, 456409. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics, 2013. CA Cancer J. Clin. 2014, 64, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Tryfonidis, K.; Senkus, E.; Cardoso, M.J.; Cardoso, F. Management of locally advanced breast cancer-perspectives and future directions. Nat. Rev. Clin. Oncol. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Knuefermann, C.; Lu, Y.; Liu, B.; Jin, W.; Liang, K.; Wu, L.; Schmidt, M.; Mills, G.B.; Mendelsohn, J.; Fan, Z. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene 2003, 22, 3205–3212. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Lu, Z.; Meng, L.; Wei, S.; Hong, K.; Zhu, W.; Huang, C. The novel agent ophiobolin O induces apoptosis and cell cycle arrest of MCF-7 cells through activation of MAPK signaling pathways. Bioorg. Med. Chem. Lett. 2012, 22, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Qin, W.; Zhu, T.; Wei, S.; Hong, K.; Zhu, W.; Chen, R.; Huang, C. Ophiobolin O isolated from Aspergillus ustus induces G1 arrest of MCF-7 cells through interaction with AKT/GSK3beta/cyclin D1 signaling. Mar. Drugs 2015, 13, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Lu, C.; Zhu, T.; Yang, X.; Wei, S.; Sun, J.; Hong, K.; Zhu, W.; Huang, C. Ophiobolin-O reverses adriamycin resistance via cell cycle arrest and apoptosis sensitization in adriamycin-resistant human breast carcinoma (MCF-7/ADR) cells. Mar. Drugs 2013, 11, 4570–4584. [Google Scholar] [CrossRef] [PubMed]
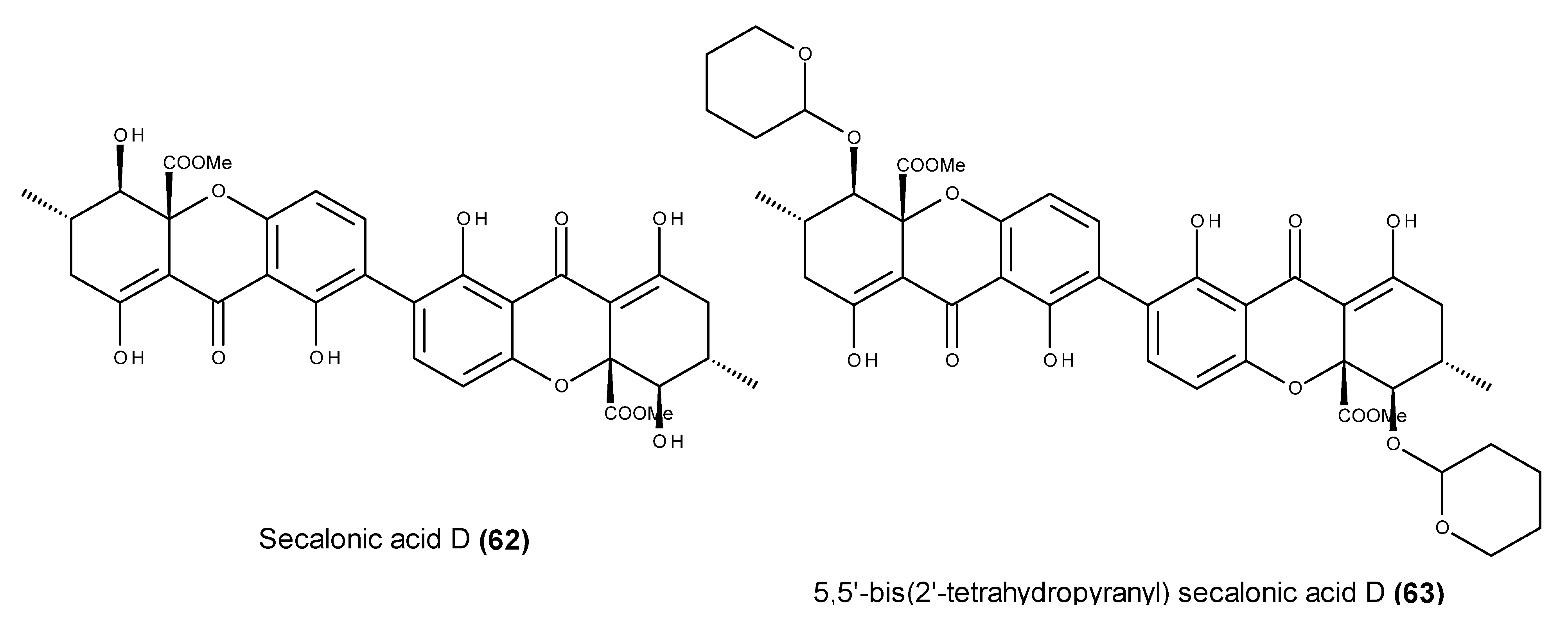
- Ren, H.; Tian, L.; Gu, Q.; Zhu, W. Secalonic acid D; a cytotoxic constituent from marine lichen-derived fungus Gliocladium sp. T31. Arch. Pharm. Res. 2006, 29, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Tao, L.Y.; Liang, Y.J.; Yan, Y.Y.; Dai, C.L.; Xia, X.K.; She, Z.G.; Lin, Y.C.; Fu, L.W. Secalonic acid D induced leukemia cell apoptosis and cell cycle arrest of G1 with involvement of GSK-3β/β-catenin/c-Myc pathway. Cell Cycle 2009, 8, 2444–2450. [Google Scholar] [CrossRef] [PubMed]
- Antia, B.S.; Aree, T.; Kasettrathat, C.; Wiyakrutta, S.; Ekpa, O.D.; Ekpe, U.J.; Mahidol, C.; Ruchirawat, S.; KIttakoop, P. Itanoic acid derivatives and diketopiperazine from the marine-derived fungus Aspergillus aculeatus CRI322-03. Phytochemistry 2011, 72, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Sun, Y.L.; Zhang, X.Y.; Han, Z.; Gao, H.C.; He, F.; Qian, P.Y.; Qi, S.H. Antifouling and antibacterial polyketides from marine gorgonian coral-associated fungus Penicillium sp. SCSGAF 0023. J. Antibiot. 2013, 66, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Jiang, Z.; Bai, J.; Wang, H.; Zhang, S.; Pei, Y. Isolation, structure determination, in vivo/vitro assay and docking study of a xanthone with antitumor activity from fungus Penicillium oxalicum. Rec. Nat. Prod. 2015, 9, 184–189. [Google Scholar]
- Kurobane, I.; Iwahashi, S.; Fukuda, A. Cytostatic activity of naturally isolated isomers of secalonic acids and their chemically rearranged dimers. Drugs Exp. Clin. Res. 1987, 13, 339–344. [Google Scholar] [PubMed]
- Hu, Y.P.; Tao, L.Y.; Wang, F.; Zhang, J.Y.; Liang, Y.J.; Fu, L.W. Secalonic acid D reduced the percentage of side populations by down-regulating the expression of ABCG2. Biochem. Pharmacol. 2013, 85, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Volk, E.I.; Farley, K.M.; Wu, Y.; Li, F.; Robey, R.W.; Schneider, E. Overexpression of wild-type breast cancer resistance protein mediates methotrexate resistance. Cancer Res. 2002, 62, 5035–5040. [Google Scholar] [PubMed]
- Hong, R. Secalonic acid D as a novel DNA topoisomerase I inhibitor from marine lichen-derived fungus Gliocladium sp. T31. Pharm. Biol. 2011, 49, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Nakamura, M.; Kataoka, T.; Iwaguchi, T. Mechanism of the antitumor activity of 5,5′-bis(2′-tetrahydropyranil) secalonic acid D against Meth-A. Cancer Chemother. Pharmacol. 1983, 11, 144–146. [Google Scholar] [CrossRef] [PubMed]
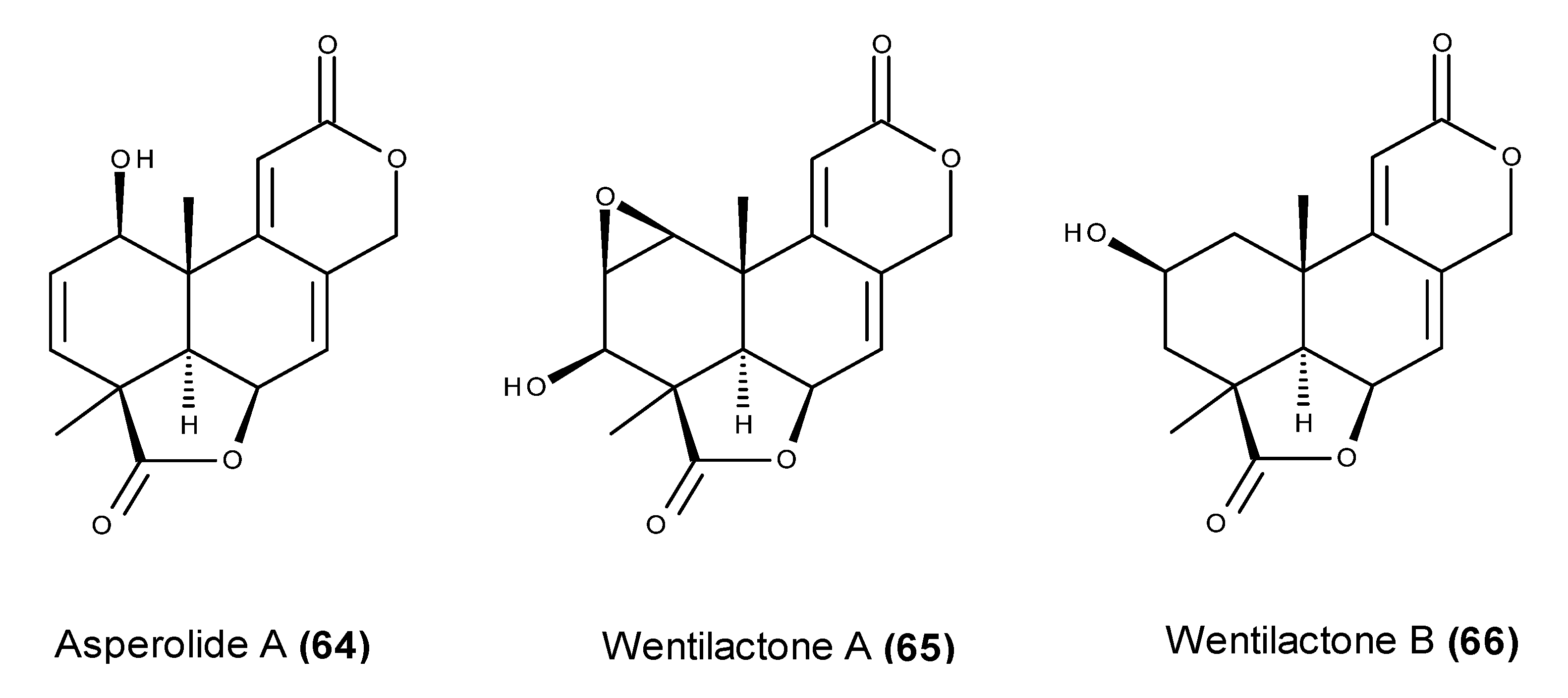
- Sun, H.F.; Li, X.M.; Meng, L.; Cui, C.M.; Gao, S.S.; Li, C.S.; Huang, C.G.; Wang, B.G. Asperolides A–C, tetranorlabdane diterpenoids from the marine alga-derived endophytic fungus Aspergillus wentii EN-48. J. Nat. Prod. 2012, 75, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Sun, W.; Sun, H.; Wei, S.; Chen, R.; Wang, B.; Huang, C. Asperolide A, a marine-derived tetranorditerpenoid, induces G2/M arrest in human NCI-H460 lung carcinoma cells, is mediated by p53-p21 stabilization and modulated by Ras/Raf/MEK/ERK signaling pathway. Mar. Drugs 2013, 11, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Hong, Y.; Miao, L.; Li, C.; Xu, G.; Wei, S.; Wang, B.; Huang, C.; Jiao, B. Wentilactone A as a novel potential antitumor agent induces apoptosis and G2/M arrest of human carcinoma cells, and is mediated by HRas-GTP accumulation to excessively activate the Ras/Raf/ERK/p53-p21 pathway. Cell Death Dis. 2013, 4, e952. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Miao, L.; Lv, C.; Sun, H.; Wei, S.; Wang, B.; Huang, C.; Jiao, B. Wentilactone B induces G2/M phase arrest and apoptosis via the Ras/Raf/MAPK signaling pathway in human hepatoma SMMC-7721 cells. Cell Death Dis. 2013, 4, e657. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Miao, L.; Sun, W.; Jiao, B.; Wang, B.; Yao, L.; Huang, C. Wentilactone B from Aspergillus wentii induces apoptosis and inhibits proliferation and migration of human hepatoma SMMC-7721 cells. Biol. Pharm. Bull. 2012, 35, 1964–1971. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.Y.S.; Abrell, L.M.; Avelar, A.; Borgeson, B.M.; Crews, P. New hirsutane based sesquiterpenes from salt water cultures of a marine sponge-derived fungus and the terrestrial fungus Coriolus consors. Tetrahedron 1998, 54, 7335–7342. [Google Scholar] [CrossRef]
- Li, H.J.; Lan, W.J.; Lam, C.K.; Yang, F.; Zhu, X.F. Hirsutane sesquiterpenoids from the marine-derived fungus Chondrostereum sp. Chem. Biodivers. 2011, 8, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Xie, Y.L.; Xie, Z.L.; Chen, Y.; Lam, C.K.; Lan, W.J. Chondrosterins A–E, triquinane-type sesquiterpenoids from soft coral-associated fungus Chondrostereum sp. Mar. Drugs 2012, 10, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chen, W.D.; Deng, R.; Li, D.D.; Wu, K.W.; Feng, G.K.; Li, H.J.; Zhu, X.F. Hirsutanol A induces apoptosis and autophagy via reactive oxygen species accumulation in breast cancer MCF-7 cells. J. Pharmacol. Sci. 2012, 119, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chen, W.D.; Deng, R.; Zhang, H.; Tang, J.; Wu, K.W.; Li, D.D.; Feng, G.K.; Lan, W.J.; Li, H.J.; et al. Hirsutanol A, a novel sesquiterpene compound from fungus Chondrostereum sp., induces apoptosis and inhibits tumor growth through mitochondrial-independent ROS production: Hirsutanol A inhibits tumor growth through ROS production. J. Trans. Med. 2013, 11, 32. [Google Scholar] [CrossRef] [PubMed]
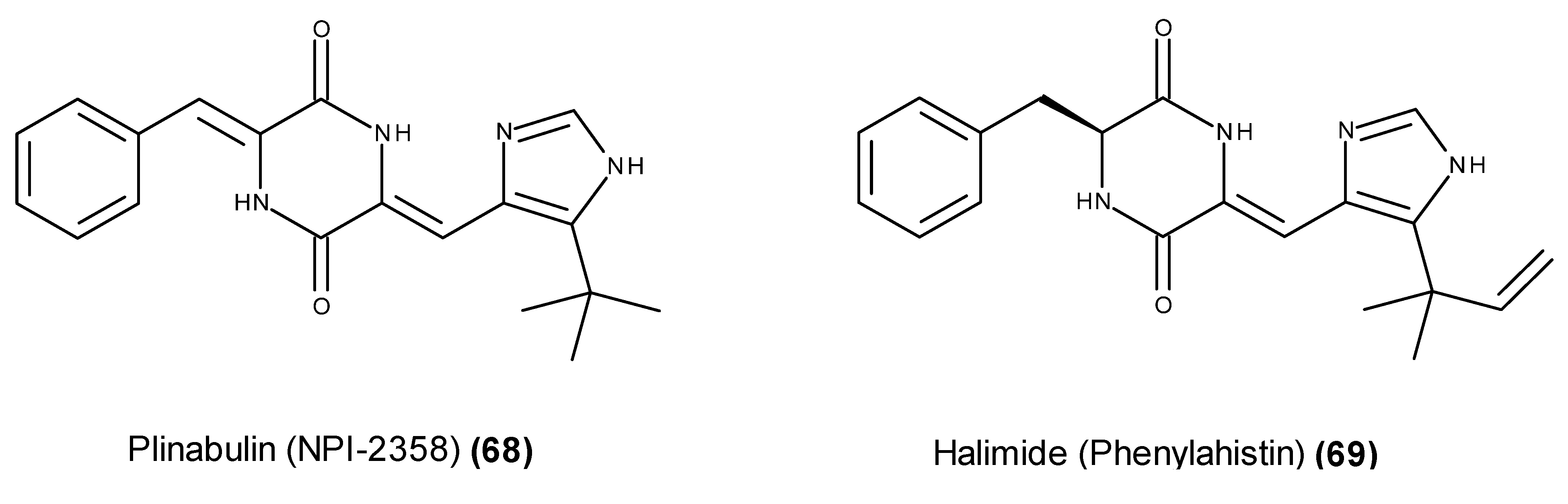
- Fenical, W.; Jensen, P.R.; Cheng, X.C. Halimide, a cytotoxic marine natural product, derivatives thereof. PCT Int. Appl. WO 1999048889 A1, 30 September 1999. [Google Scholar]
- Fenical, W.; Jensen, P.R.; Cheng, X.C. Halimide, a Cytotoxic Marine Natural Product, and Derivatives thereof. US. Patent US6069146, 30 May 2000. [Google Scholar]
- Fukumoto, K.; Kohno, S.; Kanoh, K.; Asari, T.; Kawashima, H.; Sekiya, H.; Ohmizo, K.; Harada, T. Phenylahistin and the Phenylahistin Analogs, a New Class of Anti-Tumor Compounds. US. Patent US6358957B1. US. Patent US6358957B1, 19 March 2002. [Google Scholar]
- Kanoh, K.; Kohno, S.; Asari, T.; Harada, T.; Katada, J.; Muramatsu, M.; Kawashima, H.; Sekiya, H.; Uno, I. (−)-Phenylahistin: A new mammalian cell cycle inhibitor produced by Aspergillus ustus. Bioorg. Med. Chem. Lett. 1997, 7, 2847–2852. [Google Scholar] [CrossRef]
- Kanoh, K.; Kohno, S.; Katada, J.; Takahashi, J.; Uno, I. (−)-Phenylahistin arrests cells in mitosis by inhibiting tubulin polymerization. J. Antibiot. 1999, 52, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Kanoh, K.; Kohno, S.; Katada, J.; Hayashi, Y.; Muramatsu, M.; Uno, I. Antitumor activity of phenylahistin in vitro and in vivo. Biosci. Biotechnol. Biochem. 1999, 63, 1130–1133. [Google Scholar] [CrossRef] [PubMed]
- Kanoh, K.; Kohno, S.; Katada, J.; Takahashi, J.; Uno, I.; Hayashi, Y. Synthesis and biological activities of phenylahistin derivatives. Bioorg. Med. Chem. 1999, 7, 1451–1457. [Google Scholar] [CrossRef]
- Hayashi, Y.; Orikasa, S.; Tanaka, K.; Kanoh, K.; Kiso, Y. Total synthesis of anti-microtubule diketopiperazine derivatives: Phenylahistin and aurantiamine. J. Org. Chem. 2000, 65, 8402–8405. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.; Lloyd, G.K.; Miller, B.R.; Palladino, M.A.; Kiso, Y.; Hayashi, Y.; Neuteboom, S.T. NPI-2358 is a tubulin-depolymerizing agent: In vitro evidence for activity as a tumor vascular-disrupting agent. Anticancer Drugs 2006, 17, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Sumikura, M.; Hidaka, K.; Yasui, H.; Kiso, Y.; Yakushiji, F.; Hayashi, Y. Anti-microtubule “plinabulin” chemical probe KPU-244-B3 labeled both α- and β-tubulin. Bioorg. Med. Chem. 2010, 18, 3169–3174. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Tanaka, K.; Nicholson, B.; Deyanat-Yazdi, G.; Potts, B.; Yoshida, T.; Oda, A.; Kitagawa, T.; Orikasa, S.; Kiso, Y.; et al. Synthesis and structure-activity relationship study of antimicrotubule agents phenylahistin derivatives with a didehydropiperazine-2,5-dione structure. J. Med. Chem. 2012, 55, 1056–1071. [Google Scholar] [CrossRef] [PubMed]
- Mita, M.M.; Spear, M.A.; Yee, L.K.; Mita, A.C.; Health, E.I.; Papadopoulos, K.P.; Federico, K.C.; Reich, S.D.; Romero, O.; Malburg, L.; et al. Phase 1 first-in-human trial of the vascular disrupting agent plinabulin (NPI-2358) in patients with solid tumors or lymphomas. Clin. Cancer Res. 2010, 16, 5892–5899. [Google Scholar] [CrossRef] [PubMed]
- Study of the Vascular Disrupting Agent NPI-2358 in Patients with Advanced Solid Tumors or Lymphoma. ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00322608?term=NPI-2358&rank=2 (accessed on 15 March 2015).
- Heist, R.; Aren, O.; Millward, M.; Mainwaring, P.; Mita, A.; Mita, M.; Bazhenova, L.; Blum, R.; Polikoff, J.; Gadgeel, S.; et al. Phase 1/2 study of the vascular disrupting agent (VDA) plinabulin (NPI-2358) combined with docetaxel in patients with non-small cell lung cancer (NSCLC). Mol. Cancer Ther. 2009, 8, C30. [Google Scholar] [CrossRef]
- Millward, M.; Mainwaring, P.; Mita, A.; Federico, K.; Lloyd, G.K.; Reddinger, N.; Nawrocki, S.; Mita, M.; Spear, M.A. Phase 1 study of the novel vascular disrupting agent plinabulin (NPI-2358) and docetaxel. Invest. New Drugs 2012, 30, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Phase 1/2 Study of Vascular Disrupting Agent NPI-2358 + Docetaxel in Patients with Advanced Non-Small Cell Lung Cancer. ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00630110?term=NPI-2358&rank=1 (accessed on 15 March 2015).
- Kondo, Y.; Shen, L.; Ahmed, S.; Boumber, Y.; Sekido, Y.; Haddad, B.R.; Issa, J.P.J. Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS ONE 2008, 3, e2037. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, J.; Kim, W.H.; Lee, Y.M. Hypoxic silencing of tumor suppressor RUNX3 by histone modification in gastric cancer cells. Oncogene 2009, 28, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Tang, A.J.; Castoreno, A.B.; Kuo, S.Y.; Wang, S.Y.; Wang, Q.; Kuballa, P.; Xavier, R.; Shamji, A.F.; Schreiber, S.L.; Wagner, B.K. Gossypol and an HMT G9a inhibitor act in synergy to induce cell death in pancreatic cancer cells. Cell Death Dis. 2013, 4, e690. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Takeuchi, T.; Nagayama, T.; Furihata, M. T-cadherin modulates tumor-associated molecules in gallbladder cancer cells. Cancer Invest. 2010, 28, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, A.; Remmelink, M.; D’Haene, N.; Penant, S.; Gaussin, J.F.; van Ginckel, R.; Darro, F.; Kiss, R.; Salmon, I. Development of a chemoresistant orthotopic human nonsmall cell lung carcinoma model in nude mice: Analyses of tumor heterogeneity in relation to the immunohistochemical levels of expression of cyclooxygenase-2, ornithine decarboxylase, lung-related resistance protein, prostaglandin E synthetase, and glutathione-S-transferase-alpha (GST)-alpha-, GST-mu, and GST-pi. Cancer 2004, 101, 1908–1918. [Google Scholar] [PubMed]
- Dumont, P.; Ingrassia, L.; Rouzeau, S.; Ribaucour, F.; Thomas, S.; Roland, I.; Darro, F.; Lefranc, F.; Kiss, R. The Amaryllidaceae isocarbostyril narciclasine induces apoptosis by activation of the Death Receptor and/or the mitochondrial pathways in cancer cells but not in normal fibroblasts. Neoplasia 2007, 9, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, F.; Brotchi, J.; Kiss, R. Possible future issues in the treatment of glioblastomas, with a special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J. Clin. Oncol. 2005, 23, 2411–2422. [Google Scholar] [CrossRef] [PubMed]
- Bury, M.; Girault, A.; Megalizzi, V.; Spiegl-Kreinecker, S.; Mathieu, V.; Berger, W.; Evidente, A.; Kornienko, A.; Gailly, P.; Vandier, C.; et al. Ophiobolin A induces paraptosis-like cell death in human glioblastoma cells by decreasing BKCa channel activity. Cell Death Dis. 2013, 4, e561. [Google Scholar] [CrossRef] [PubMed]
- Chu, E. An update on the current and emerging targeted agents in metastatic colorectal cancer. Clin. Colorectal Cancer 2012, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ravaud, A.; Gross-Goupil, M.; Bellmunt, J. Combination therapy in metastatic renal cell cancer. Semin. Oncol. 2013, 40, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Saksena, R.; Wong, S.T. Clinical evidence of the efficacy of everolimus and its potential in the treatment of breast cancer. Breast Cancer (Dove Med. Press) 2013, 5, 27–35. [Google Scholar] [PubMed]
- Beck, J.T.; Ismail, A.; Tolomeo, C. Targeting the phosphatidylinositol 3-kinase (PI3K)AKT/mammalian target of rapamycin (mTOR) pathway: An emerging treatment strategy for squamous cell lung carcinoma. Cancer Treat. Rev. 2014, 40, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Soengas, M.S.; Lowe, S.W. Apoptosis and melanoma chemoresistance. Oncogene 2003, 22, 3138–3151. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, V.; Mijatovic, T.; van Damme, M.; Kiss, R. Gastrin exerts pleitropic effects on human melanoma cell biology. Neoplasia 2005, 7, 930–943. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P.; Giannoni, E. Anoikis: A necessary death program for anchorage-dependent cells. Biochem. Pharmacol. 2008, 76, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.D.; Anyiwe, K.; Schimmer, A.D. Anoikis resistance and tumor metastasis. Cancer Lett. 2008, 272, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.H.; Lemoine, N.R. Pancreatic cancer: Molecular pathogenesis and new therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Bussink, J.; van Herpen, C.; Kaanders, J.; Oyen, W. PET-CT for response assessment and treatment adaptation in head and neck cancer. Lancet Oncol. 2010, 11, 661–669. [Google Scholar] [CrossRef]
- Kennedy, B.; Gargoum, F.; Bystricky, B.R.; Curran, D.M.; O’Connor, T. Novel agents in the management of lung cancer. Curr. Med. Chem. 2010, 17, 4291–4325. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, N.G.M.; Lefranc, F.; Kijjoa, A.; Kiss, R. Can Some Marine-Derived Fungal Metabolites Become Actual Anticancer Agents? Mar. Drugs 2015, 13, 3950-3991. https://doi.org/10.3390/md13063950
Gomes NGM, Lefranc F, Kijjoa A, Kiss R. Can Some Marine-Derived Fungal Metabolites Become Actual Anticancer Agents? Marine Drugs. 2015; 13(6):3950-3991. https://doi.org/10.3390/md13063950
Chicago/Turabian StyleGomes, Nelson G. M., Florence Lefranc, Anake Kijjoa, and Robert Kiss. 2015. "Can Some Marine-Derived Fungal Metabolites Become Actual Anticancer Agents?" Marine Drugs 13, no. 6: 3950-3991. https://doi.org/10.3390/md13063950
APA StyleGomes, N. G. M., Lefranc, F., Kijjoa, A., & Kiss, R. (2015). Can Some Marine-Derived Fungal Metabolites Become Actual Anticancer Agents? Marine Drugs, 13(6), 3950-3991. https://doi.org/10.3390/md13063950