
Chiral Alkyl Halides: Underexplored Motifs in Medicine

Abstract
:1. Introduction
2. Alkyl Halides in Medicine
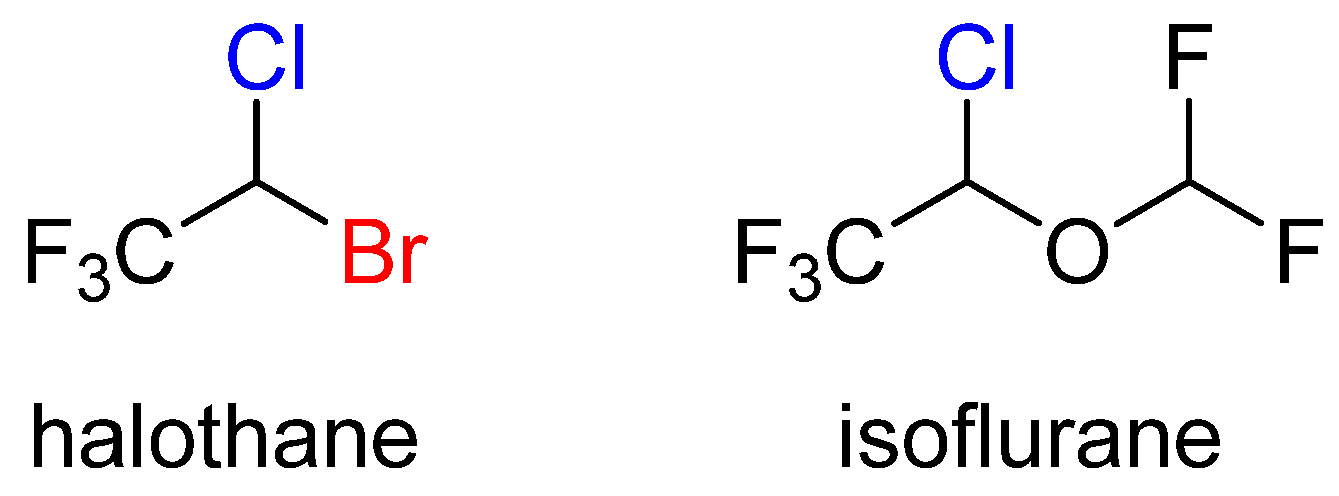
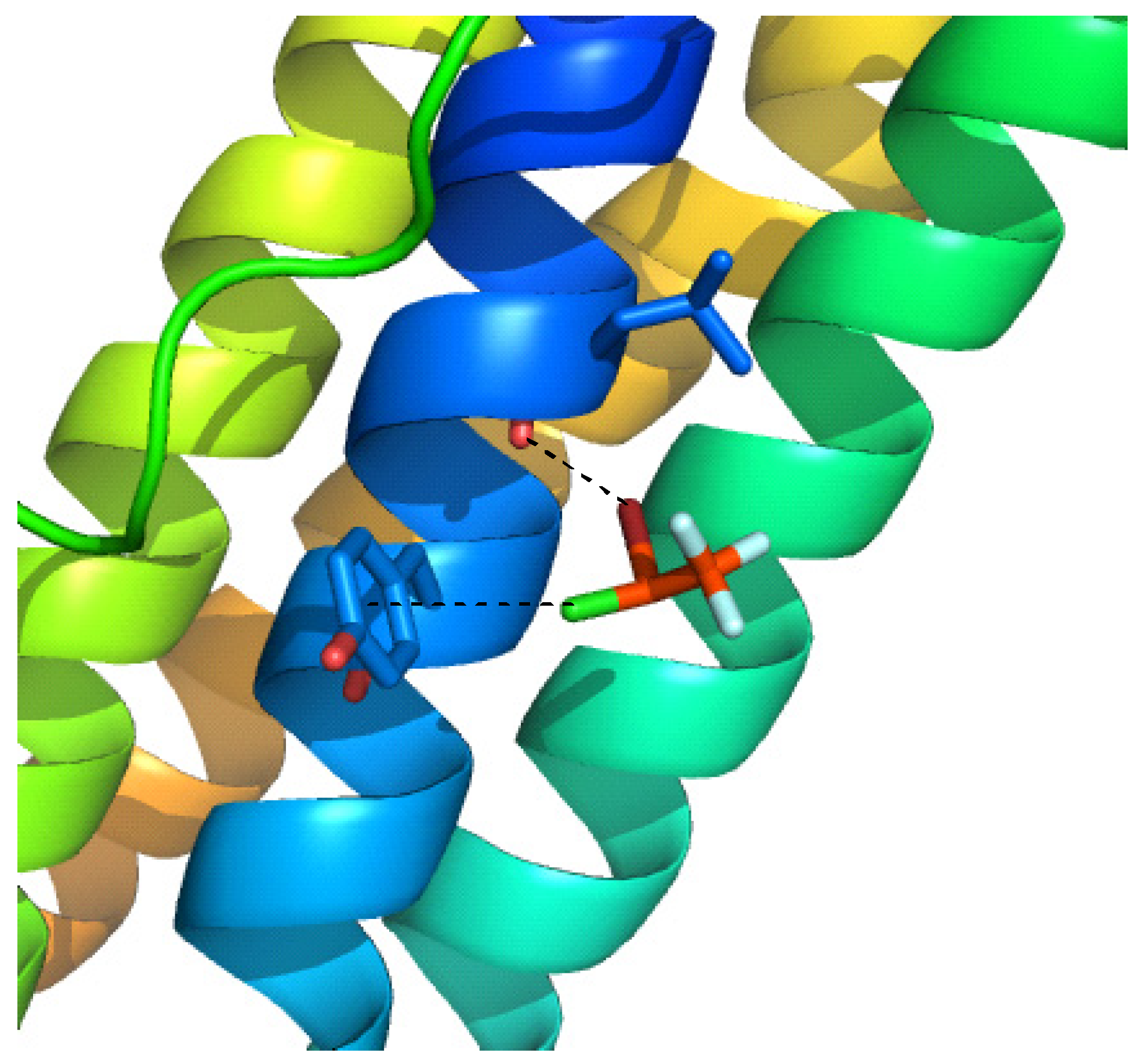
2.1. Anesthetics
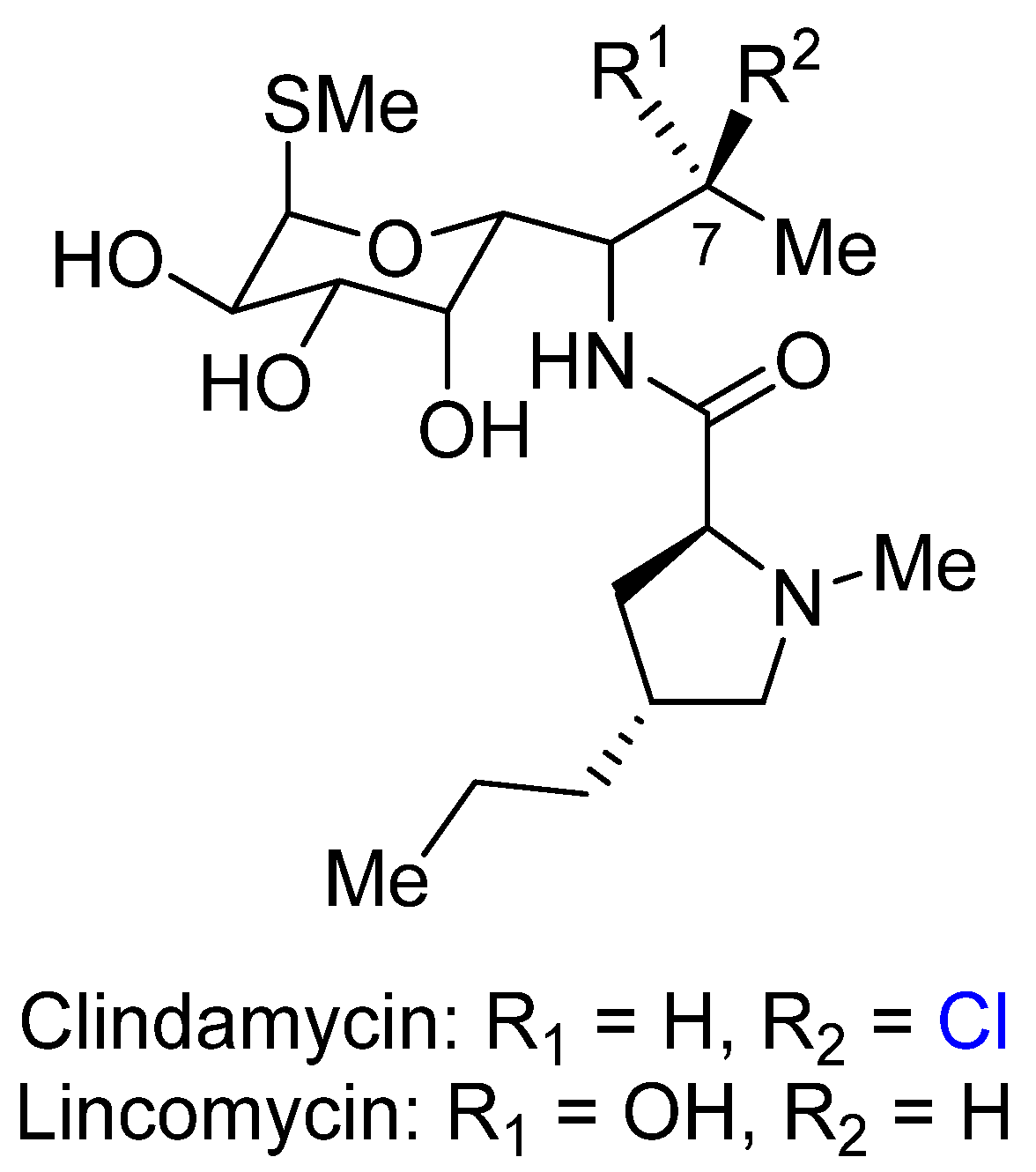
2.2. Clindamycin
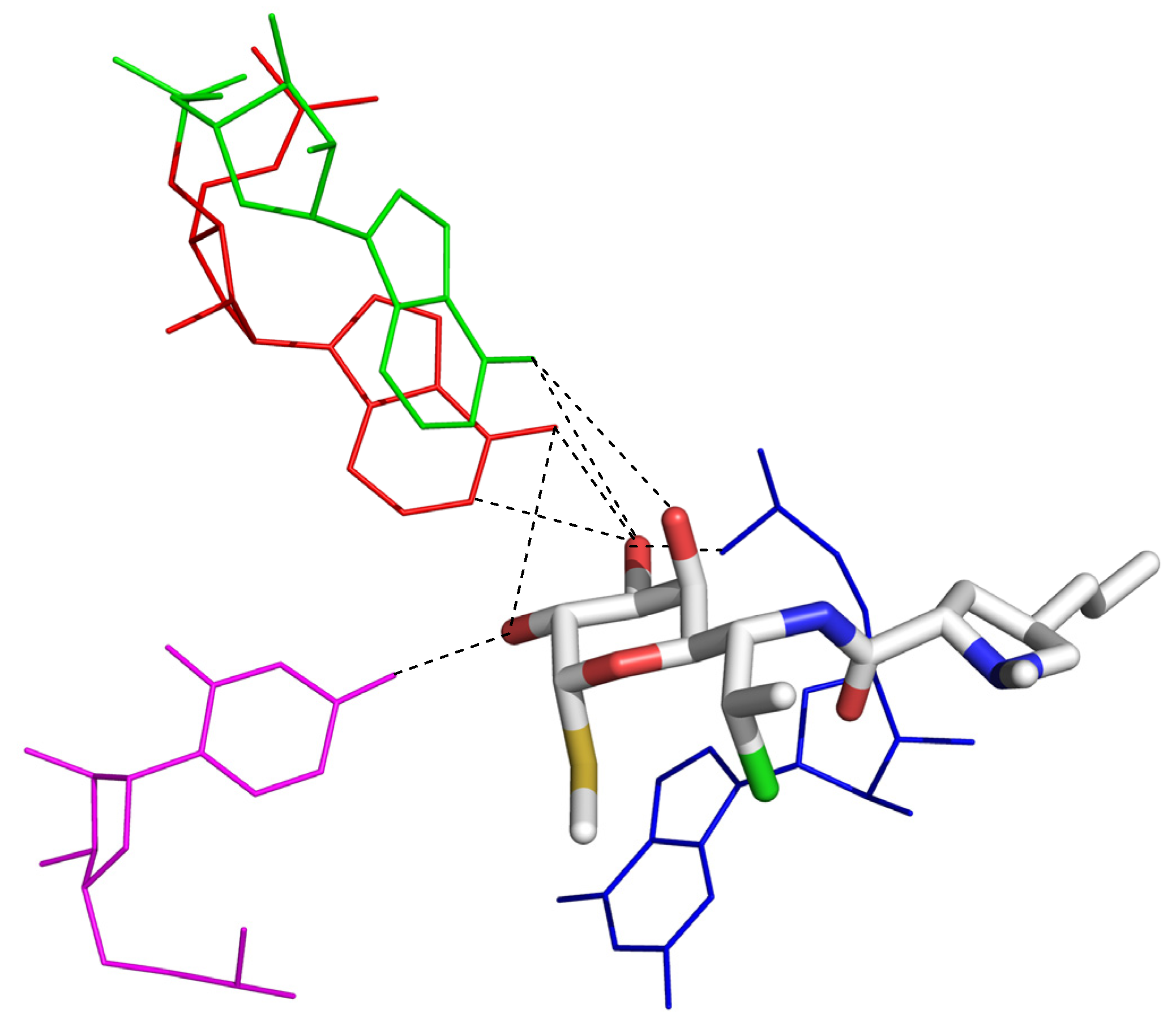
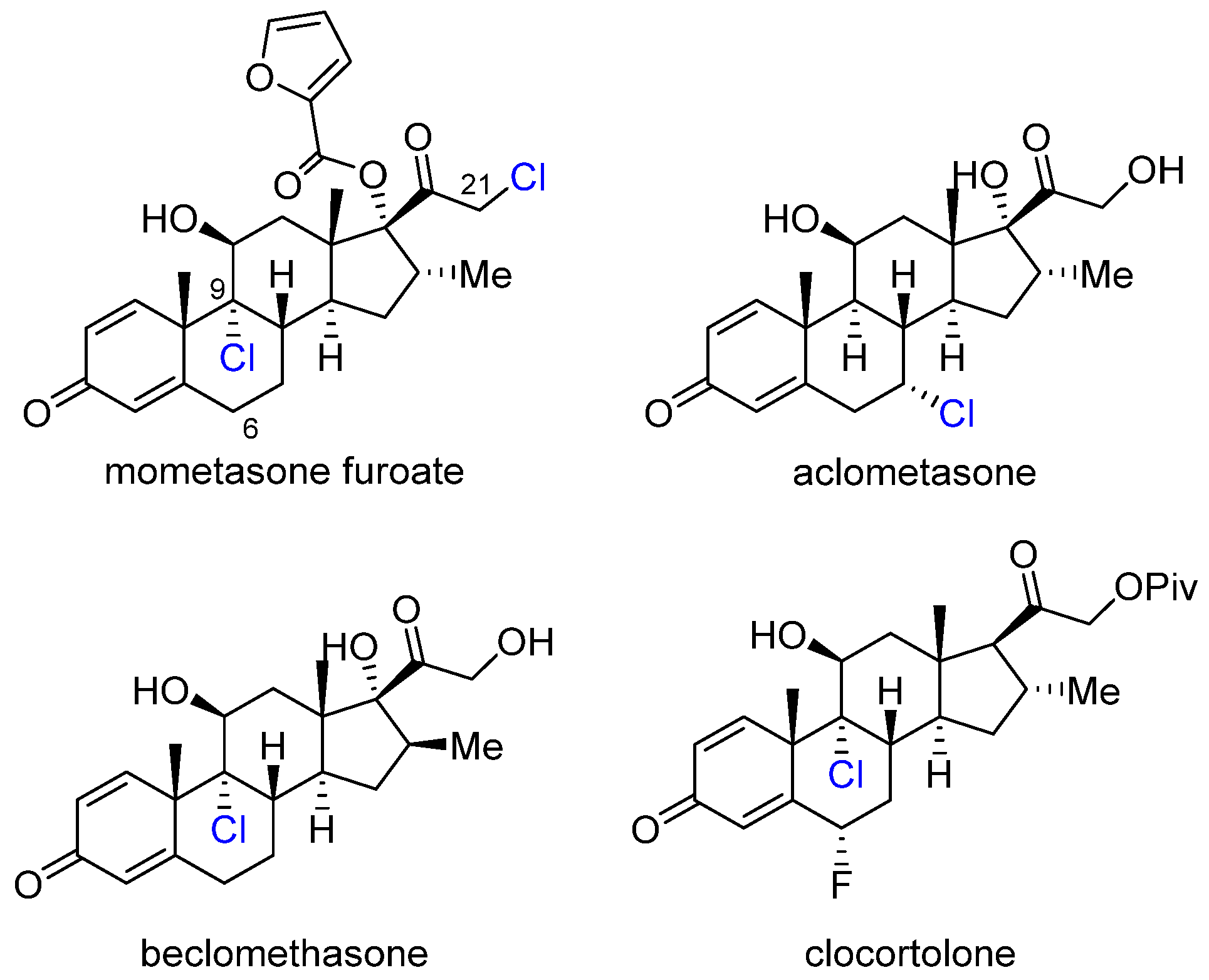
2.3. Corticosteroids
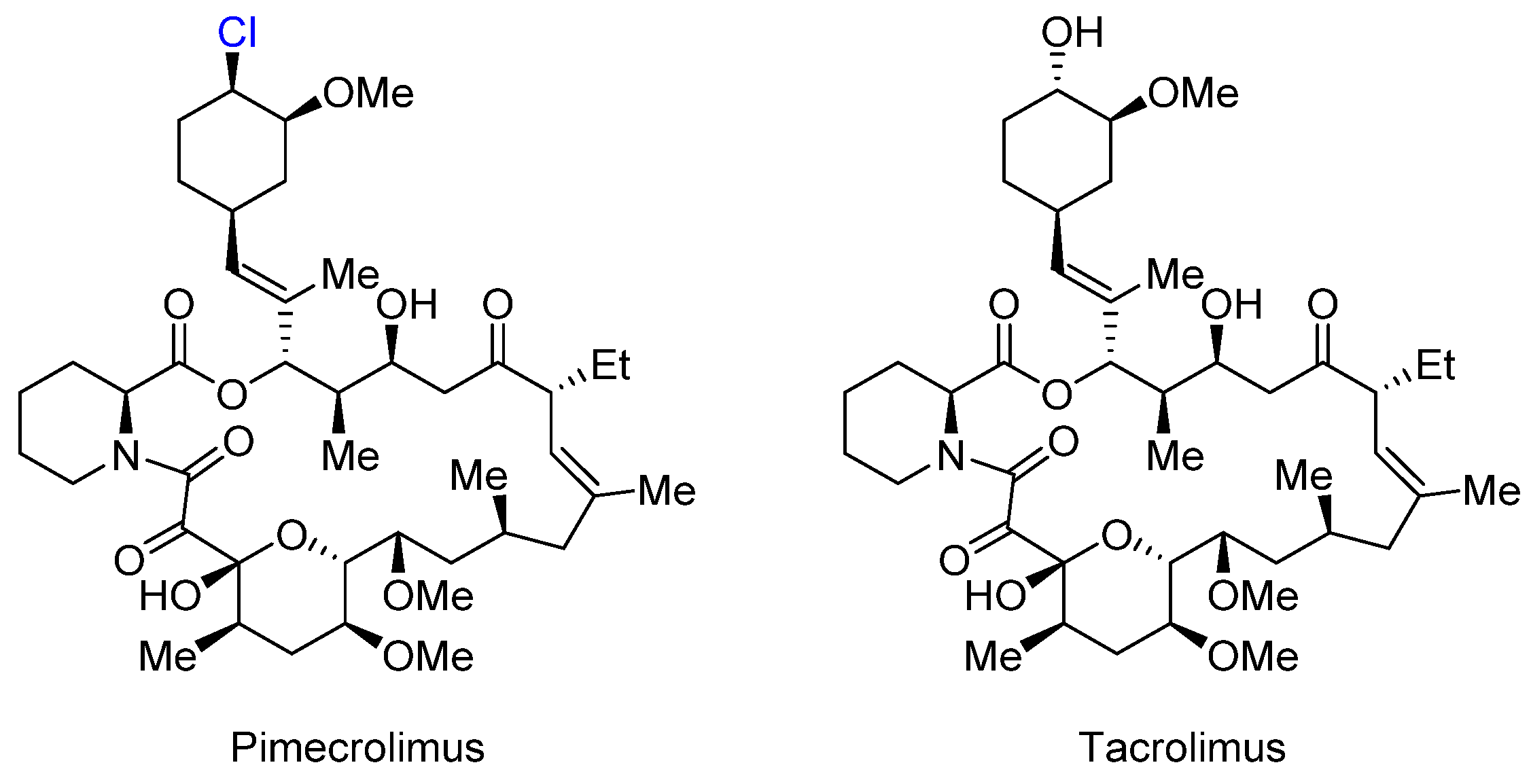
2.4. Pimecrolimus
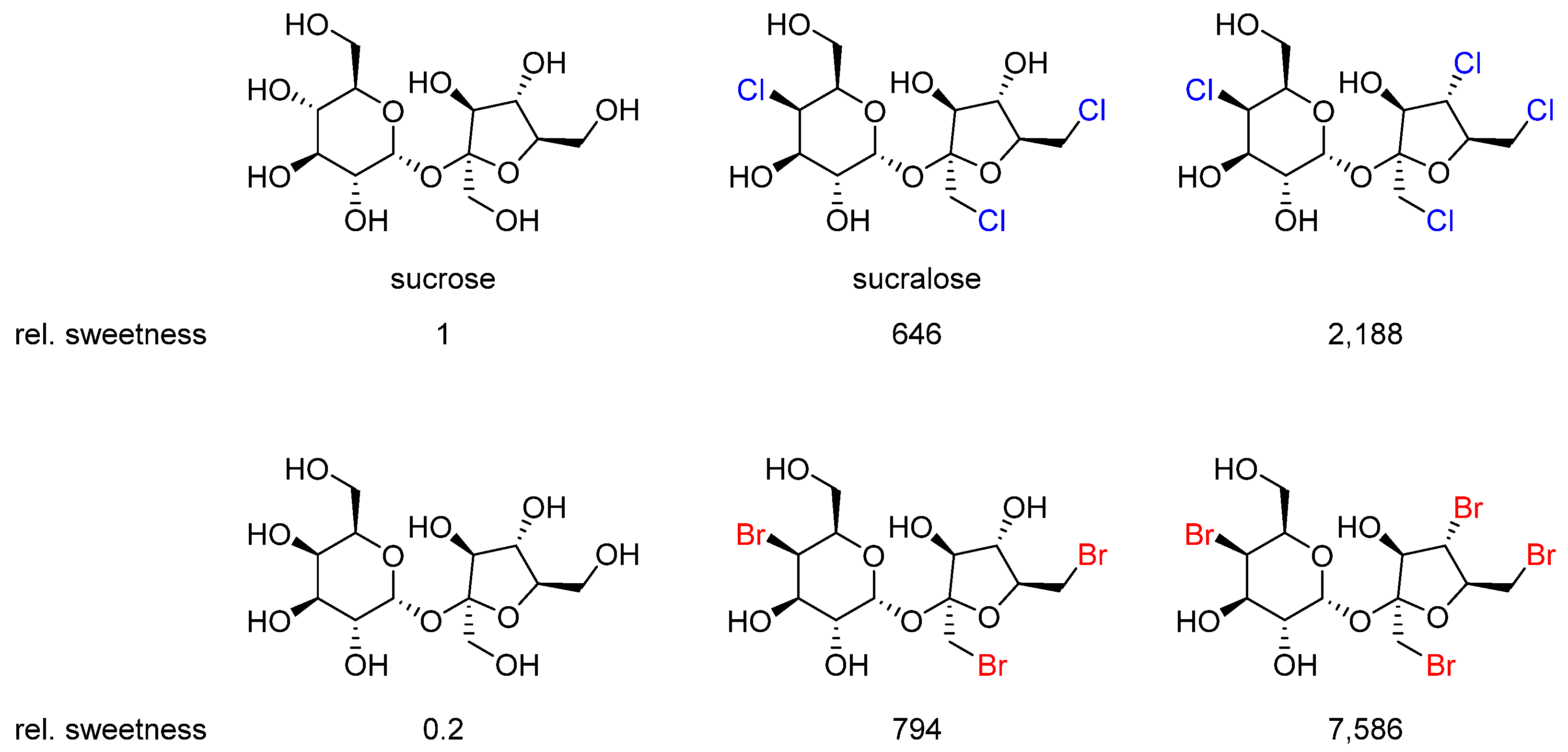
2.5. Sucralose
3. Alkyl Halide Natural Products
3.1. Halomon
3.2. Lapachone
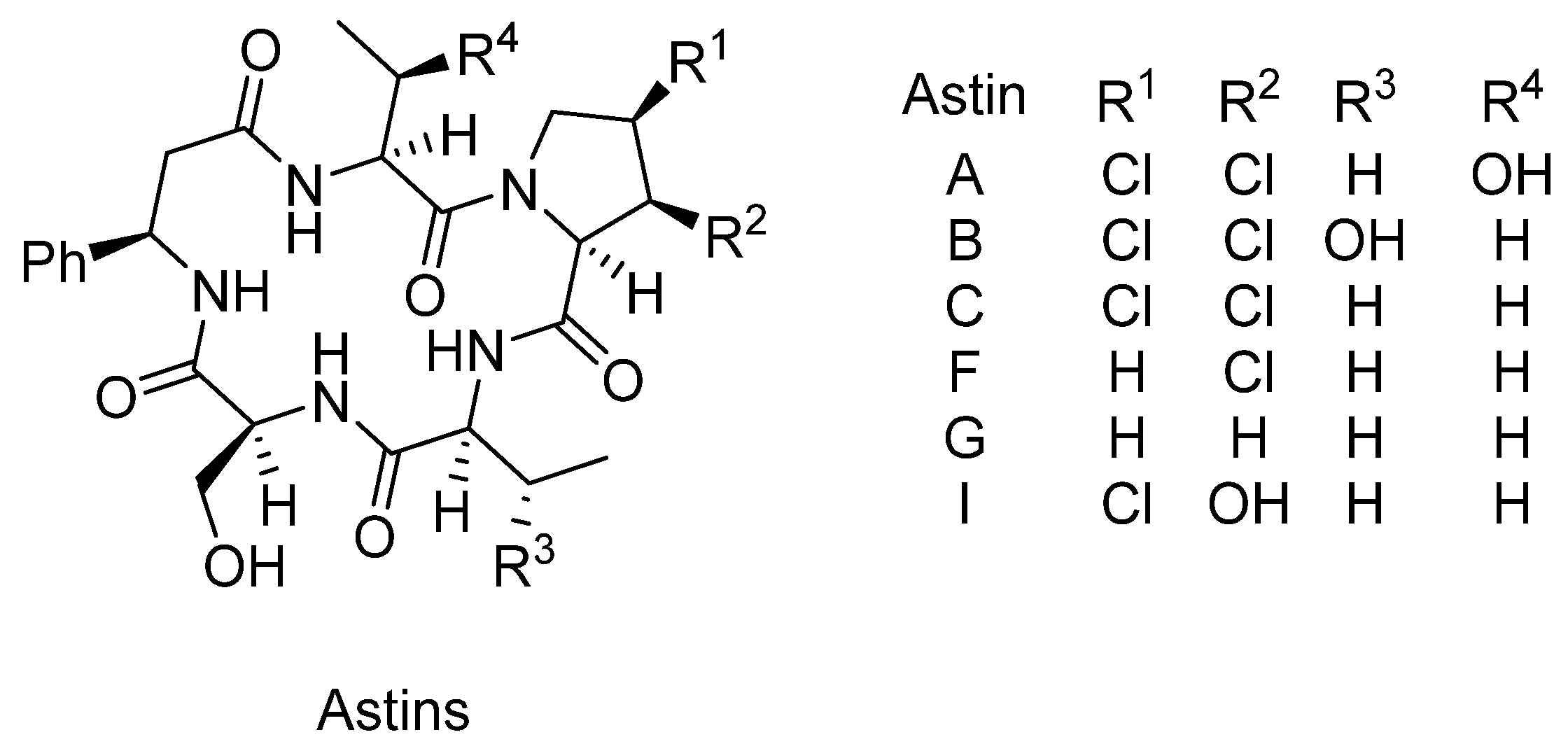
3.3. Astins
3.4. Forazoline
4. Summary and Outlook
Acknowledgments
Conflicts of Interest
References
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry, Part B: Reaction and Synthesis, 5th ed.; Springer: New York, NY, USA, 2007; pp. 217–223. [Google Scholar]
- Kambe, N.; Iwasaki, T.; Terao, J. Pd-catalyzed cross-coupling reactions of alkyl halides. Chem. Soc. Rev. 2011, 40, 4937–4947. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Edmonds, D.J.; Bulger, P.G. Cascade Reactions in Total Synthesis. Angew. Chem. Int. Ed. 2006, 45, 7134–7186. [Google Scholar] [CrossRef] [PubMed]
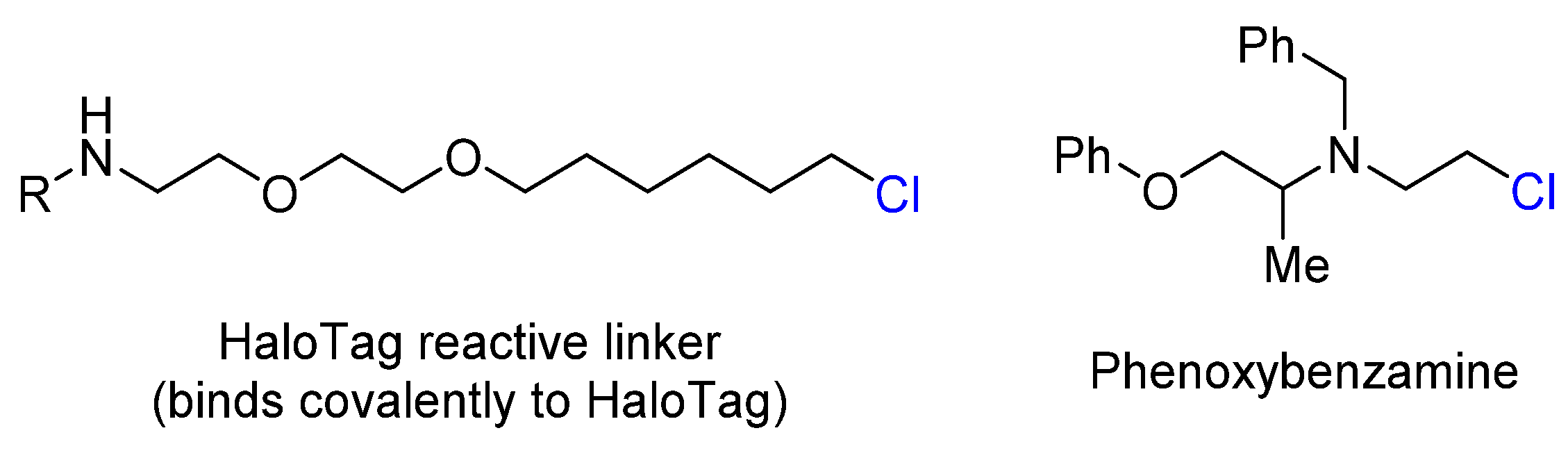
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Friedman Ohana, R.; Urh, M.; et al. HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Frang, H. Phenoxybenzamine Binding Reveals the Helical Orientation of the Third Transmembrane Domain of Adrenergic Receptors. J. Biol. Chem. 2001, 276, 31279–31284. [Google Scholar] [CrossRef] [PubMed]
- WHO Model List of Essential Medicines. Available online: http://www.who.int/medicines/publications/essentialmedicines/en/index.html (accessed on 1 August 2016).
- Smith, D.A.; van de Waterbeemd, H. Pharmacokinetics and metabolism in early drug discovery. Curr. Opin. Chem. Biol. 1999, 3, 373–378. [Google Scholar] [CrossRef]
- Gerebtzoff, G.; Li-Blatter, X.; Fischer, H.; Frentzel, A.; Seelig, A. Halogenation of Drugs Enhances Membrane Binding and Permeation. ChemBioChem 2004, 5, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Gentry, C.L.; Egleton, R.D.; Gillespie, T.; Abbruscato, T.J.; Bechowski, H.B.; Hruby, V.J.; Davis, T.P. The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides 1999, 20, 1229–1238. [Google Scholar] [CrossRef]
- Xu, Z.; Yang, Z.; Liu, Y.; Lu, Y.; Chen, K.; Zhu, W. Halogen Bond: Its Role beyond Drug-Target Binding Affinity for Drug Discovery and Development. J. Chem. Inf. Model. 2014, 54, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Franks, N.P. General Anaesthesia: From Molecular Targets to Neural Pathways of Sleep and Arousal. Nat. Rev. Neurosci. 2008, 9, 370–386. [Google Scholar] [CrossRef] [PubMed]
- Franks, N.P.; Lieb, W.R. Stereospecific Effects of Inhalational General Anesthetic Optical Isomers on Nerve Ion Channels. Science 1991, 254, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Loll, P.J.; Eckenhoff, R.G. Structural Bases for High-Affinity Volatile Anesthetic Binding in a Natural 4-Helix Bundle Protein. FASEB J. 2005, 19, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Rensati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisini, E.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen bonding in halocarbon-protein complexes: A structural survey. Chem. Soc. Rev. 2011, 40, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Clindamycin Hydrochloride. Available online: https://www.drugs.com/monograph/clindamycin-hydrochloride.html (accessed on 1 August 2016).
- Gale, E.F.; Cundliffe, E.; Reynolds, P.E.; Richmond, M.H.; Waring, M.J. The Molecular Basis of Antibiotic Action, 2nd ed.; John Wiley & Sons: New York, NY, USA, 1981; pp. 478–480. [Google Scholar]
- Le Goffic, F. Structure Activity Relationships in Lincosamide and Streptogramin Antibiotics. J. Antimicrob. Chemother. 1985, 16 (Suppl. A), 13–21. [Google Scholar] [CrossRef] [PubMed]
- Douthwaite, S. Interaction of the Antibiotics Clindamycin and Lincomycin with Escherichia coli 23S Ribosomal RNA. Nucleic Acids Res. 1992, 20, 4717–4720. [Google Scholar] [CrossRef] [PubMed]
- Schlünzen, F.; Zarivach, R.; Harms, J.; Bashan, A.; Tocilj, A.; Albrecht, R.; Yonath, A.; Franceschi, F. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature 2001, 413, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Therapeutic strategies for allergic diseases. Nature 1999, 402 (Suppl. 6760), B31–B38. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Thompson, E.B. The structure of the nuclear hormone receptors. Steroids 1999, 64, 310–319. [Google Scholar] [CrossRef]
- Kohn, J.A.; Deshpande, K.; Ortlund, E.A. Deciphering modern glucocorticoid cross-pharmacology using ancestral corticosteroid receptors. J. Biol. Chem. 2012, 287, 16267–16275. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.E.; Baxter, J.D.; Kollman, P.A.; Lee, D.L.; Kuntz, I.D.; Bloom, E.; Matulich, D.T.; Morris, J. Nature of steroid-glucocorticoid receptor interactions: Thermodynamic analysis of the binding reaction. Biochemistry 1978, 17, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, P. Glucocorticoid receptor binding: A biphasic dependence on molecular size as revealed by the bilinear LinBiExp mode. Steroids 2008, 73, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.R.; Lakhanpaul, M.; Morris, A.; Lateo, S.; Davies, T.; Scott, G.; Cardno, M.; Ebelin, M.E.; Burtin, P.; Stephenson, T.J. Systemic exposure, tolerability, and efficacy of pimecrolimus cream 1% in atopic dermatitis patients. Arch. Dis. Child. 2003, 88, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.M.; Fresneau, M.; Moenius, T.; Stuetz, A.; Billich, A. Binding of Pimecrolimus and Tacrolimus to Skin and Plasma Proteins: Implications for Systemic Exposure after Topical Application. Drug Metab. Dispos. 2008, 336, 1812–1818. [Google Scholar] [CrossRef] [PubMed]
- Knight, I. The development and applications of sucralose, a new high-intensity sweetener. Can. J. Physiol. Pharmacol. 1994, 72, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Renwick, A.G.; Sims, J.; Snodin, D.J. Sucralose metabolism and pharmacokinetics in man. Food Chem. Toxicol. 2000, 38 (Suppl. 2), S31–S41. [Google Scholar] [CrossRef]
- Schallenberger, R.S.; Acree, T.E. Molecular theory of sweet taste. Nature 1967, 216, 480–482. [Google Scholar] [CrossRef]
- Schallenberger, R.S.; Acree, T.E. Molecular structure and sweet taste. J. Acric. Food Chem. 1969, 17, 701–703. [Google Scholar] [CrossRef]
- Kier, L.B. Molecular theory of sweet taste. J. Pharm. Sci. 1972, 61, 1394–1397. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W. Biological Activity of Recently Discovered Halogenated Marine Natural Products. Mar. Drugs 2015, 13, 4044–4136. [Google Scholar] [CrossRef] [PubMed]
- Burreson, J.B.; Woolard, F.X.; Moore, R.E. Evidence for the biogenesis of halogenated myrcenes from the red alga Chondrococcus hornemanni. Chem. Lett. 1975, 4, 1111–1114. [Google Scholar] [CrossRef]
- Fuller, R.W.; Cardellinall, J.H.; Kato, Y.; Brinen, L.S.; Clardy, J.; Snader, K.M.; Boyd, M.R. A pentahalogenated monoterpene from the red alga Portieria hornemannii produces a novel cytotoxicity profile against a diverse panel of human tumor cell lines. J. Med. Chem. 1992, 35, 3007–3011. [Google Scholar] [CrossRef] [PubMed]
- Fuller, R.W.; Cardellina, J.H.; Jurek, J.; Scheuer, P.J.; Alvarado-Lindner, B.; McGuire, M.; Gray, G.N.; Steiner, J.R.; Clardy, J. Isolation and Structure/Activity Features of Halomon-Related Antitumor Monoterpenes from the Red Alga Portieria hornemannii. J. Med. Chem. 1994, 37, 4407–4411. [Google Scholar] [CrossRef] [PubMed]
- Andrianasolo, E.H.; France, D.; Cornell-Kennon, S.; Gerwick, W.H. DNA Methyl Transferase Inhibiting Halogenated Monoterpenes from the Madagascar Red Marine Alga Portieria hornemannii. J. Nat. Prod. 2006, 69, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Paull, K.D.; Shoemaker, R.H.; Hodes, L.; Monks, A.; Scudiero, D.A.; Rubinstein, L.; Plowman, J.; Boyd, M.R. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: Development of mean graph and COMPARE algorithm. J. Natl. Cancer Inst. 1989, 81, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Bucher, C.; Deans, R.M.; Burns, N.Z. Highly Selective Synthesis of Halomon, Plocamenone, and Isoplocamenone. J. Am. Chem. Soc. 2015, 137, 12784–12787. [Google Scholar] [CrossRef] [PubMed]
- Bucher, C.; Valeriote, F.A.; Burns, N.Z.; Department of Chemistry, Stanford University. 2016; Unpublished work.
- Guiraud, P.; Steiman, R.; Campos-Takaki, G.-M.; Seigle-Murandi, F.; de Buochberg, M. Comparison of Antibacterial and Antifungal Activities of Lapachol and β-Lapachone. Planta Med. 1994, 60, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Sacau, E.; Estévez-Braun, A.; Ravelo, Á.G.; Gutiérrez Yapu, D.; Giménez Turba, A. Antiplasmodial Activity of Naphthoquinones Related to Lapachol and β-Lapachone. Chem. Biodivers. 2005, 2, 264–274. [Google Scholar] [CrossRef] [PubMed]
- De Andrade-Neto, V.F.; Goulart, M.O.; da Silva Filho, J.F.; da Silva, M.J.; Pinto Mdo, C.; Pinto, A.V.; Zalis, M.G.; Carvalho, L.H.; Krettli, A.U. Antimalarial activity of phenazines from lapachol, β-lapachone and its derivatives against Plasmodium falciparum in vitro and Plasmodium berghei in vivo. Bioorg. Med. Chem. Lett. 2004, 14, 1145–1149. [Google Scholar] [CrossRef] [PubMed]
- Pardee, A.B.; Li, Y.Z.; Li, C.J. Cancer therapy with β-lapachone. Curr. Cancer Drug Targets 2002, 2, 224–227. [Google Scholar] [CrossRef]
- Pérez-Sacau, E.; Díaz-Peñate, R.G.; Estévez-Braun, A.; Ravelo, A.G.; García-Castellano, J.M.; Pardo, L.; Campillo, M. Synthesis and pharmacophore modeling of naphthoquinone derivatives with cytotoxic activity in human promyelocytic leukemia HL-60 cell line. J. Med. Chem. 2007, 50, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Nagashima, S.; Uchiumi, Y.; Kuroki, O.; Takeya, K.; Itokawa, H. Cyclic peptides from higher plants. XXVIII. Antitumor activity and hepatic microsomal biotransformation of cyclic pentapeptides, astins, from Aster tataricus. Chem. Pharm. Bull. 1996, 5, 1026–1032. [Google Scholar] [CrossRef]
- Wyche, T.P.; Piotrowski, J.S.; Hou, Y.; Braun, D.; Deshpande, R.; McIlwain, S.; Ong, I.M.; Myers, C.L.; Guzei, I.A.; Westler, W.M.; et al. Forazoline A: marine-derived polyketide with antifungal in vivo efficacy. Angew. Chem. Int. Ed. Engl. 2014, 53, 11583–11586. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
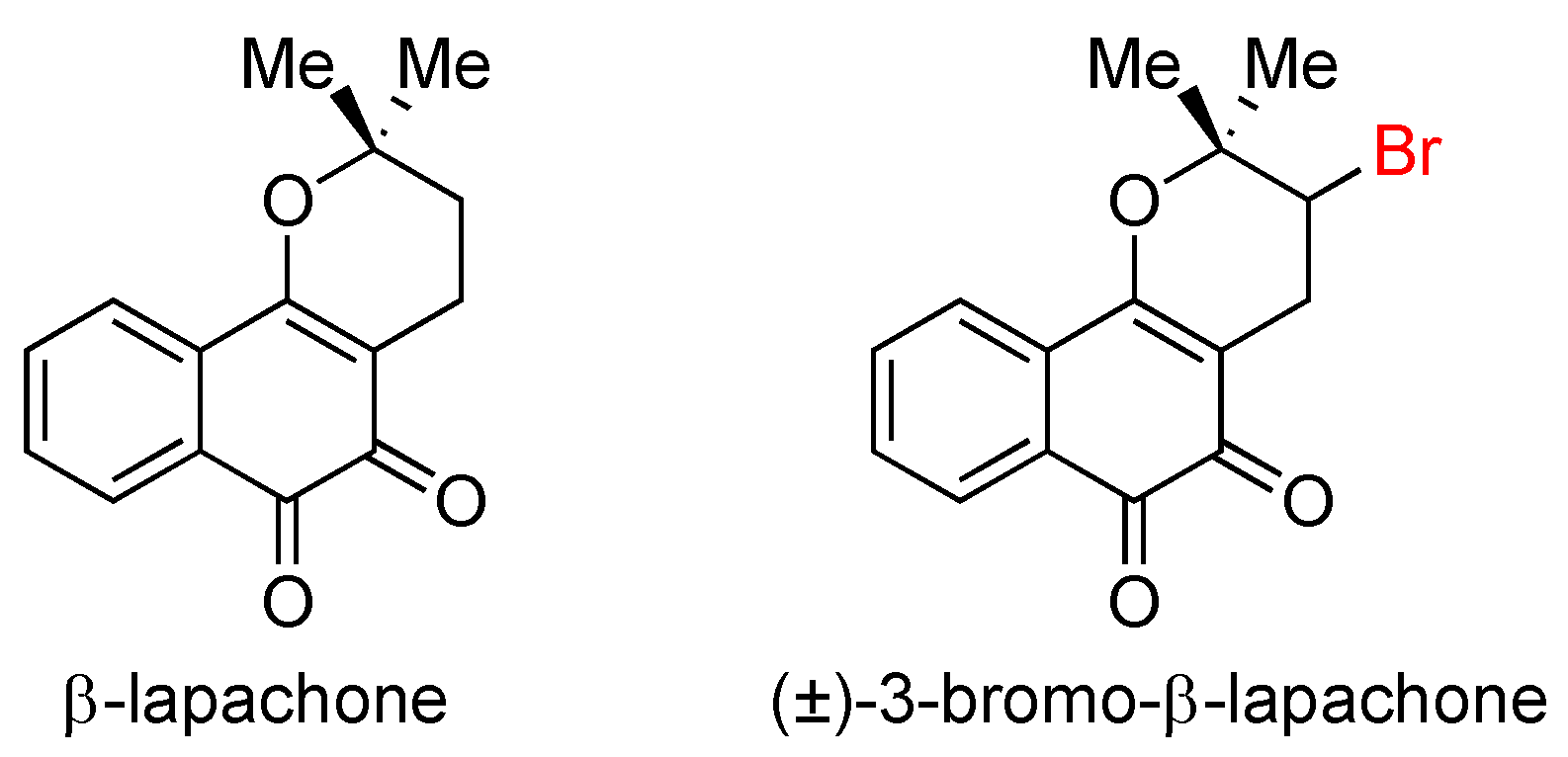{kind=link}
{kind=link}
{kind=link}
| Compound | Straph. aureus | Str. faecalis |
|---|---|---|
| R1 = OH, R2 = H | 0.4 | 12.5 |
| R1 = H, R2 = OH | 1.6 | 25 |
| R1 = Cl, R2 = H | 0.8 | 12.5 |
| R1 = H, R2 = Cl | 0.1 | 6.2 |
| R1 = H, R2 = Br | 0.05 | 6.2 |
| R1 = H, R2 = I | 0.05 | 3.2 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gál, B.; Bucher, C.; Burns, N.Z. Chiral Alkyl Halides: Underexplored Motifs in Medicine. Mar. Drugs 2016, 14, 206. https://doi.org/10.3390/md14110206
Gál B, Bucher C, Burns NZ. Chiral Alkyl Halides: Underexplored Motifs in Medicine. Marine Drugs. 2016; 14(11):206. https://doi.org/10.3390/md14110206
Chicago/Turabian StyleGál, Bálint, Cyril Bucher, and Noah Z. Burns. 2016. "Chiral Alkyl Halides: Underexplored Motifs in Medicine" Marine Drugs 14, no. 11: 206. https://doi.org/10.3390/md14110206
APA StyleGál, B., Bucher, C., & Burns, N. Z. (2016). Chiral Alkyl Halides: Underexplored Motifs in Medicine. Marine Drugs, 14(11), 206. https://doi.org/10.3390/md14110206