The Oxepane Motif in Marine Drugs



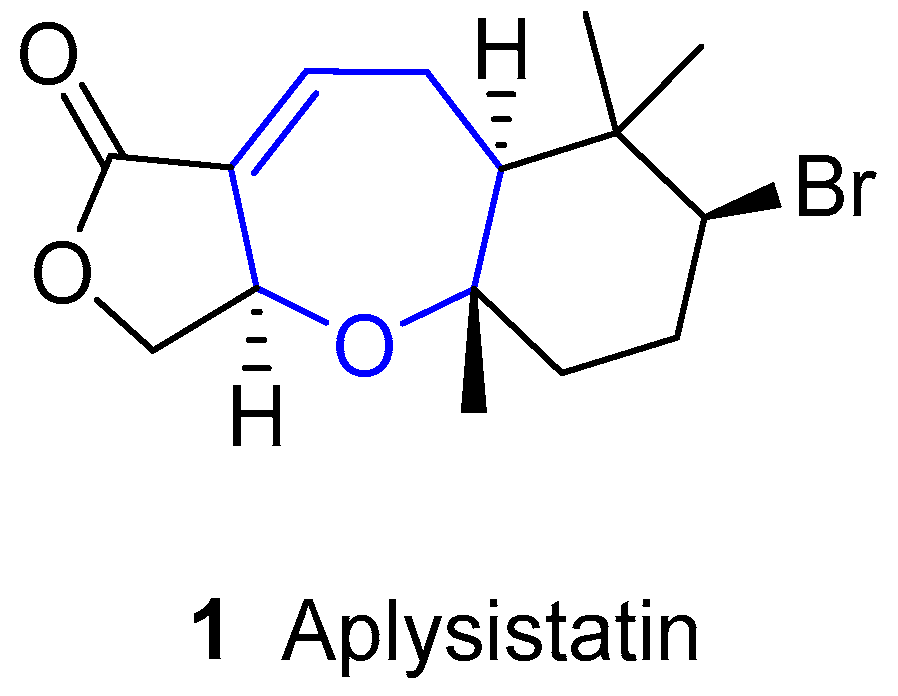{kind=link}
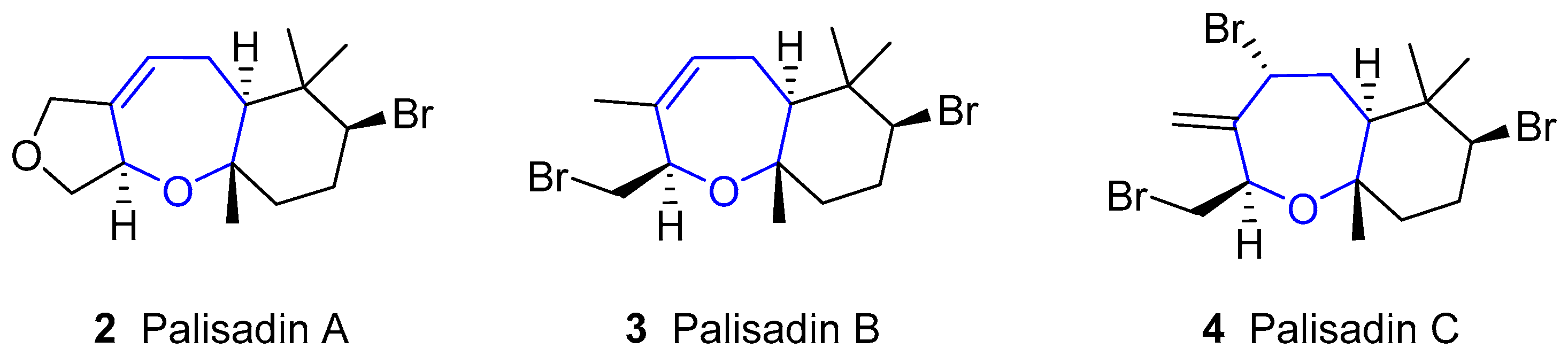{kind=link}
{kind=link}
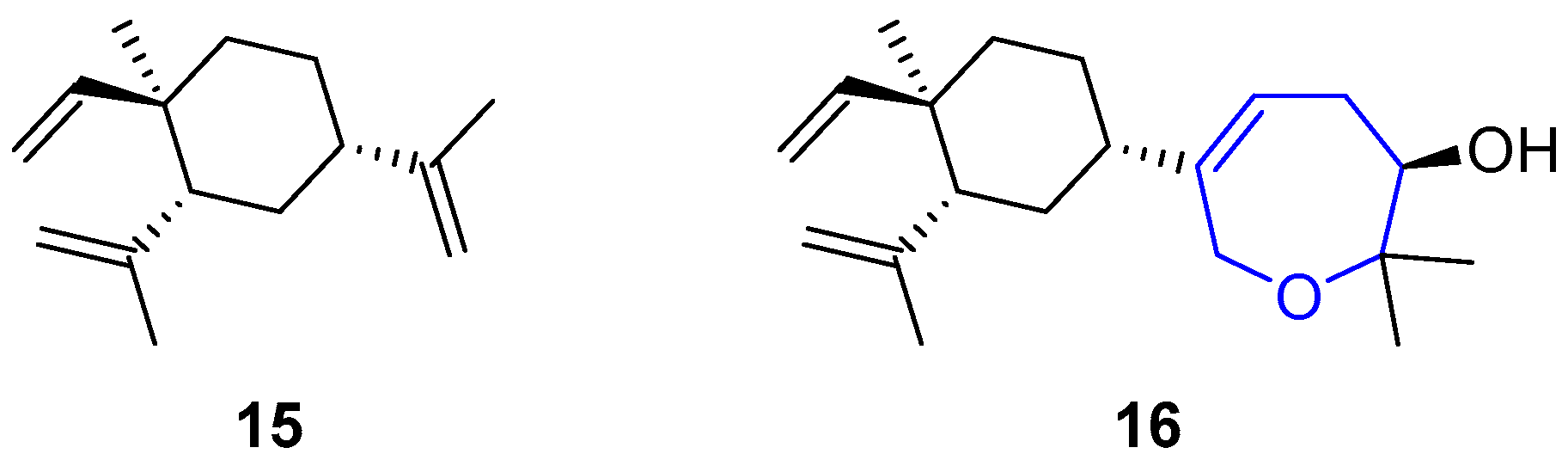{kind=link}
{kind=link}
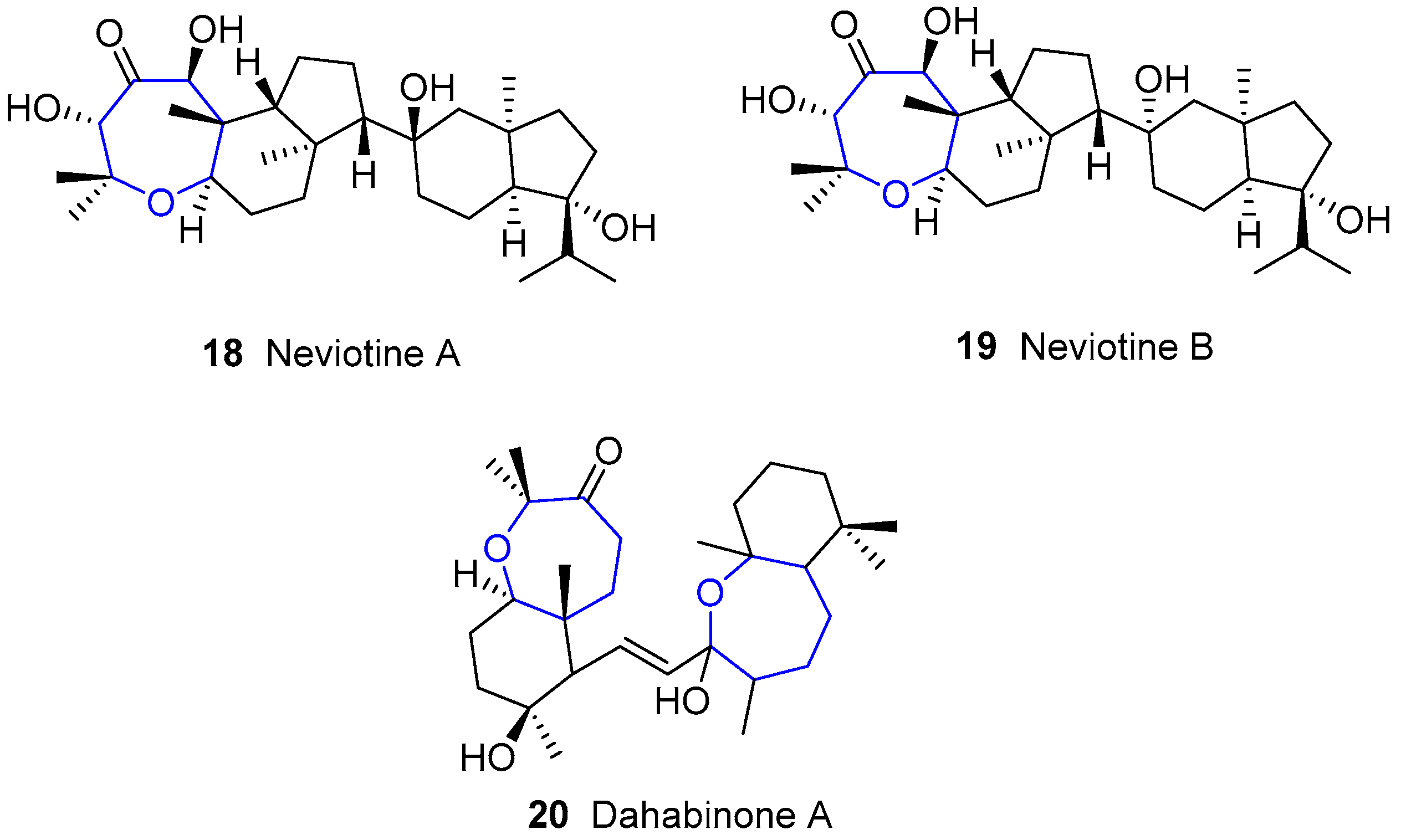{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
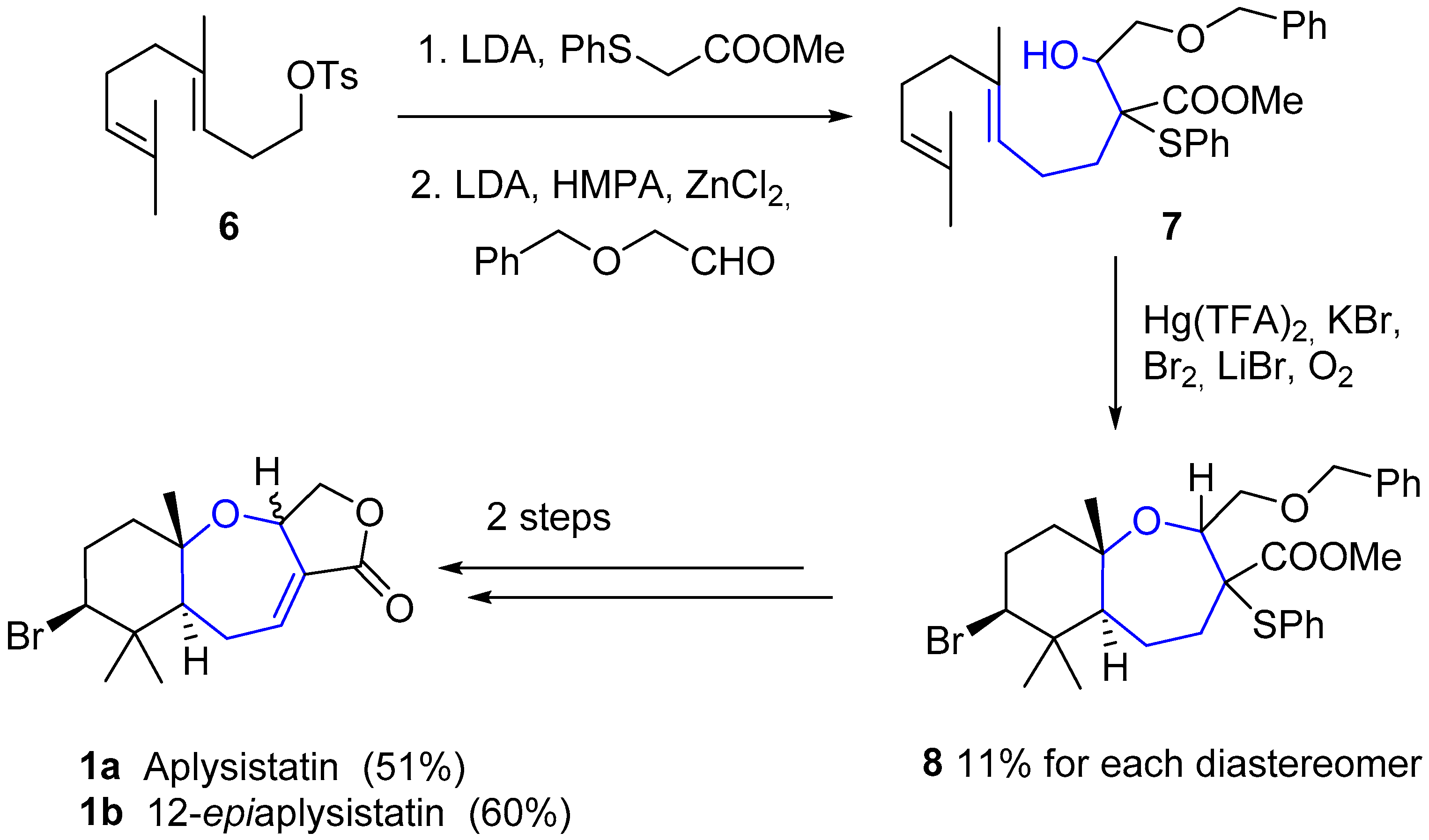{kind=link}
{kind=link}
{kind=link}
{kind=link}
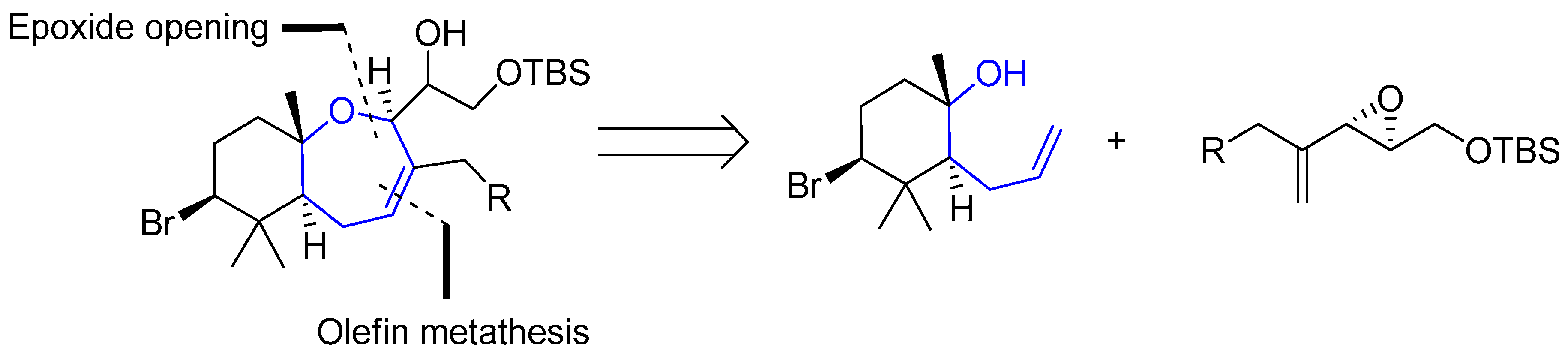
{kind=link}
{kind=link}
{kind=link}
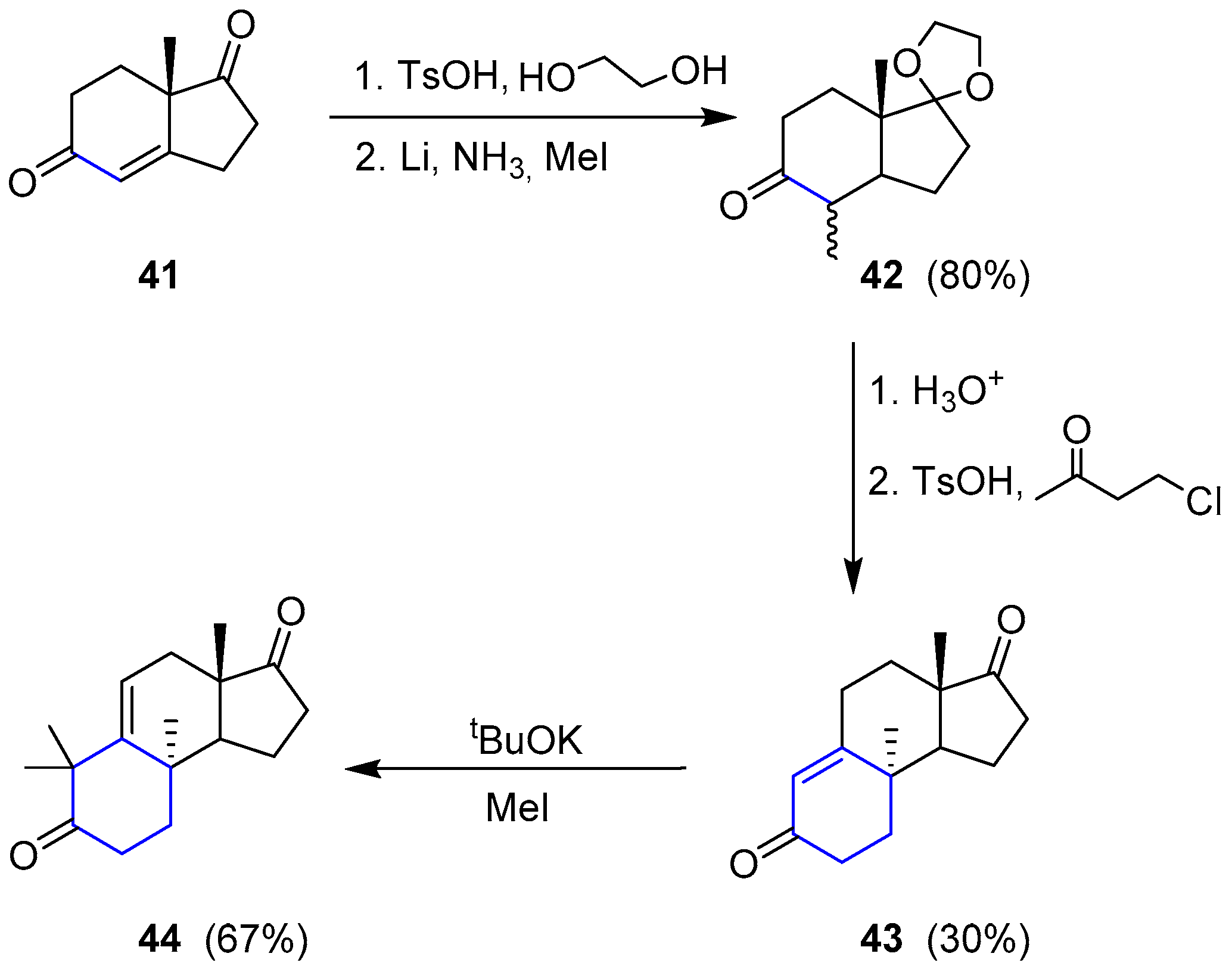
{kind=link}
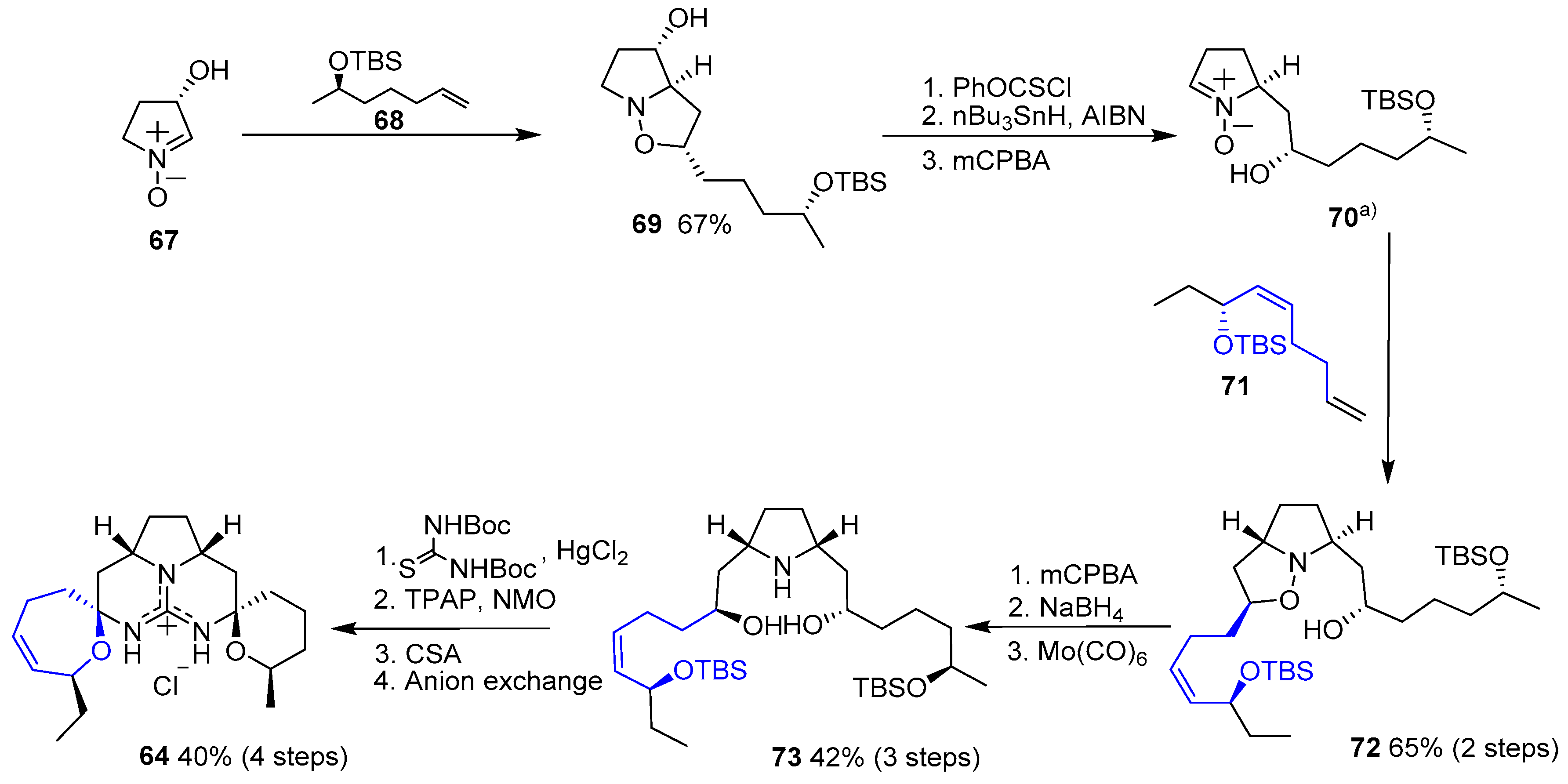{kind=link}
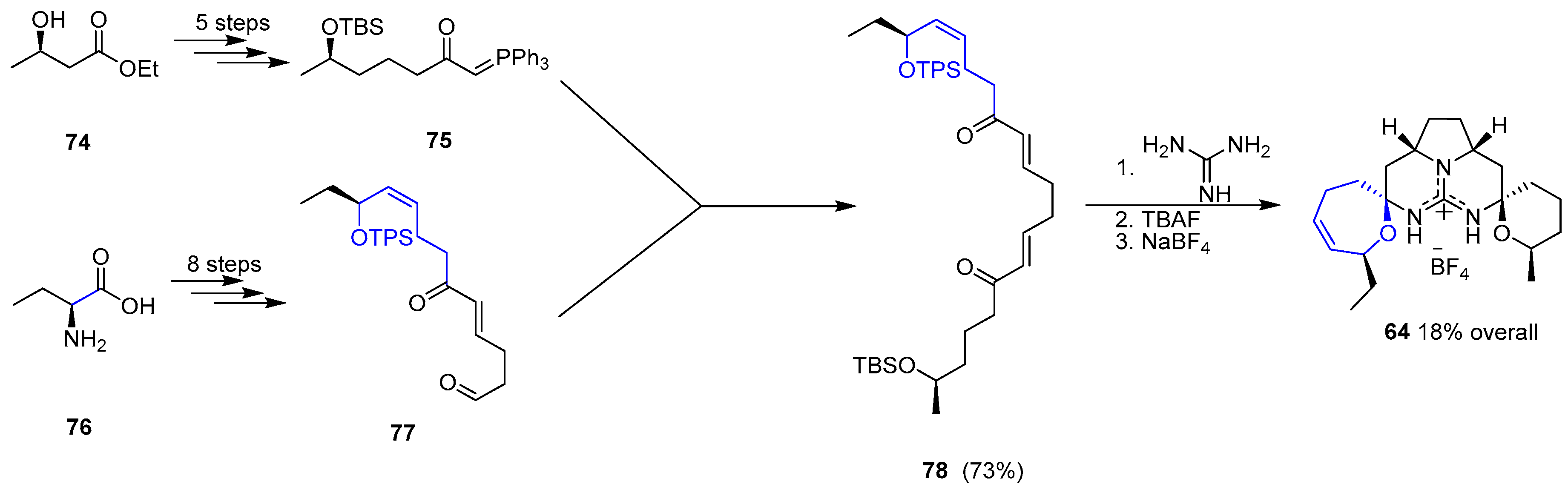{kind=link}
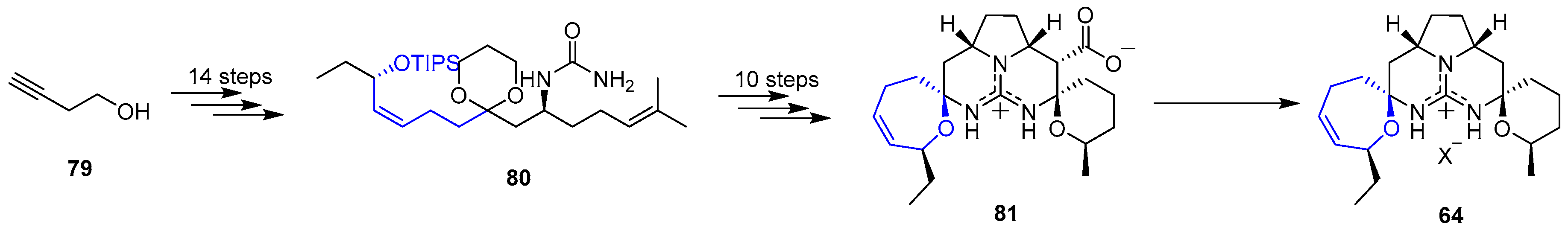{kind=link}
{kind=link}
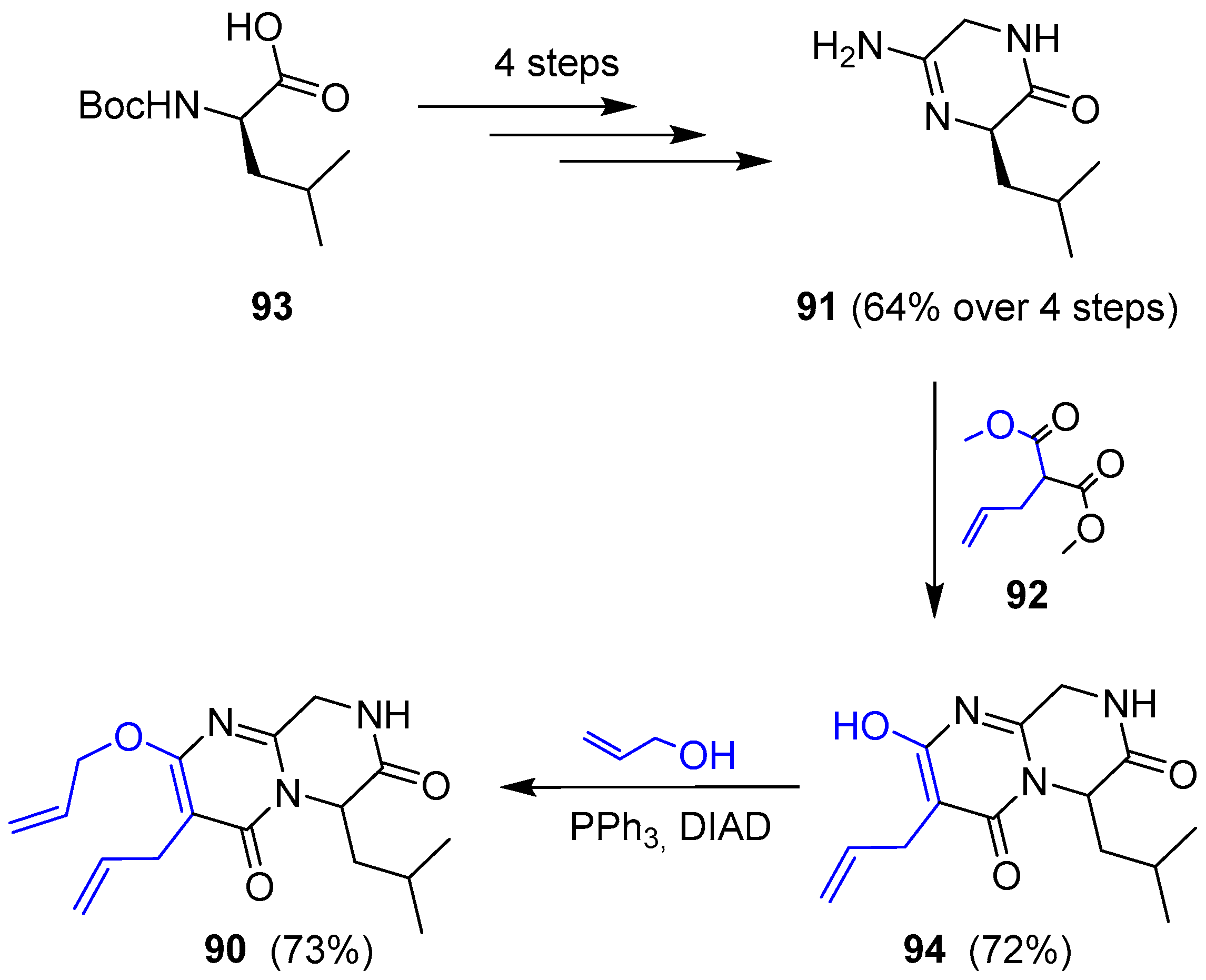{kind=link}
{kind=link}
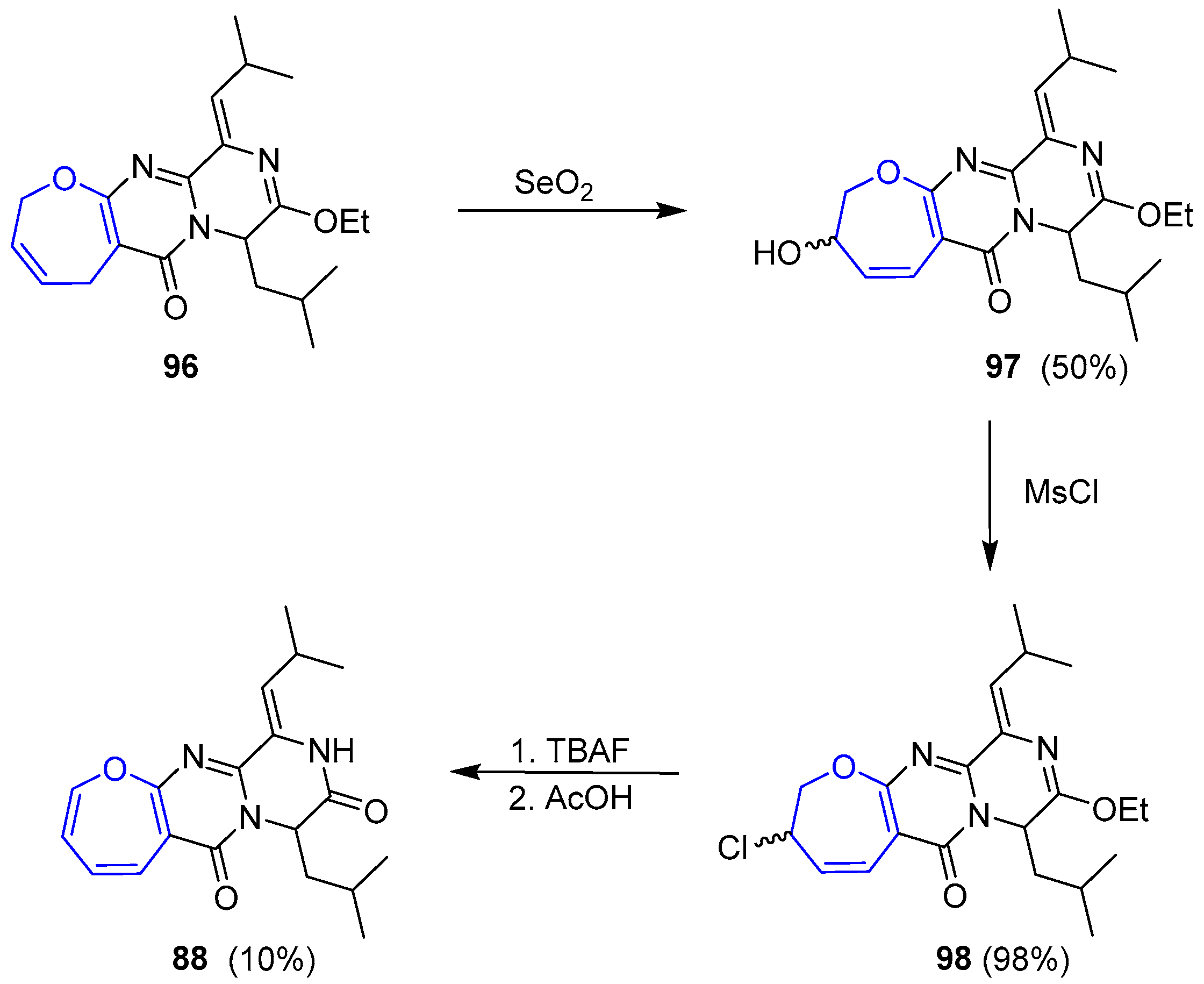{kind=link}
{kind=link}
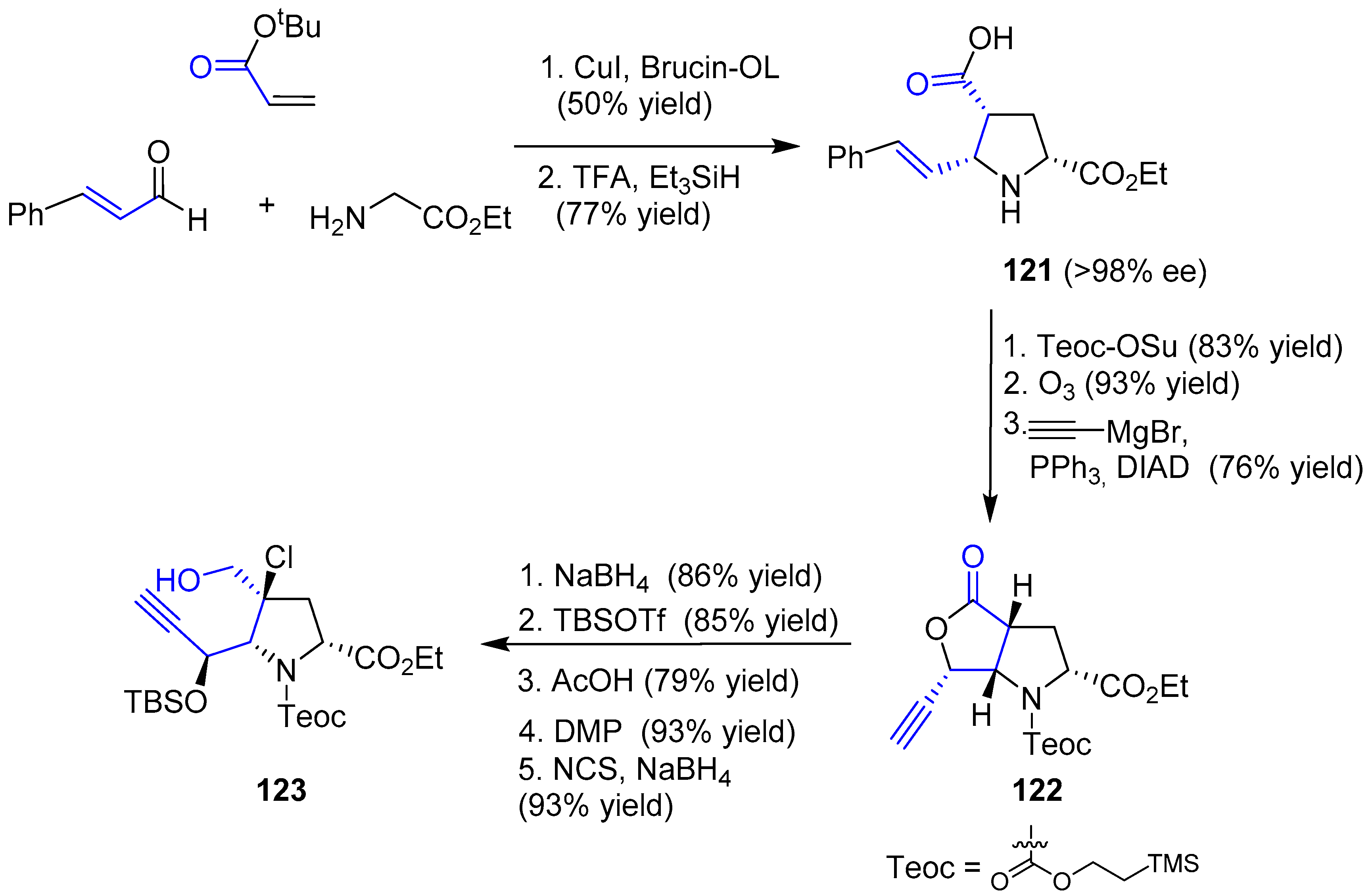{kind=link}
{kind=link}
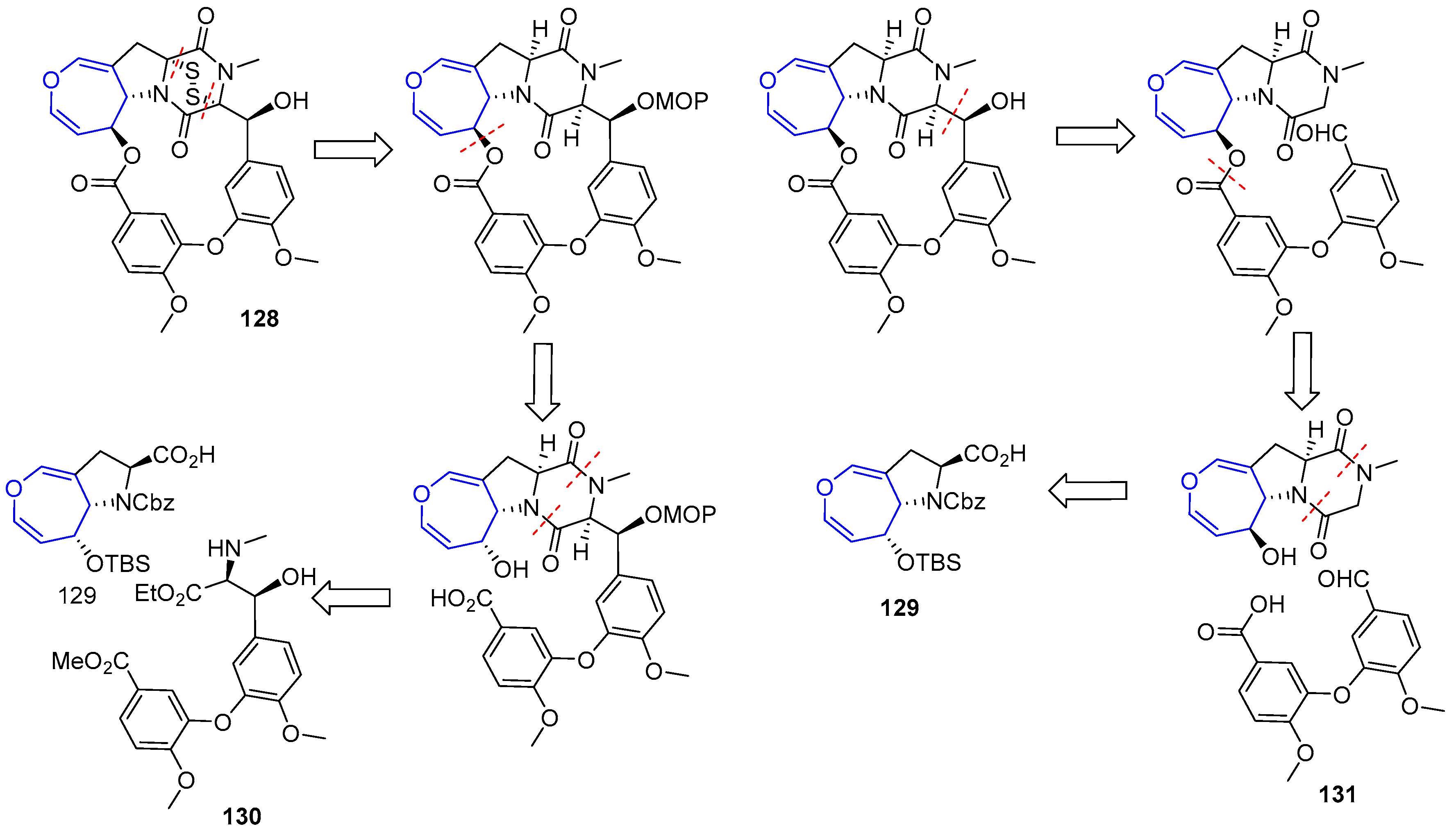{kind=link}
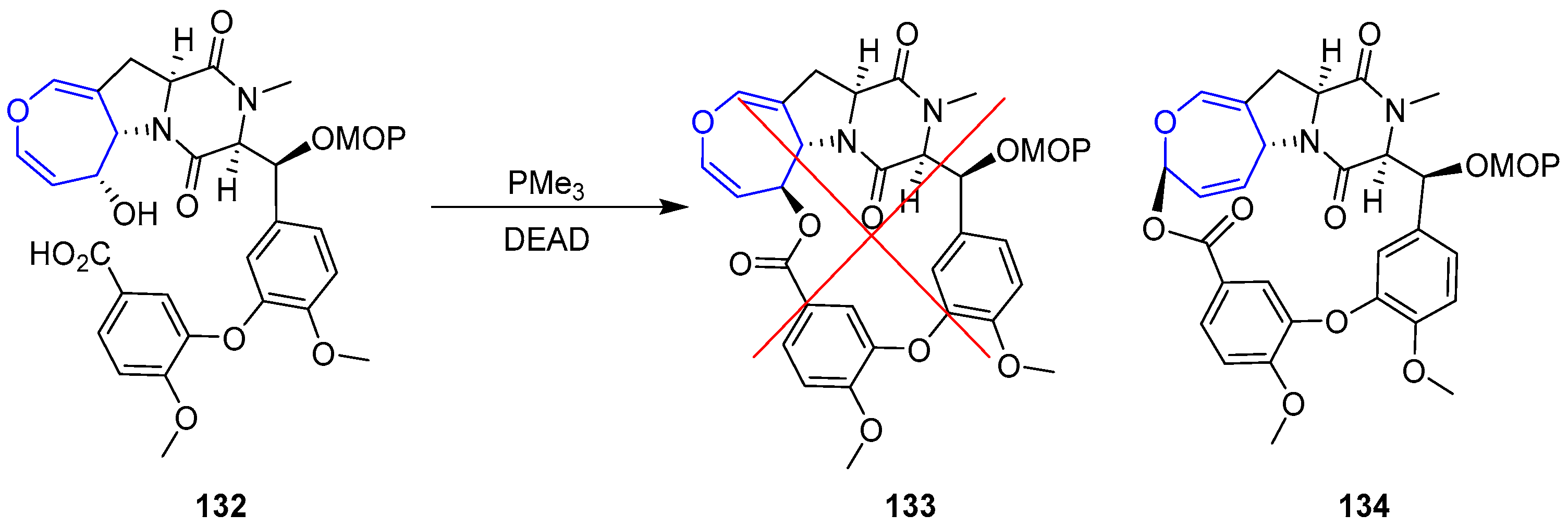{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Halogenated Sesquiterpenoids: Aplysistatin and Palisadins
3. Marine Diterpenes: Oxepin Lobatrienol
4. Marine Triterpenes
4.1. Sipholenols Family
4.2. Neviotanes and Dahabanes
4.3. Sodwanones
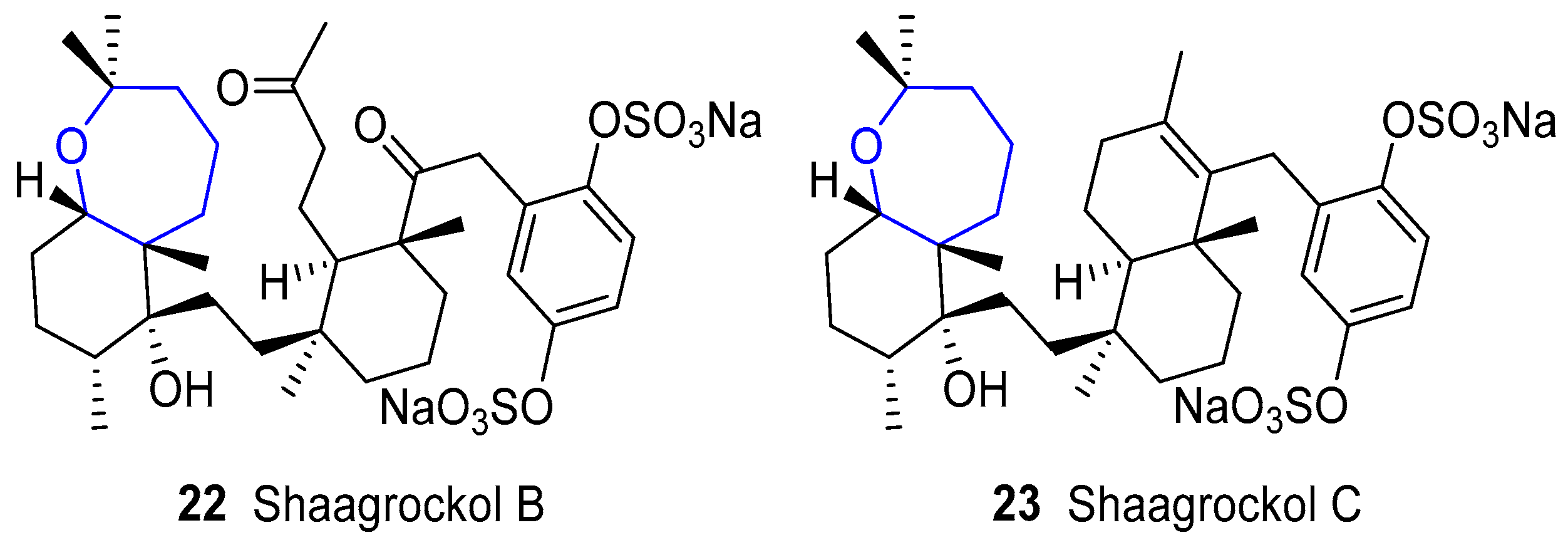
4.4. Shaagrockols
4.5. Raspacionins
5. Meroterpenoids: Austalides
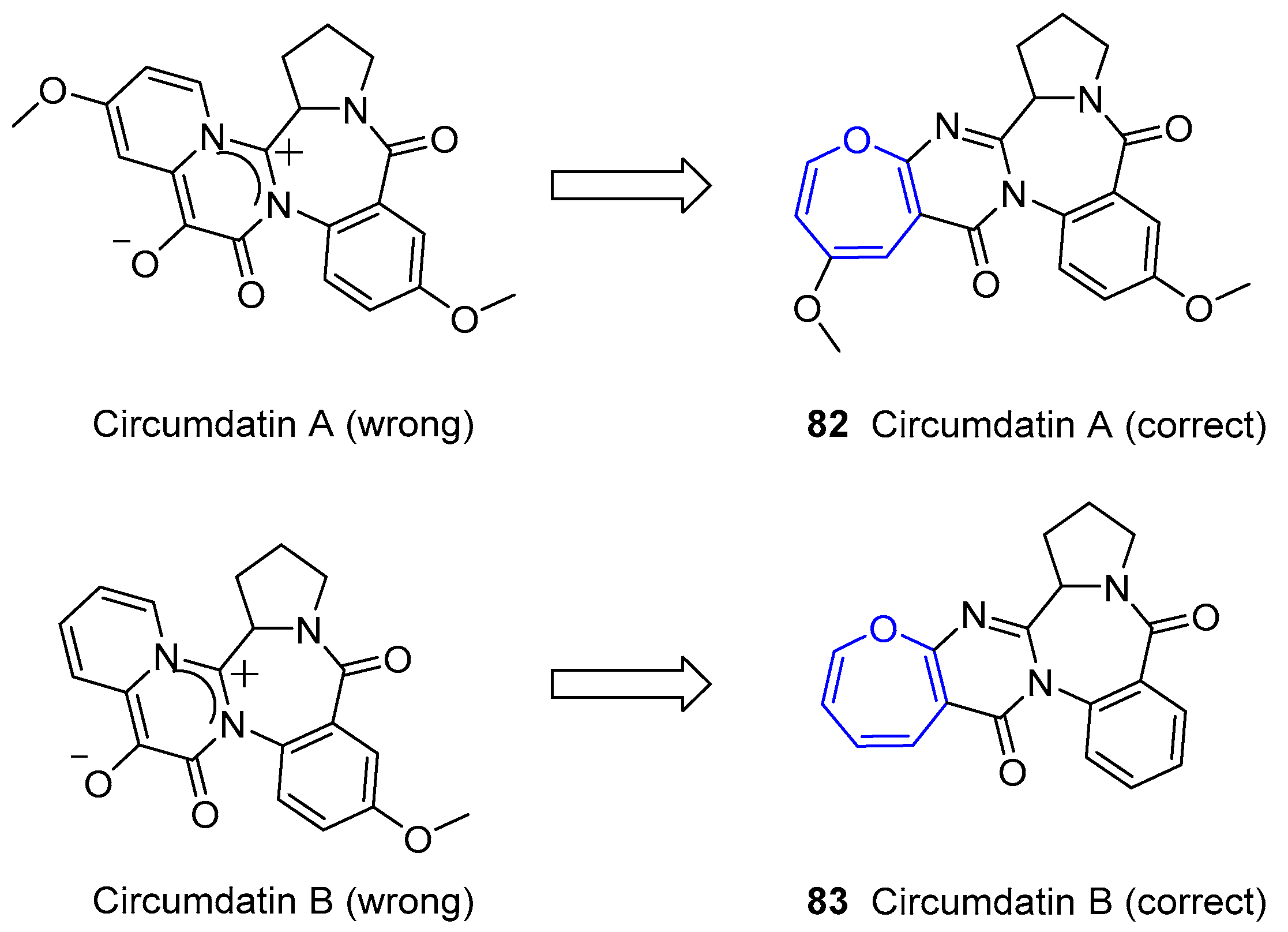
6. Alkaloids
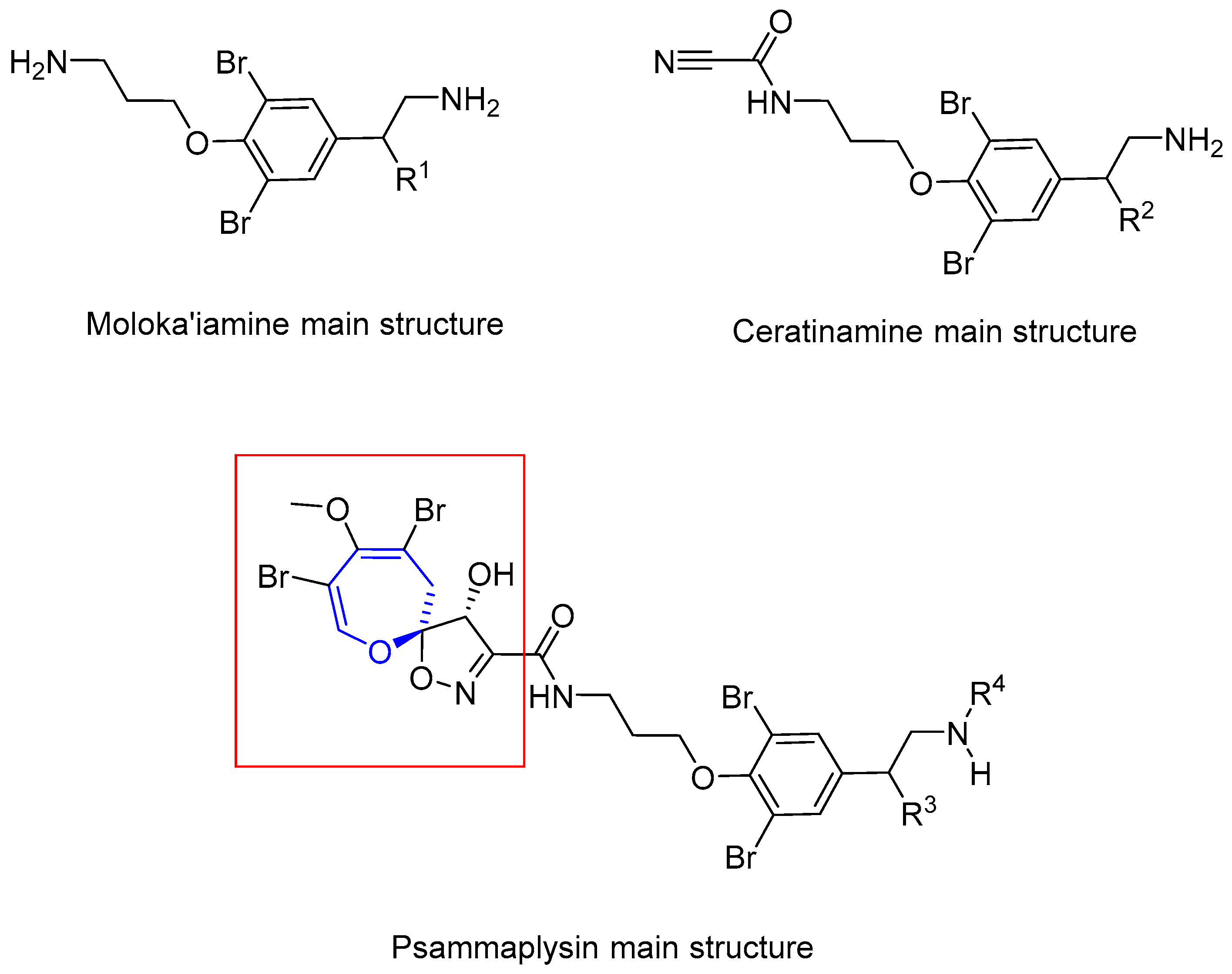
6.1. Bromotyrosine Alkaloids: Psammaplysins and Ceratinamides
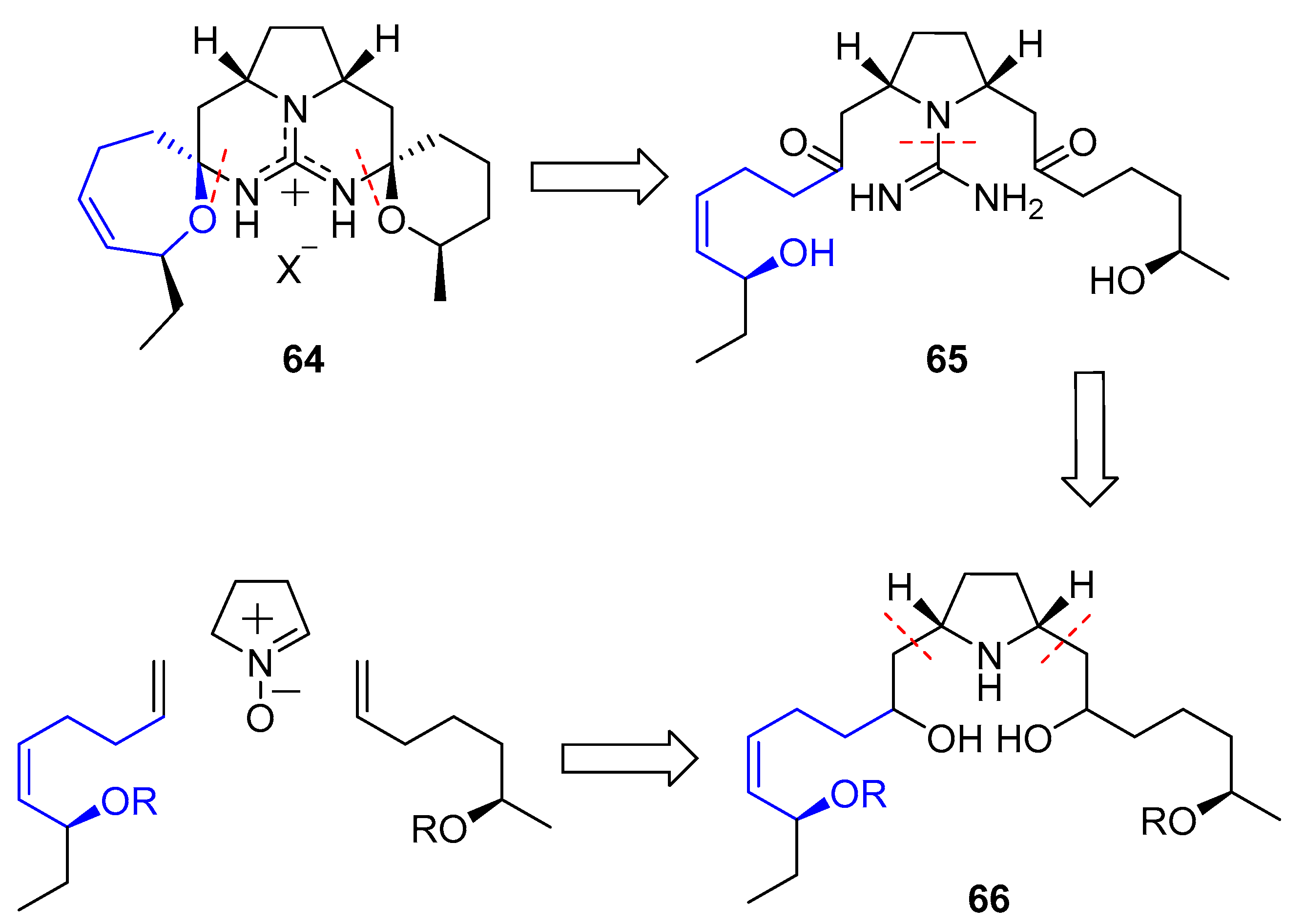
6.2. Guanidinium Alkaloids
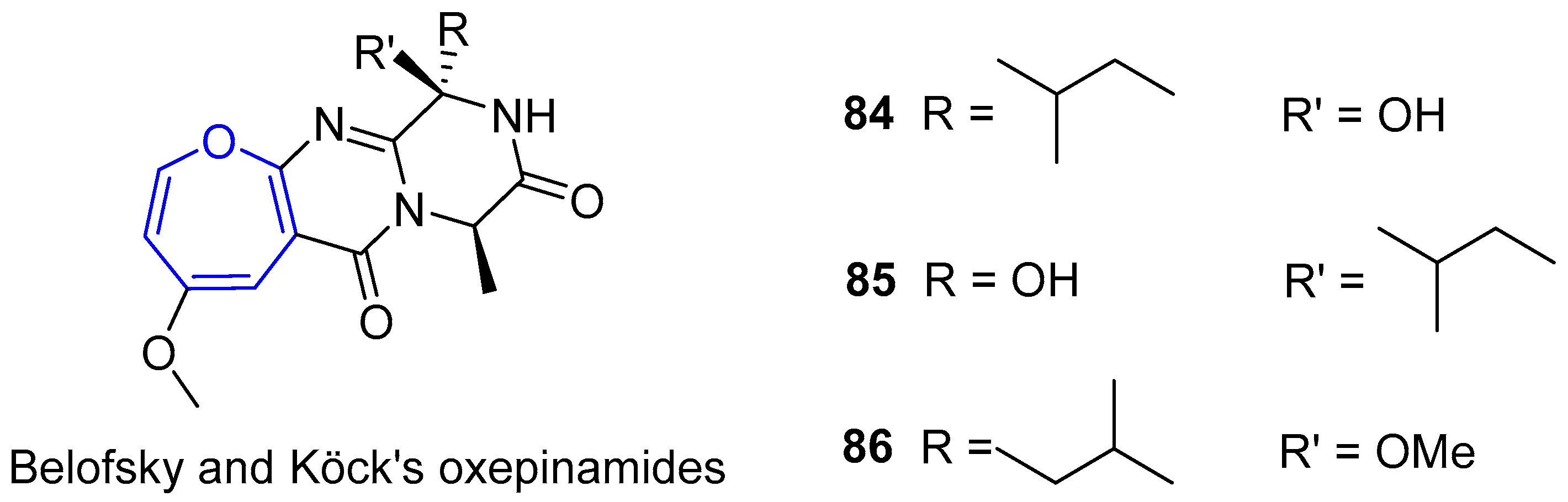
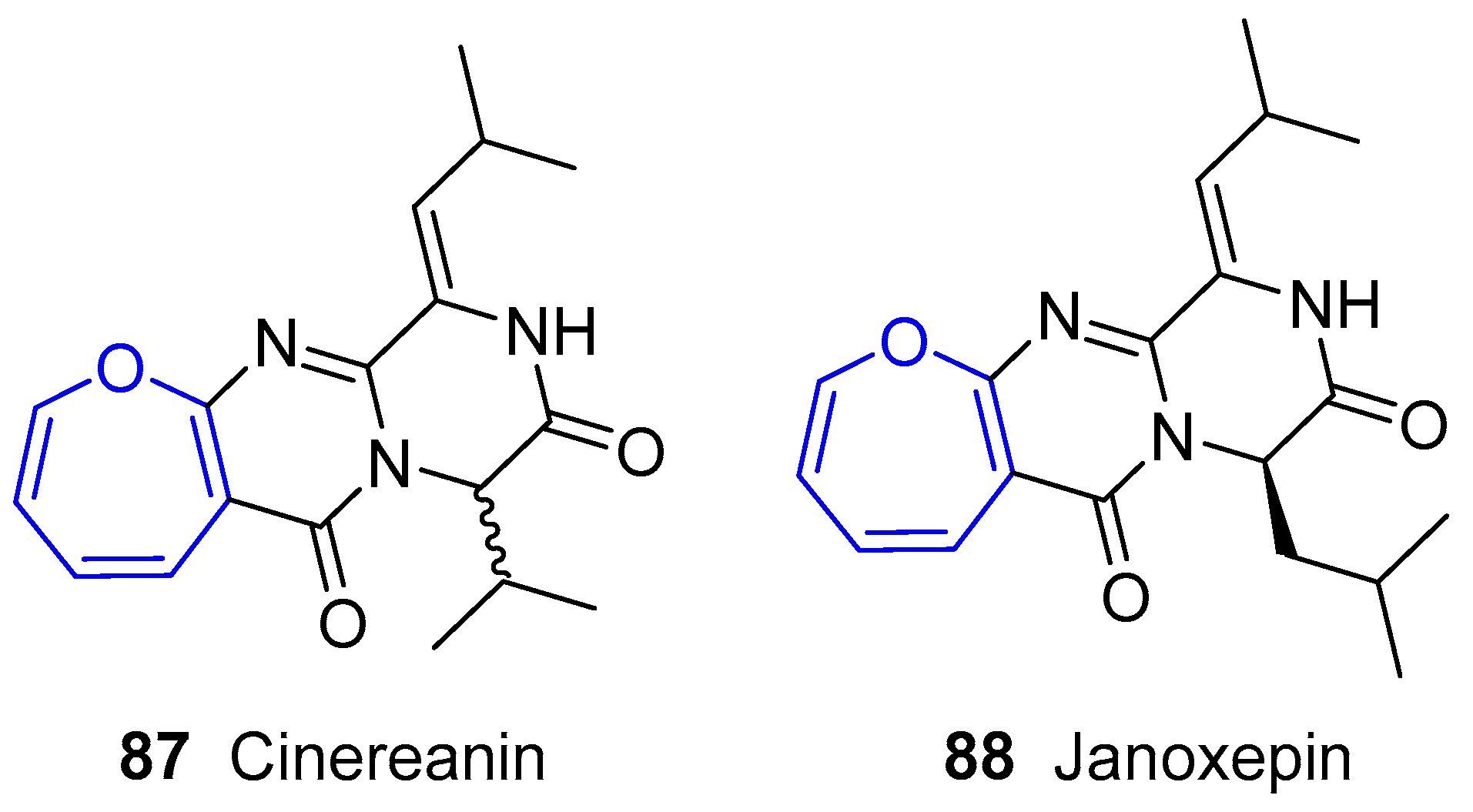
6.3. Oxepinamides Family
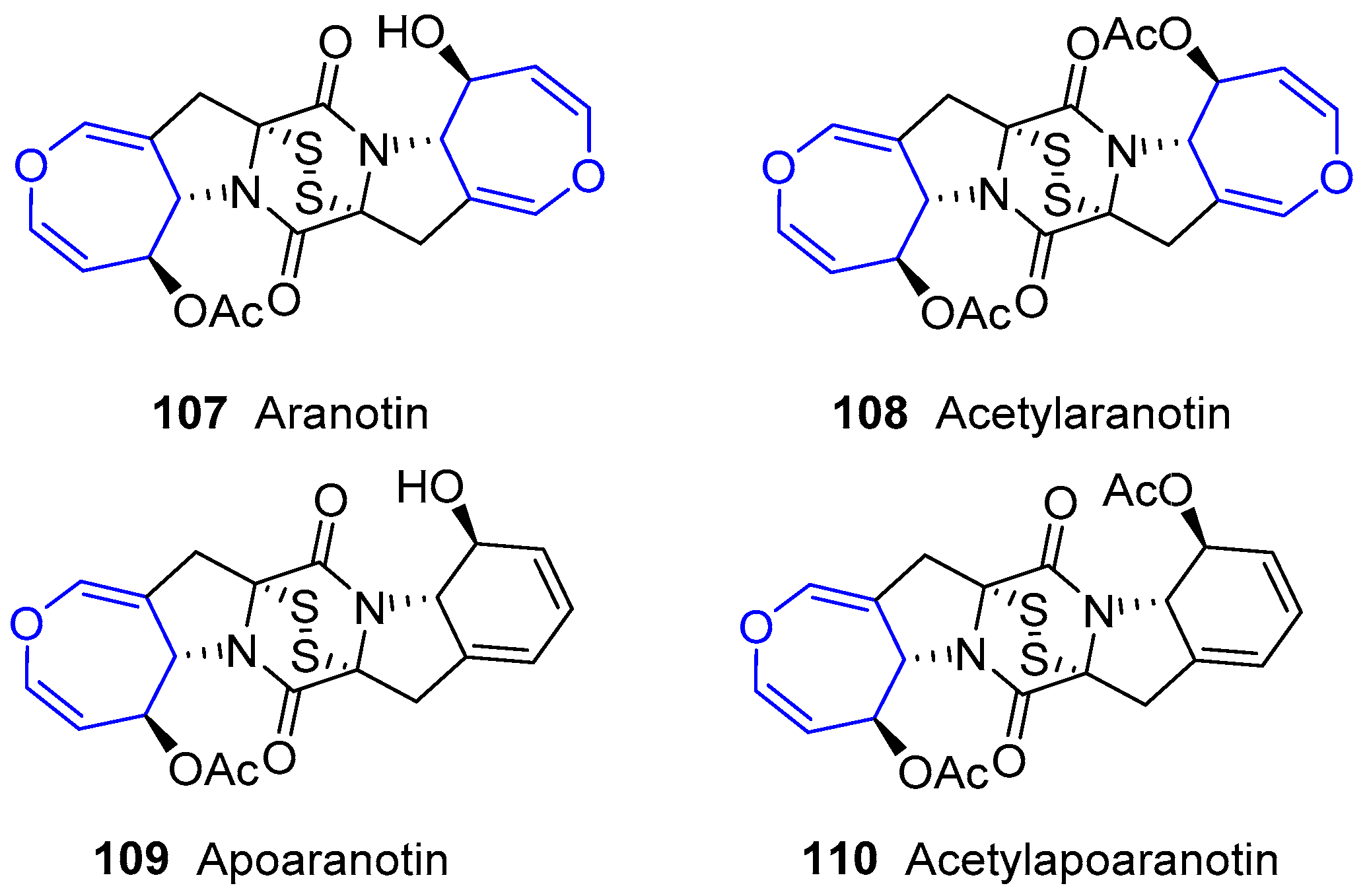
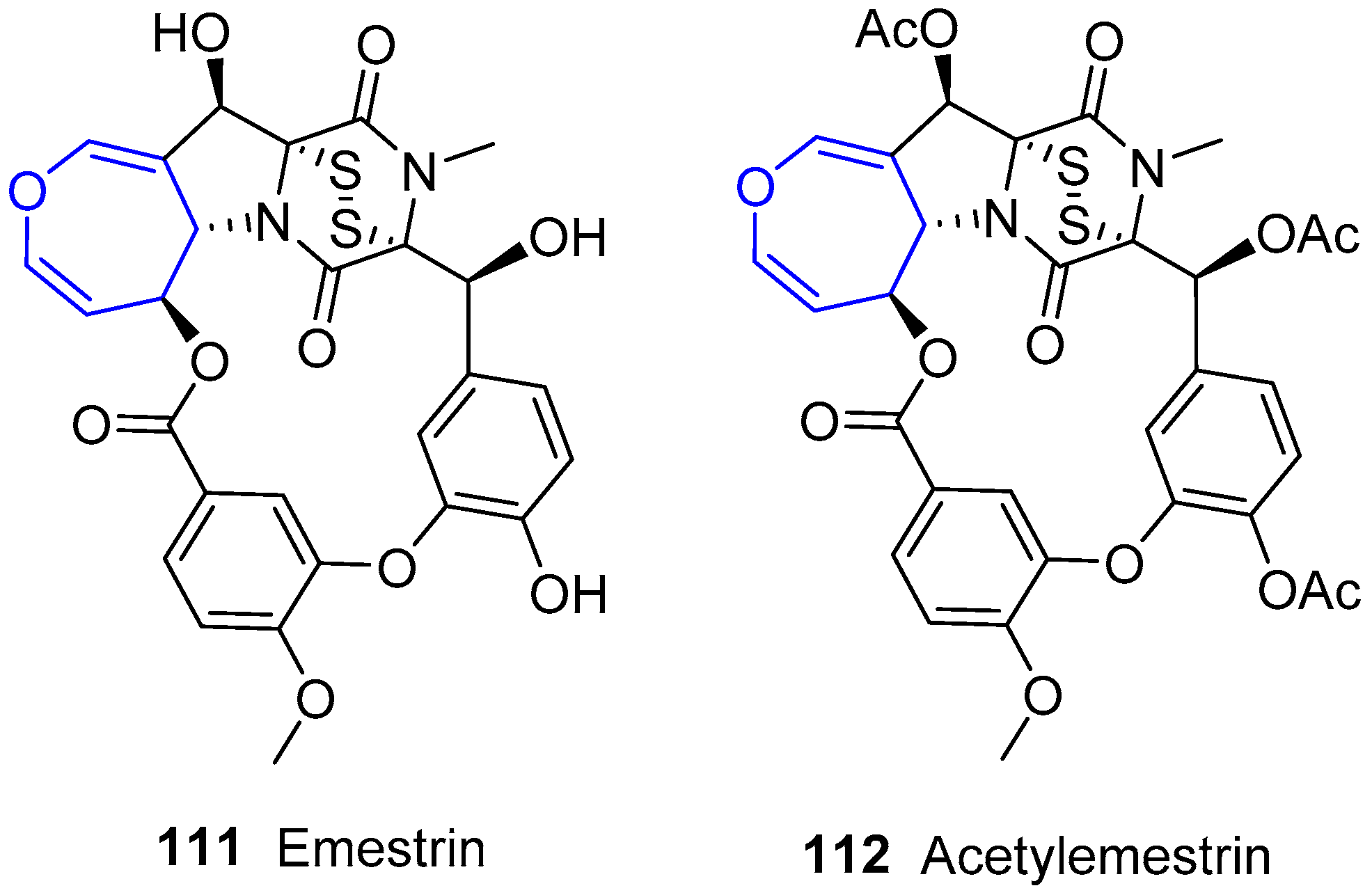
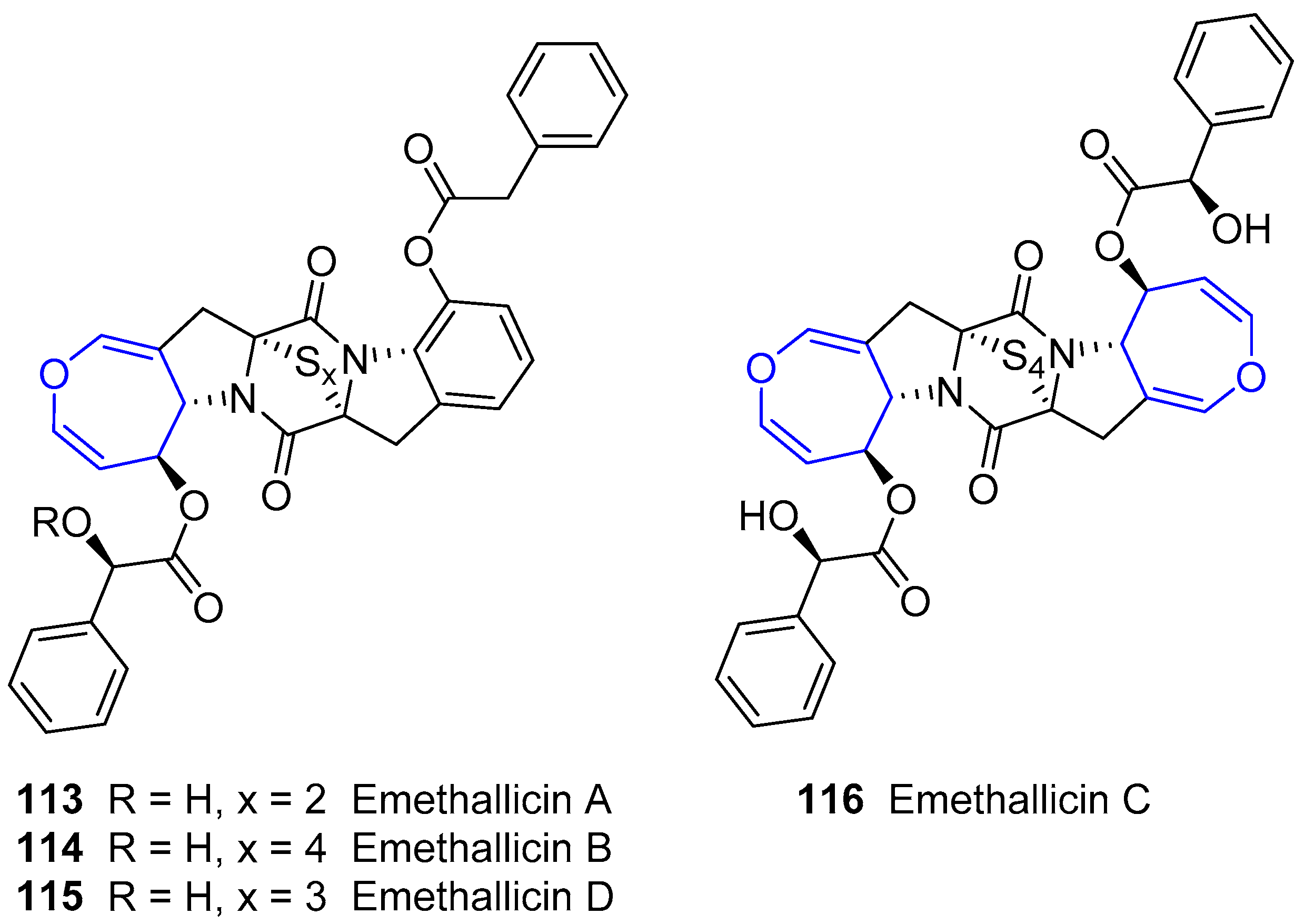
6.4. Diketopiperazine Sulfides
Acknowledgments
Conflicts of Interest
References
- Blunt, J.W.; Copp, B.R.; Hu, W.-P.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2008, 25, 35–94. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K. Total Synthesis of Medium-Ring Ethers from Laurencia Red Algae. In Marine Natural Products; Kiyota, H., Ed.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 97–148. [Google Scholar]
- Nicolaou, K.C.; Frederick, M.O.; Aversa, R.J. The Continuing Saga of the Marine Polyether Biotoxins. Angew. Chem. Int. Ed. 2008, 47, 7182–7225. [Google Scholar] [CrossRef] [PubMed]
- Vilotijevic, I.; Jamison, F.T. Synthesis of Marine Polycyclic Polyethers via Endo-Selective Epoxide-Opening Cascades. Mar. Drugs 2010, 8, 763–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.T. Biogenetic Relationships of Bioactive Sponge Merotriterpenoids. Mar. Drugs 2017, 15, 285. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.J.; Souto, M.L.; Norte, M. Marine polyether triterpenes. Nat. Prod. Rep. 2000, 17, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, D.J. Marine natural products. Nat. Prod. Rep. 1994, 11, 355–394. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Herald, C.L.; Allen, M.S.; Von Dreele, R.B.; Vanell, L.D.; Kao, J.P.Y.; Blake, W. Antineoplastic agents. 48. The isolation and structure of aplysistatin. J. Am. Chem. Soc. 1977, 99, 262–263. [Google Scholar] [CrossRef] [PubMed]
- Palaniveloo, K.; Vairappan, C.S. Chemical relationship between red algae genus Laurencia and sea hare (Aplysia dactylomela Rang) in the North Borneo Island. J. Appl. Phycol. 2014, 26, 1199–1205. [Google Scholar] [CrossRef]
- De Nys, R.; Steinberg, P.D.; Rogers, C.N.; Charlton, T.S.; Duncan, M.W. Quantitative variation of secondary metabolites in the sea hare Aplysia parvula and its host plant, Delisea pulchra. Mar. Ecol. Prog. Ser. 1996, 130, 135–146. [Google Scholar] [CrossRef]
- Rogers, C.N.; De Nys, R.; Charlton, T.S.; Steinberg, P.D. Dynamics of Algal Secondary Metabolites in Two Species of Sea Hare. J. Chem. Ecol. 2000, 26, 721–744. [Google Scholar] [CrossRef]
- Pennings, S.C.; Paul, V.J. Sequestration of dietary secondary metabolites by three species of sea hares: Location, specificity and dynamics. Mar. Biol. 1993, 117, 535–546. [Google Scholar] [CrossRef]
- Paul, V.J.; Fenical, W. Palisadins A, B and related monocyclofarnesol-derived sesquiterpenoids from the red marine alga Laurencia cf. palisada. Tetrahedron Lett. 1980, 21, 2787–2790. [Google Scholar] [CrossRef]
- Capon, R.; Ghisalberti, E.L.; Jefferies, P.R.; Skelton, B.W.; White, A.H. Sesquiterpene metabolites from laurencia filiformis. Tetrahedron 1981, 37, 1613–1621. [Google Scholar] [CrossRef]
- Wright, A.D.; König, G.M.; Sticher, O. New Sesquiterpenes and C15 Acetogenins from the Marine Red Alga Laurencia implicata. J. Nat. Prod. 1991, 54, 1025–1033. [Google Scholar] [CrossRef]
- De Nys, R.; Wright, A.D.; König, G.M.; Sticher, O.; Alino, P.M. Five New Sesquiterpenes from the Red Alga Laurencia flexilis. J. Nat. Prod. 1993, 56, 877–883. [Google Scholar] [CrossRef]
- Su, J.-Y.; Zhong, Y.-L.; Zeng, L.-M.; Wu, H.-M.; Ma, K. Terpenoids from Laurencia karlae. Phytochemistry 1995, 40, 195–197. [Google Scholar] [CrossRef]
- Kuniyoshi, M.; Marma, M.S.; Higa, T.; Bernardinelli, G.; Jefford, C.W. New Bromoterpenes from the Red Alga Laurencia luzonensis. J. Nat. Prod. 2001, 64, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yuan, Z.-H.; Li, J.; Guo, S.-J.; Deng, L.-P.; Han, L.-J.; Zhu, X.-B.; Shi, D.-Y. Sesquiterpenes from the Marine Red Alga Laurencia saitoi. Helv. Chim. Acta 2009, 92, 1291–1297. [Google Scholar] [CrossRef]
- Su, H.; Shi, D.-Y.; Li, J.; Guo, S.-J.; Li, L.-L.; Yuan, Z.-H.; Zhu, X.-B. Sesquiterpenes from Laurencia similis. Molecules 2009, 14, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Lee Tan, K.; Matsunaga, S.; Vairappan, C.S. Halogenated chamigranes of red alga Laurencia snackeyi (Weber-van Bosse) Masuda from Sulu-Sulawesi Sea. Biochem. Syst. Ecol. 2011, 39, 213–215. [Google Scholar] [CrossRef]
- Von Dreele, R.B.; Kao, J.P.Y. The structure of the sesquiterpene aplysistatin. Acta Crystallogr. B 1980, 36, 2695–2698. [Google Scholar] [CrossRef]
- König, G.M.; Wright, A.D.; Sticher, O.; Angerhofer, C.K.; Pezzuto, J.M. Biological Activities of Selected Marine Natural Products. Planta Med. 1994, 60, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Vairappan, C.S.; Kamada, T.; Lee, W.-W.; Jeon, Y.-J. Anti-inflammatory activity of halogenated secondary metabolites of Laurencia snackeyi (Weber-van Bosse) Masuda in LPS-stimulated RAW 264.7 macrophages. J. Appl. Phycol. 2013, 25, 1805–1813. [Google Scholar] [CrossRef]
- Wijesinghe, W.A.; Kim, E.A.; Kang, M.C.; Lee, W.W.; Lee, H.S.; Vairappan, C.S.; Jeon, Y.J. Assessment of anti-inflammatory effect of 5beta-hydroxypalisadin B isolated from red seaweed Laurencia snackeyi in zebrafish embryo in vivo model. Environ. Toxicol. Pharmacol. 2014, 37, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Hoye, T.R.; Kurth, M.J. Total synthesis of dl-aplysistatin. J. Am. Chem. Soc. 1979, 101, 5065–5067. [Google Scholar] [CrossRef]
- Hoye, T.R.; Caruso, A.J.; Dellaria, J.F.; Kurth, M.J. Two syntheses of dl-aplysistatin. J. Am. Chem. Soc. 1982, 104, 6704–6709. [Google Scholar] [CrossRef]
- Shieh, H.-M.; Prestwich, G.D. Chiral, biomimetic total synthesis of (−)-aplysistatin. Tetrahedron Lett. 1982, 23, 4643–4646. [Google Scholar] [CrossRef]
- White, J.D.; Nishiguchi, T.; Skeean, R.W. Stereoselective, biogenetically patterned synthesis of (.+-.)-aplysistatin. J. Am. Chem. Soc. 1982, 104, 3923–3928. [Google Scholar] [CrossRef]
- Gosselin, P.; Rouessac, F. Polycyclisations cationiques de polyenes via leurs bromohydrines—II synthese de la (±) aplysistatine. Tetrahedron Lett. 1983, 24, 5515–5518. [Google Scholar] [CrossRef]
- Kraus, G.A.; Gottschalk, P. A direct synthesis of .beta.-hydroxybutyrolactones. Total synthesis of dendrolasin and formal total synthesis of aplysistatin. J. Org. Chem. 1983, 48, 5356–5357. [Google Scholar] [CrossRef]
- Tanaka, A.; Otsuka, S.; Yamashita, K. Biomimetic Total Synthesis of (−)-Aplysistatin. Agric. Biol. Chem. 1984, 48, 2535–2540. [Google Scholar] [CrossRef]
- Tanaka, A.; Suzuki, M.; Yamashita, K. Total Synthesis of (+)-Palisadin A and (+)-12-Hydroxypalisadin B. Agric. Biol. Chem. 1986, 50, 1069–1071. [Google Scholar] [CrossRef]
- Couladouros, E.A.; Vidali, V.P. Novel Stereocontrolled Approach to syn- and anti-Oxepene-Cyclogeranyl trans-Fused Polycyclic Systems: Asymmetric Total Synthesis of (−)-Aplysistatin, (+)-Palisadin A, (+)-Palisadin B, (+)-12-Hydroxy-Palisadin B, and the AB Ring System of Adociasulfate-2 and Toxicol A. Chem. Eur. J. 2004, 10, 3822–3835. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, R.; Wells, R. Isolation of Some Novel Diterpenes from a Soft Coral of the Genus Lobophytum. Aust. J. Chem. 1979, 32, 1345–1351. [Google Scholar] [CrossRef]
- Raju, B.L.; Subbaraju, G.V.; Rao, C.B.; Trimurtulu, G. Two New Oxygenated Lobanes from a Soft Coral of Lobophytum Species of the Andaman and Nicobar Coasts. J. Nat. Prod. 1993, 56, 961–966. [Google Scholar] [CrossRef]
- Shin, J.; Fenical, W. Fuscosides A-D: Anti-inflammatory diterpenoid glycosides of new structural classes from the caribbean gorgonian Eunicea fusca. J. Org. Chem. 1991, 56, 3153–3158. [Google Scholar] [CrossRef]
- Marchbank, D.H.; Berrue, F.; Kerr, R.G. Eunicidiol, an Anti-inflammatory Dilophol Diterpene from Eunicea fusca. J. Nat. Prod. 2012, 75, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Hamada, T.; Kusumi, T.; Ishitsuka, M.O.; Kakisawa, H. Structures and Absolute Configuration of New Lobane Diterpenoids from the Okinawan Soft Coral Sinularia flexibilis. Chem. Lett. 1992, 21, 33–36. [Google Scholar] [CrossRef]
- Kusumi, T.; Hamada, T.; Ishitsuka, M.O.; Ohtani, I.; Kakisawa, H. Elucidation of the relative and absolute stereochemistry of lobatriene, a marine diterpene, by a modified Mosher method. J. Org. Chem. 1992, 57, 1033–1035. [Google Scholar] [CrossRef]
- Chai, M.-C.; Wang, S.-K.; Dai, C.-F.; Duh, C.-Y. A Cytotoxic Lobane Diterpene from the Formosan Soft Coral Sinularia inelegans. J. Nat. Prod. 2000, 63, 843–844. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, I.; Jhaumeer-Laulloo, S.B.; Bontemps, N.; Banaigs, B.; Aknin, M. New Lobane and Cembrane Diterpenes from Two Comorian Soft Corals. Mar. Drugs 2010, 8, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Edrada, R.A.; Proksch, P.; Wray, V.; Witte, L.; van Ofwegen, L. Four New Bioactive Lobane Diterpenes of the Soft Coral Lobophytum pauciflorum from Mindoro, Philippines. J. Nat. Prod. 1998, 61, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, H.; Yamabe, O.; Kato, M. Synthetic study of marine lobane diterpenes: Efficient synthesis of (+)-fuscol. J. Chem. Soc. Perkin Trans. 1 1998, 217–222. [Google Scholar] [CrossRef]
- Kato, M.; Kosugi, H.; Ichiyanagi, T.; Yamabe, O. Synthetic study of marine lobane diterpenes. Enantioselective syntheses of lobatrienolide and lobatrientriol from (+)-nopinone. J. Chem. Soc. Perkin Trans. 1 1999, 783–788. [Google Scholar] [CrossRef]
- Marchbank, D.H.; Kerr, R.G. Semisynthesis of fuscoside B analogues and eunicosides, and analysis of anti-inflammatory activity. Tetrahedron 2011, 67, 3053–3061. [Google Scholar] [CrossRef]
- Edrada, R.A.; Wray, V.; Handayani, D.; Schupp, P.; Balbin-Oliveros, M.; Proksch, P. Structure-activity relationships of bioactive metabolites from some Indo-Pacific marine invertebrates. Stud. Nat. Prod. Chem. 2000, 21, 251–292. [Google Scholar] [CrossRef]
- Menna, M.; Imperatore, C.; D’Aniello, F.; Aiello, A. Meroterpenes from Marine Invertebrates: Structures, Occurrence, and Ecological Implications. Mar. Drugs 2013, 11, 1602–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegazy, M.; Mohamed, T.; Alhammady, M.; Shaheen, A.; Reda, E.; Elshamy, A.; Aziz, M.; Paré, P. Molecular Architecture and Biomedical Leads of Terpenes from Red Sea Marine Invertebrates. Mar. Drugs 2015, 13, 3154–3181. [Google Scholar] [CrossRef] [PubMed]
- Shmueli, U.; Carmely, S.; Groweiss, A.; Kashman, Y. Sipholenol and sipholenone, two new triterpenes from the marine sponge siphonochalina siphonella (levi). Tetrahedron Lett. 1981, 22, 709–712. [Google Scholar] [CrossRef]
- Carmely, S.; Kashman, Y. The sipholanes, a novel group of triterpenes from the marine sponge Siphonochalina siphonella. J. Org. Chem. 1983, 48, 3517–3525. [Google Scholar] [CrossRef]
- Carmely, S.; Loya, Y.; Kashman, Y. Siphenellinol, a new triterpene from the marine sponge siphonochalinasiphonella. Tetrahedron Lett. 1983, 24, 3673–3676. [Google Scholar] [CrossRef]
- Kashman, Y.; Yosief, T.; Carmeli, S. New Triterpenoids from the Red Sea Sponge Siphonochalina siphonella. J. Nat. Prod. 2001, 64, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Abraham, I.; Carvalho, P.; Kuang, Y.-H.; Shaala, L.A.; Youssef, D.T.A.; Avery, M.A.; Chen, Z.-S.; El Sayed, K.A. Sipholane Triterpenoids: Chemistry, Reversal of ABCB1/P-Glycoprotein-Mediated Multidrug Resistance, and Pharmacophore Modeling. J. Nat. Prod. 2009, 72, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Al-Lihaibi, S.S.; Abdel-Lateff, A.; Alarif, W.M.; Nogata, Y.; Ayyad, S.-E.N.; Okino, T. Potent Antifouling Metabolites from Red Sea Organisms. Asian J. Chem. 2015, 27, 2252–2256. [Google Scholar] [CrossRef]
- Li, Y.-X.; Himaya, S.; Kim, S.-K. Triterpenoids of Marine Origin as Anti-Cancer Agents. Molecules 2013, 18, 7886–7909. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Laphookhieo, S.; Shi, Z.; Fu, L.-W.; Akiyama, S.-I.; Chen, Z.-S.; Youssef, D.T.A.; van Soest, R.W.M.; El Sayed, K.A. Reversal of P-Glycoprotein-Mediated Multidrug Resistance by Sipholane Triterpenoids. J. Nat. Prod. 2007, 70, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Jain, S.; Kim, I.-W.; Peng, X.-X.; Abraham, I.; Youssef, D.T.A.; Fu, L.-W.; El Sayed, K.; Ambudkar, S.V.; Chen, Z.-S. Sipholenol A, a marine-derived sipholane triterpene, potently reverses P-glycoprotein (ABCB1)-mediated multidrug resistance in cancer cells. Cancer Sci. 2007, 98, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Abraham, I.; Jain, S.; Wu, C.-P.; Khanfar, M.A.; Kuang, Y.; Dai, C.-L.; Shi, Z.; Chen, X.; Fu, L.; Ambudkar, S.V.; et al. Marine sponge-derived sipholane triterpenoids reverse P-glycoprotein (ABCB1)-mediated multidrug resistance in cancer cells. Biochem. Pharmacol. 2010, 80, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Abraham, I.; El Sayed, K.; Chen, Z.-S.; Guo, H. Current Status on Marine Products with Reversal Effect on Cancer Multidrug Resistance. Mar. Drugs 2012, 10, 2312–2321. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Lateff, A.; Al-Abd Ahmed, M.; Alahdal Abdulrahman, M.; Alarif Walied, M.; Ayyad Seif-Eldin, N.; Al-Lihaibi Sultan, S.; Hegazy Mohamed, E.; Al Mohammadi, A.; Abdelghany Tamer, M.; Abdel-Naim Ashraf, B.; et al. Antiproliferative effects of triterpenoidal derivatives, obtained from the marine sponge Siphonochalina sp. on human hepatic and colorectal cancer cells. Z. Naturforsch. C J. Biosci. 2016, 71, 29. [Google Scholar] [CrossRef] [PubMed]
- Carmely, S.; Kashman, Y. Neviotine-A, a new triterpene from the red sea sponge Siphonochalina siphonella. J. Org. Chem. 1986, 51, 784–788. [Google Scholar] [CrossRef]
- Angawi, R.; Saqer, E.; Abdel-Lateff, A.; Badria, F.; Ayyad, S.-E. Cytotoxic neviotane triterpene-type from the red sea sponge Siphonochalina siphonella. Pharmacogn. Mag. 2014, 10, 334–341. [Google Scholar] [CrossRef]
- Rudi, A.; Goldberg, I.; Stein, Z.; Benayahu, Y.; Schleyer, M.; Kashman, Y. Sodwanones A–C, three new triterpenoids from a marine sponge. Tetrahedron Lett. 1993, 34, 3943–3944. [Google Scholar] [CrossRef]
- Rudi, A.; Kashman, Y.; Benayahu, Y.; Schleyer, M. Sodwanones A–F, New Triterpenoids from the Marine Sponge Axinella weltneri. J. Nat. Prod. 1994, 57, 1416–1423. [Google Scholar] [CrossRef]
- Rudi, A.; Stein, Z.; Goldberg, I.; Yosief, T.; Kashman, Y.; Schleyer, M. Yardenone and abudinol two new triterpenes from the marine sponge Ptilocaulis spiculifer. Tetrahedron Lett. 1998, 39, 1445–1448. [Google Scholar] [CrossRef]
- Carletti, I.; Long, C.; Funel, C.; Amade, P. Yardenone A and B: New Cytotoxic Triterpenes from the Indian Ocean Sponge Axinella cf. bidderi. J. Nat. Prod. 2003, 66, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Rudi, A.; Aknin, M.; Gaydou, E.M.; Kashman, Y. Sodwanones K, L, and M; New Triterpenes from the Marine Sponge Axinella weltneri. J. Nat. Prod. 1997, 60, 700–703. [Google Scholar] [CrossRef] [PubMed]
- Rudi, A.; Goldberg, I.; Stein, Z.; Kashman, Y.; Benayahu, Y.; Schleyer, M.; Gravalos, M.D.G. Sodwanones G, H, and I, New Cytotoxic Triterpenes from a Marine Sponge. J. Nat. Prod. 1995, 58, 1702–1712. [Google Scholar] [CrossRef] [PubMed]
- Rudi, A.; Yosief, T.; Schleyer, M.; Kashman, Y. Several new isoprenoids from two marine sponges of the family Axinellidae. Tetrahedron 1999, 55, 5555–5566. [Google Scholar] [CrossRef]
- Funel-Le Bon, C.; Berrué, F.; Thomas, O.P.; Reyes, F.; Amade, P. Sodwanone S, a Triterpene from the Marine Sponge Axinella weltneri. J. Nat. Prod. 2005, 68, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Fishback, J.A.; Zhou, Y.-D.; Nagle, D.G. Sodwanone and Yardenone Triterpenes from a South African Species of the Marine Sponge Axinella Inhibit Hypoxia-Inducible Factor-1 (HIF-1) Activation in Both Breast and Prostate Tumor Cells. J. Nat. Prod. 2006, 69, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Whibley, C.E.; Keyzers, R.A.; Soper, A.G.; Davies-Coleman, M.T.; Samaai, T.; Hendricks, D.T. Antiesophageal Cancer Activity from Southern African Marine Organisms. Ann. N. Y. Acad. Sci. 2005, 1056, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, S.; Kashman, Y. Shaagrockol B and C; two hexaprenylhydroquinone disulfates from the red sea sponge toxiclona toxius. Tetrahedron Lett. 1992, 33, 2227–2230. [Google Scholar] [CrossRef]
- Loya, S.; Tal, R.; Hizi, A.; Issacs, S.; Kashman, Y.; Loya, Y. Hexaprenoid Hydroquinones, Novel Inhibitors of the Reverse Transcriptase of Human Immunodeficiency Virus Type 1. J. Nat. Prod. 1993, 56, 2120–2125. [Google Scholar] [CrossRef] [PubMed]
- Kornprobst, J.-M.; Sallenave, C.; Barnathan, G. Sulfated Compounds from Marine Organisms. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1998, 119, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Prinsep, M.R. Sulfur-Containing Natural Products from Marine Invertebrates. In Studies in Natural Products Chemistry; Rahman, A.-U., Ed.; Elsevier: Amsterdam, The Nederlands, 2003; Volume 28, pp. 617–751. [Google Scholar]
- Bokesch, H.R.; Stull, A.C.; Pannell, L.K.; McKee, T.C.; Boyd, M.R. A new pentacyclic sulfated hydroquinone from the marine sponge Haliclona sp. Tetrahedron Lett. 2002, 43, 3079–3081. [Google Scholar] [CrossRef]
- Menhour, B.; Aclinou, P.; Pete, J.-P. Synthesis of bicyclic oxepanes: An enantioselective approach to the western part of Shaagrockol C. Med. J. Chem. 2013, 2, 522–540. [Google Scholar] [CrossRef]
- Puliti, R.; Trivellone, E.; Crispino, A.; Cimino, G. Raspacionin, a New Tetracyclic Triterpenoid from the Sponge Raspaciona aculeata: A Structure Containing Disordered Solvent. Acta Crystallogr. C 1991, 47, 2609–2612. [Google Scholar] [CrossRef]
- Cimino, G.; Crispino, A.; Epifanio, R.d.A.; Madaio, A.; Mattia, C.A.; Mazzarella, L.; Puliti, R.; Enrico, T.; Maria, U. Raspacionin-A: A novel rearranged triterpenoid from the mediterranean sponge raspaciona aculeata. Tetrahedron 1992, 48, 9013–9022. [Google Scholar] [CrossRef]
- Cimino, G.; Crispino, A.; Madaio, A.; Trivellone, E.; Uriz, M. Raspacionin B, a Further Triterpenoid from the Mediterranean Sponge Raspaciona aculeata. J. Nat. Prod. 1993, 56, 534–538. [Google Scholar] [CrossRef]
- Cimino, G.; Epifanio, R.D.A.; Madaio, A.; Puliti, R.; Trivellone, E. Absolute Stereochemistry of Raspacionin, the Main Triterpenoid from the Marine Sponge Raspaciona Aculeata. J. Nat. Prod. 1993, 56, 1622–1626. [Google Scholar] [CrossRef]
- Cimino, G.; Madaio, A.; Trivellone, E.; Uriz, M. Minor Triterpenoids from the Mediterranean Sponge, Raspaciona aculeata. J. Nat. Prod. 1994, 57, 784–790. [Google Scholar] [CrossRef]
- Ciavatta, M.L.; Scognamiglio, G.; Trivellone, E.; Bisogno, T.; Cimino, G. New additional triterpenoids from the Mediterranean sponge Raspaciona aculeata. Tetrahedron 2002, 58, 4943–4948. [Google Scholar] [CrossRef]
- Horak, R.M.; Steyn, P.S.; Van Rooyen, P.H.; Vleggaar, R.; Rabie, C.J. Structures of the austalides A–E, five noval toxic metabolites from Aspergillus ustus. J. Chem. Soc. Chem. Commun. 1981, 1265–1267. [Google Scholar] [CrossRef]
- Horak, R.M.; Steyn, P.S.; Vleggaar, R.; Rabie, C.J. Metabolites of Aspergillus ustus. Part 3. Structure elucidation of austalides G–L. J. Chem. Soc. Perkin Trans. 1 1985, 363–367. [Google Scholar] [CrossRef]
- Zhou, Y.; Mándi, A.; Debbab, A.; Wray, V.; Schulz, B.; Müller, W.E.G.; Lin, W.; Proksch, P.; Kurtán, T.; Aly, A.H. New Austalides from the Sponge-Associated Fungus Aspergillus sp. Eur. J. Org. Chem. 2011, 2011, 6009–6019. [Google Scholar] [CrossRef]
- Zhou, Y.; Debbab, A.; Wray, V.; Lin, W.; Schulz, B.; Trepos, R.; Pile, C.; Hellio, C.; Proksch, P.; Aly, A.H. Marine bacterial inhibitors from the sponge-derived fungus Aspergillus sp. Tetrahedron Lett. 2014, 55, 2789–2792. [Google Scholar] [CrossRef] [Green Version]
- Zhuravleva, O.I.; Sobolevskaya, M.P.; Leshchenko, E.V.; Kirichuk, N.N.; Denisenko, V.A.; Dmitrenok, P.S.; Dyshlovoy, S.A.; Zakharenko, A.M.; Kim, N.Y.; Afiyatullov, S.S. Meroterpenoids from the Alga-Derived Fungi Penicillium thomii Maire and Penicillium lividum Westling. J. Nat. Prod. 2014, 77, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.-G.; Wu, Z.-Y.; Pang, W.-W.; Ma, L.-F.; Ying, Y.-M.; Zhan, Z.-J. α-Glucosidase Inhibitors from the Fungus Aspergillus terreus 3.05358. Chem. Biodivers. 2015, 12, 1718–1724. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhang, X.; Wang, W.; Zhu, T.; Gu, Q.; Li, D. Austalides S–U, New Meroterpenoids from the Sponge-Derived Fungus Aspergillus aureolatus HDN14-107. Mar. Drugs 2016, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, A.E.; Horak, R.M.; Steyn, P.S.; Vleggaar, R. Biosynthesis of austalide D, a meroterpenoid mycotoxin from Aspergillus ustus. J. Chem. Soc. Chem. Commun. 1983, 716–718. [Google Scholar] [CrossRef]
- De Jesus, A.E.; Horak, R.M.; Steyn, P.S.; Vleggaar, R. Metabolites of Aspergillus ustus. Part 4. Stable-isotope labelling studies on the biosynthesis of the austalides. J. Chem. Soc. Perkin Trans. 1 1987, 2253–2257. [Google Scholar] [CrossRef]
- Dillen, J.L.M.; Horak, R.M.; Maharaj, V.J.; Marais, S.F.; Vleggaar, R. Absolute configuration and biosynthesis of the austalides, meroterpenoid metabolites of Aspergillus ustus: Mode of cyclisation of the farnesyl moiety. J. Chem. Soc. Chem. Commun. 1989, 393–394. [Google Scholar] [CrossRef]
- Paquette, L.A.; Wang, T.-Z.; Sivik, M.R. Enantioselective synthesis of natural (−)-austalide B, an unusual ortho ester metabolite produced by toxigenic cultures of Aspergillus ustus. J. Am. Chem. Soc. 1994, 116, 2665–2666. [Google Scholar] [CrossRef]
- Paquette, L.A.; Wang, T.-Z.; Sivik, M.R. Total Synthesis of (−)-Austalide B. A Generic Solution to Elaboration of the Pyran/p-Cresol/Butenolide Triad. J. Am. Chem. Soc. 1994, 116, 11323–11334. [Google Scholar] [CrossRef]
- Rotem, M.; Carmely, S.; Kashman, Y.; Loya, Y. Two new antibiotics from the red sea sponge Psammaplysilla purpurea. Tetrahedron 1983, 39, 667–676. [Google Scholar] [CrossRef]
- Roll, D.M.; Chang, C.W.J.; Scheuer, P.J.; Gray, G.A.; Shoolery, J.N.; Matsumoto, G.K.; Van Duyne, G.D.; Clardy, J. Structure of the psammaplysins. J. Am. Chem. Soc. 1985, 107, 2916–2920. [Google Scholar] [CrossRef]
- Mándi, A.; Mudianta, I.W.; Kurtán, T.; Garson, M.J. Absolute Configuration and Conformational Study of Psammaplysins A and B from the Balinese Marine Sponge Aplysinella strongylata. J. Nat. Prod. 2015, 78, 2051–2056. [Google Scholar] [CrossRef] [PubMed]
- Copp, B.R.; Ireland, C.M.; Barrows, L.R. Psammaplysin C: A New Cytotoxic Dibromotyrosine-Derived Metabolite from the Marine Sponge Druinella (=Psammaplysilla) purpurea. J. Nat. Prod. 1992, 55, 822–823. [Google Scholar] [CrossRef] [PubMed]
- Ichiba, T.; Scheuer, P.J.; Kelly-Borges, M. Three bromotyrosine derivatives, one terminating in an unprecedented diketocyclopentenylidene enamine. J. Org. Chem. 1993, 58, 4149–4150. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Kato, H.; Hirota, H.; Fusetani, N. Ceratinamides A and B: New antifouling dibromotyrosine derivatives from the marine sponge Pseudoceratina purpurea. Tetrahedron 1996, 52, 8181–8186. [Google Scholar] [CrossRef]
- Liu, S.; Fu, X.; Schmitz, F.J.; Kelly-Borges, M. Psammaplysin F, a New Bromotyrosine Derivative from a Sponge, Aplysinella sp. J. Nat. Prod. 1997, 60, 614–615. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, G.M.; Eckman, L.L.; Ray, S.; Hughes, R.O.; Pfefferkorn, J.A.; Barluenga, S.; Nicolaou, K.C.; Bewley, C.A. Bromotyrosine-Derived Natural and Synthetic Products as Inhibitors of Mycothiol-S-Conjugate Amidase. Bioorg. Med. Chem. Lett. 2002, 12, 2487–2490. [Google Scholar] [CrossRef]
- Nicholas, G.M.; Eckman, L.L.; Newton, G.L.; Fahey, R.C.; Ray, S.; Bewley, C.A. Inhibition and kinetics of mycobacterium tuberculosis and mycobacterium smegmatis mycothiol-S-conjugate amidase by natural product inhibitors. Bioorg. Med. Chem. 2003, 11, 601–608. [Google Scholar] [CrossRef]
- Yang, X.; Davis, R.A.; Buchanan, M.S.; Duffy, S.; Avery, V.M.; Camp, D.; Quinn, R.J. Antimalarial Bromotyrosine Derivatives from the Australian Marine Sponge Hyattella sp. J. Nat. Prod. 2010, 73, 985–987. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Andrews, K.T.; Birrell, G.W.; Tran, T.L.; Camp, D.; Davis, R.A.; Quinn, R.J. Psammaplysin H, a new antimalarial bromotyrosine alkaloid from a marine sponge of the genus Pseudoceratina. Bioorg. Med. Chem. Lett. 2011, 21, 846–848. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.D.; Schupp, P.J.; Schrör, J.-P.; Engemann, A.; Rohde, S.; Kelman, D.; de Voogd, N.; Carroll, A.; Motti, C.A. Twilight Zone Sponges from Guam Yield Theonellin Isocyanate and Psammaplysins I and J. J. Nat. Prod. 2012, 75, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Mudianta, I.W.; Skinner-Adams, T.; Andrews, K.T.; Davis, R.A.; Hadi, T.A.; Hayes, P.Y.; Garson, M.J. Psammaplysin Derivatives from the Balinese Marine Sponge Aplysinella strongylata. J. Nat. Prod. 2012, 75, 2132–2143. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Han, S.; Lee, H.-S.; Kang, J.S.; Yun, J.; Sim, C.J.; Shin, H.J.; Lee, J.S. Cytotoxic Psammaplysin Analogues from a Suberea sp. Marine Sponge and the Role of the Spirooxepinisoxazoline in Their Activity. J. Nat. Prod. 2013, 76, 1731–1736. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Burtoloso, A.C.B.; Kossuga, M.H. The chemistry and biology of organic guanidine derivatives. Nat. Prod. Rep. 2008, 25, 919–954. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, I.; Kusumi, T.; Kakisawa, H.; Kashman, Y.; Hirsh, S. Structure and chemical properties of ptilomycalin A. J. Am. Chem. Soc. 1992, 114, 8472–8479. [Google Scholar] [CrossRef]
- Kashman, Y.; Hirsh, S.; McConnell, O.J.; Ohtani, I.; Kusumi, T.; Kakisawa, H. Ptilomycalin A: A novel polycyclic guanidine alkaloid of marine origin. J. Am. Chem. Soc. 1989, 111, 8925–8926. [Google Scholar] [CrossRef]
- Jares-Erijman, E.A.; Sakai, R.; Rinehart, K.L. Crambescidins: New antiviral and cytotoxic compounds from the sponge Crambe crambe. J. Org. Chem. 1991, 56, 5712–5715. [Google Scholar] [CrossRef]
- Jares-Erijman, E.A.; Ingrum, A.L.; Carney, J.R.; Rinehart, K.L.; Sakai, R. Polycyclic guanidine-containing compounds from the Mediterranean sponge Crambe crambe: The structure of 13,14,15-isocrambescidin 800 and the absolute stereochemistry of the pentacyclic guanidine moieties of the crambescidins. J. Org. Chem. 1993, 58, 4805–4808. [Google Scholar] [CrossRef]
- Palagiano, E.; De Marino, S.; Minale, L.; Riccio, R.; Zollo, F.; Iorizzi, M.; Carré, J.B.; Debitus, C.; Lucarain, L.; Provost, J. Ptilomycalin A, crambescidin 800 and related new highly cytotoxic guanidine alkaloids from the starfishes Fromia monilis and Celerina heffernani. Tetrahedron 1995, 51, 3675–3682. [Google Scholar] [CrossRef]
- Venkateswarlu, Y.; Reddy, M.V.R.; Ramesh, P.; Rao, J.V. Neofolitispates, pentacyclic guanidine alkaloids from the sponge Neofolitispa dianchora. Indian J. Chem. 1999, 38B, 254–256. [Google Scholar] [CrossRef]
- Braekman, J.C.; Daloze, D.; Tavares, R.; Hajdu, E.; Van Soest, R.W.M. Novel Polycyclic Guanidine Alkaloids from Two Marine Sponges of the Genus Monanchora. J. Nat. Prod. 2000, 63, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Makarieva, T.N.; Tabakmaher, K.M.; Guzii, A.G.; Denisenko, V.A.; Dmitrenok, P.S.; Shubina, L.K.; Kuzmich, A.S.; Lee, H.-S.; Stonik, V.A. Monanchocidins B–E: Polycyclic Guanidine Alkaloids with Potent Antileukemic Activities from the Sponge Monanchora pulchra. J. Nat. Prod. 2011, 74, 1952–1958. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Moazami, Y.; Pierce, J.G. Structure, synthesis and biological properties of the pentacyclic guanidinium alkaloids. Bioorg. Med. Chem. 2017, 25, 2817–2824. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, J.E.H.; Nitcheu, J.; Mahmoudi, N.; Ibana, J.A.; Mangalindan, G.C.; Black, G.P.; Howard-Jones, A.G.; Moore, C.G.; Thomas, D.A.; Mazier, D.; et al. Antimalarial Activity of Crambescidin 800 and Synthetic Analogues against Liver and Blood Stage of Plasmodium sp. J. Antibiot. 2006, 59, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, J.; Ternon, E.; López-Alonso, H.; Thomas, O.; Vega, F.; Vieytes, M.; Botana, L. Crambescidin-816 Acts as a Fungicidal with More Potency than Crambescidin-800 and -830, Inducing Cell Cycle Arrest, Increased Cell Size and Apoptosis in Saccharomyces cerevisiae. Mar. Drugs 2013, 11, 4419–4434. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Kong, D.; Matsui, K.; Kobayashi, M. Erythroid Differentiation in K562 Chronic Myelogenous Cells Induced by Crambescidin 800, a Pentacyclic Guanidine Alkaloid. Anticancer Res. 2004, 24, 2325–2330. [Google Scholar] [PubMed]
- Aron, Z.D.; Pietraszkiewicz, H.; Overman, L.E.; Valeriote, F.; Cuevas, C. Synthesis and anticancer activity of side chain analogs of the crambescidin alkaloids. Bioorg. Med. Chem. Lett. 2004, 14, 3445–3449. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.S.; Gustafson, K.R. Marine pharmacology in 2003–2004: Anti-tumour and cytotoxic compounds. Eur. J. Cancer 2006, 42, 2241–2270. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, J.A.; López-Alonso, H.; Roel, M.; Vieytes, M.R.; Thomas, O.; Ternon, E.; Vega, F.V.; Botana, L.M. Mechanism of cytotoxic action of crambescidin-816 on human liver-derived tumour cells. Br. J. Pharmacol. 2014, 171, 1655–1667. [Google Scholar] [CrossRef] [PubMed]
- Mendez, A.G.; Juncal, A.B.; Silva, S.B.L.; Thomas, O.P.; Martín Vázquez, V.; Alfonso, A.; Vieytes, M.R.; Vale, C.; Botana, L.M. The Marine Guanidine Alkaloid Crambescidin 816 Induces Calcium Influx and Cytotoxicity in Primary Cultures of Cortical Neurons through Glutamate Receptors. ACS Chem. Neurosci. 2017, 8, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Braekman, J.C.; Daloze, D.; Bruno, I.; Riccio, R.; Ferri, S.; Spampinato, S.; Speroni, E. Polycyclic Guanidine Alkaloids from the Marine Sponge Crambe crambe and Ca++ Channel Blocker Activity of Crambescidin 816. J. Nat. Prod. 1993, 56, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Ohizumi, Y.; Sasaki, S.; Kusumi, T.; Ohtani, I.I. Ptilomycalin A, a novel Na(+), K(+)- or Ca2(+)-ATPase inhibitor, competitively interacts with ATP at its binding site. Eur. J. Pharmacol. 1996, 310, 95–98. [Google Scholar] [CrossRef]
- Suna, H.; Aoki, S.; Setiawan, A.; Kobayashi, M. Crambescidin 800, a pentacyclic guanidine alkaloid, protects a mouse hippocampal cell line against glutamate-induced oxidative stress. J. Nat. Med. 2007, 61, 288–295. [Google Scholar] [CrossRef]
- Donahue, M.G. Chapter 1—Recent Developments in the Synthesis of Cyclic Guanidine Alkaloids. In Progress in Heterocyclic Chemistry; Gribble, G.W., Joule, J.A., Eds.; Elsevier: Amsterdam, The Nederlands, 2014; Volume 26, pp. 1–28. [Google Scholar]
- Ma, Y.; De, S.; Chen, C. Syntheses of cyclic guanidine-containing natural products. Tetrahedron 2015, 71, 1145–1173. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.J.; Williams, H.L. Synthesis of a pentacyclic model of ptilomycalin A. J. Chem. Soc. Chem. Commun. 1994, 819–820. [Google Scholar] [CrossRef]
- Murphy, P.J.; Williams, H.L.; Hursthouse, M.B.; Malik, K.M.A. Synthetic studies towards ptilomycalin a using A biomimetic approach. J. Chem. Soc. Chem. Commun. 1994, 119–120. [Google Scholar] [CrossRef]
- Black, G.P.; Murphy, P.J.; Walshe, N.D.A.; Hibbs, D.E.; Hursthouse, M.B.; Abdul Malik, K.M. A short synthetic route to the tricyclic guanidinium core of the batzelladine alkaloids. Tetrahedron Lett. 1996, 37, 6943–6946. [Google Scholar] [CrossRef]
- Murphy, P.J.; Harri Lloyd, W.; Hibbs, D.E.; Hursthouse, M.B.; Abdul Malik, K.M. Biomimetic model studies towards ptilomycalin A. Tetrahedron 1996, 52, 8315–8332. [Google Scholar] [CrossRef]
- Murphy, P.J.; Williams, H.L.; Hibbs, D.E.; Hursthouse, M.B.; Malik, K.M.A. Crystallographic evidence for the proposed host behaviour of ptilomycalin A. Chem. Commun. 1996, 445–447. [Google Scholar] [CrossRef]
- Black, G.P.; Murphy, P.J.; Walshe, N.D.A. A short synthetic route to the tricyclic guanidinium core of the batzelladine alkaloids. Tetrahedron 1998, 54, 9481–9488. [Google Scholar] [CrossRef]
- Black, G.P.; Murphy, P.J.; Thornhill, A.J.; Walshe, N.D.A.; Zanetti, C. Synthesis of the left hand unit of batzelladine F; Revision of the reported relative stereochemistry. Tetrahedron 1999, 55, 6547–6554. [Google Scholar] [CrossRef]
- Overman, L.E.; Rabinowitz, M.H.; Renhowe, P.A. Enantioselective Total Synthesis of (−)-Ptilomycalin A. J. Am. Chem. Soc. 1995, 117, 2657–2658. [Google Scholar] [CrossRef]
- Coffey, D.S.; McDonald, A.I.; Overman, L.E.; Rabinowitz, M.H.; Renhowe, P.A. A Practical Entry to the Crambescidin Family of Guanidine Alkaloids. Enantioselective Total Syntheses of Ptilomycalin A, Crambescidin 657 and Its Methyl Ester (Neofolitispates 2), and Crambescidin 800. J. Am. Chem. Soc. 2000, 122, 4893–4903. [Google Scholar] [CrossRef]
- Coffey, D.S.; Overman, L.E.; Stappenbeck, F. Enantioselective Total Syntheses of 13,14,15-Isocrambescidin 800 and 13,14,15-Isocrambescidin 657. J. Am. Chem. Soc. 2000, 122, 4904–4914. [Google Scholar] [CrossRef]
- Overman, L.E.; Rhee, Y.H. Total Synthesis of (−)-Crambidine and Definition of the Relative Configuration of Its Unique Tetracyclic Guanidinium Core. J. Am. Chem. Soc. 2005, 127, 15652–15658. [Google Scholar] [CrossRef] [PubMed]
- Perl, N.R.; Ide, N.D.; Prajapati, S.; Perfect, H.H.; Durón, S.G.; Gin, D.Y. Annulation of Thioimidates and Vinyl Carbodiimides to Prepare 2-Aminopyrimidines, Competent Nucleophiles for Intramolecular Alkyne Hydroamination. Synthesis of (−)-Crambidine. J. Am. Chem. Soc. 2010, 132, 1802–1803. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, K.; Georgieva, A.; Koshino, H.; Nakata, T.; Kita, T.; Hashimoto, Y. Total Synthesis of Crambescidin 359. Org. Lett. 2002, 4, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.G.; Murphy, P.J.; Williams, H.L.; McGown, A.T.; Smith, N.K. A synthesis of crambescidin 359. Tetrahedron Lett. 2003, 44, 251–254. [Google Scholar] [CrossRef]
- Aron, Z.D.; Overman, L.E. Total Synthesis and Properties of the Crambescidin Core Zwitterionic Acid and Crambescidin 359. J. Am. Chem. Soc. 2005, 127, 3380–3390. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.; Lefranc, F.; Kijjoa, A.; Kiss, R. Can Some Marine-Derived Fungal Metabolites Become Actual Anticancer Agents? Mar. Drugs 2015, 13, 3950–3991. [Google Scholar] [CrossRef] [PubMed]
- Cutler, H.G.; Springer, J.P.; Arrendale, R.F.; Arison, B.H.; Cole, P.D.; Roberts, R.G. Cinereain: A Novel Metabolite with Plant Growth Regulating Properties from Botrytis cinerea. Agric. Biol. Chem. 1988, 52, 1725–1733. [Google Scholar] [CrossRef]
- Rahbæk, L.; Breinholt, J.; Frisvad, J.C.; Christophersen, C. Circumdatin A, B, and C: Three New Benzodiazepine Alkaloids Isolated from a Culture of the Fungus Aspergillus ochraceus. J. Org. Chem. 1999, 64, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Ookura, R.; Kito, K.; Ooi, T.; Namikoshi, M.; Kusumi, T. Structure Revision of Circumdatins A and B, Benzodiazepine Alkaloids Produced by Marine Fungus Aspergillus ostianus, by X-ray Crystallography. J. Org. Chem. 2008, 73, 4245–4247. [Google Scholar] [CrossRef] [PubMed]
- González-Jartı́n, J.M.; Alfonso, A.; Sainz, M.J.; Vieytes, M.R.; Botana, L.M. UPLC-MS-IT-TOF Identification of Circumdatins Produced by Aspergillus ochraceus. J. Agric. Food. Chem. 2017, 65, 4843–4852. [Google Scholar] [CrossRef] [PubMed]
- Belofsky, G.N.; Anguera, M.; Jensen, P.R.; Fenical, W.; Köck, M. Oxepinamides A–C and Fumiquinazolines H–I: Bioactive Metabolites from a Marine Isolate of a Fungus of the Genus Acremonium. Chem. Eur. J. 2000, 6, 1355–1360. [Google Scholar] [CrossRef]
- Sprogøe, K.; Manniche, S.; Larsen, T.O.; Christophersen, C. Janoxepin and brevicompanine B: Antiplasmodial metabolites from the fungus Aspergillus janus. Tetrahedron 2005, 61, 8718–8721. [Google Scholar] [CrossRef]
- Doveston, R.G.; Steendam, R.; Jones, S.; Taylor, R.J.K. Total Synthesis of an Oxepine Natural Product, (±)-Janoxepin. Org. Lett. 2012, 14, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.U.; Asami, Y.; Lee, D.; Jang, J.-H.; Ahn, J.S.; Oh, H. Protuboxepins A and B and Protubonines A and B from the Marine-Derived Fungus Aspergillus sp. SF-5044. J. Nat. Prod. 2011, 74, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Asami, Y.; Jang, J.-H.; Soung, N.-K.; He, L.; Moon, D.O.; Kim, J.W.; Oh, H.; Muroi, M.; Osada, H.; Kim, B.Y.; et al. Protuboxepin A, a marine fungal metabolite, inducing metaphase arrest and chromosomal misalignment in tumor cells. Bioorg. Med. Chem. 2012, 20, 3799–3806. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Mándi, A.; Li, X.-M.; Du, F.-Y.; Wang, J.-N.; Li, X.; Kurtán, T.; Wang, B.-G. Varioxepine A, a 3H-Oxepine-Containing Alkaloid with a New Oxa-Cage from the Marine Algal-Derived Endophytic Fungus Paecilomyces variotii. Org. Lett. 2014, 16, 4834–4837. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, X.-M.; Wang, J.-N.; Wang, B.-G. Oxepine-Containing Diketopiperazine Alkaloids from the Algal-Derived Endophytic Fungus Paecilomyces variotii EN-291. Helv. Chim. Acta 2015, 98, 800–804. [Google Scholar] [CrossRef]
- Wang, J.; He, W.; Huang, X.; Tian, X.; Liao, S.; Yang, B.; Wang, F.; Zhou, X.; Liu, Y. Antifungal New Oxepine-Containing Alkaloids and Xanthones from the Deep-Sea-Derived Fungus Aspergillus versicolor SCSIO 05879. J. Agric. Food. Chem. 2016, 64, 2910–2916. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Shi, Y.; Chen, X.; Chen, C.-T.A.; Tao, X.; Wu, B. New compounds from a hydrothermal vent crab-associated fungus Aspergillus versicolor XZ-4. Org. Biomol. Chem. 2017, 15, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, D.M.; Waring, P.; Howlett, B.J. The epipolythiodioxopiperazine (ETP) class of fungal toxins: distribution, mode of action, functions and biosynthesis. Microbiology 2005, 151, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Isaka, M.; Kittakoop, P.; Kirtikara, K.; Hywel-Jones, N.L.; Thebtaranonth, Y. Bioactive Substances from Insect Pathogenic Fungi. Acc. Chem. Res. 2005, 38, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, A.D. 2,5-Diketopiperazines: Synthesis, Reactions, Medicinal Chemistry, and Bioactive Natural Products. Chem. Rev. 2012, 112, 3641–3716. [Google Scholar] [CrossRef] [PubMed]
- Munday, R. Harmful and Beneficial Effects of Organic Monosulfides, Disulfides, and Polysulfides in Animals and Humans. Chem. Res. Toxicol. 2012, 25, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Bladt, T.; Frisvad, J.; Knudsen, P.; Larsen, T. Anticancer and Antifungal Compounds from Aspergillus, Penicillium and Other Filamentous Fungi. Molecules 2013, 18, 11338–11376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.-M.; Yi, X.-X.; Zhou, Y.; Su, X.; Peng, Y.; Gao, C.-H. An Update on 2,5-Diketopiperazines from Marine Organisms. Mar. Drugs 2014, 12, 6213–6235. [Google Scholar] [CrossRef] [PubMed]
- Kuppusamy, P.; Yusoff, M.M.; Maniam, G.P.; Ichwan, S.J.A.; Soundharrajan, I.; Govindan, N. Nutraceuticals as potential therapeutic agents for colon cancer: A review. Acta Pharm. Sin. B 2014, 4, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Welch, T.R.; Williams, R.M. Epidithiodioxopiperazines. occurrence, synthesis and biogenesis. Nat. Prod. Rep. 2014, 31, 1376–1404. [Google Scholar] [CrossRef] [PubMed]
- Nadumane, V.K.; Venkatachalam, P.; Gajaraj, B. Chapter 19—Aspergillus Applications in Cancer Research A2. In New and Future Developments in Microbial Biotechnology and Bioengineering; Vijai, G., Ed.; Elsevier: Amsterdam, The Nederlands, 2016; pp. 243–255. [Google Scholar]
- Nagarajan, R.; Huckstep, L.L.; Lively, D.H.; DeLong, D.C.; Marsh, M.M.; Neuss, N. Aranotin and related metabolites from Arachniotus aureus. I. Determination of structure. J. Am. Chem. Soc. 1968, 90, 2980–2982. [Google Scholar] [CrossRef]
- Neuss, N.; Nagarajan, R.; Molloy, B.B.; Huckstep, L.L. Aranotin and related metabolites. II. Isolation, characterization, and structures of two new metabolites. Tetrahedron Lett. 1968, 9, 4467–4471. [Google Scholar] [CrossRef]
- Nagarajan, R.; Woody, R.W. Circular dichroism of gliotoxin and related epidithiapiperazinediones. J. Am. Chem. Soc. 1973, 95, 7212–7222. [Google Scholar] [CrossRef] [PubMed]
- Murdock, K.C. Antiviral agents. Chemical modifications of a disulfide antibiotic, acetylaranotin. J. Med. Chem. 1974, 17, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Kamata, S.; Sakai, H.; Hirota, A. Isolation of Acetylaranotin, Bisdethiodi(methylthio)-acetylaranotin and Terrein as Plant Growth Inhibitors from a Strain of Aspergillus terreus. Agric. Biol. Chem. 1983, 47, 2637–2638. [Google Scholar] [CrossRef]
- Guo, C.-J.; Yeh, H.-H.; Chiang, Y.-M.; Sanchez, J.F.; Chang, S.-L.; Bruno, K.S.; Wang, C.C.C. Biosynthetic Pathway for the Epipolythiodioxopiperazine Acetylaranotin in Aspergillus terreus Revealed by Genome-Based Deletion Analysis. J. Am. Chem. Soc. 2013, 135, 7205–7213. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Park, J.S.; Kim, Y.J.; Jung, J.H.; Lee, J.K.; Kwon, H.C.; Yang, H.O. Apoptosis-inducing effect of diketopiperazine disulfides produced by Aspergillus sp. KMD 901 isolated from marine sediment on HCT116 colon cancer cell lines. J. Appl. Microbiol. 2011, 110, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Seya, H.; Nakajima, S.; Kawai, K.-I.; Udagawa, S.-I. Structure and absolute configuration of emestrin, a new macrocyclic epidithiodioxopiperazine from Emericell striata. J. Chem. Soc. Chem. Commun. 1985, 657–658. [Google Scholar] [CrossRef]
- Seya, H.; Nozawa, K.; Nakajima, S.; Kawai, K.-I.; Udagawa, S.-I. Studies on fungal products. Part 8. Isolation and structure of emestrin, a novel antifungal macrocyclic epidithiodioxopiperazine from Emericella striata. X-ray molecular structure of emestrin. J. Chem. Soc. Perkin Trans. 1 1986, 109–116. [Google Scholar] [CrossRef]
- Seya, H.; Nozawa, K.; Udagawa, S.-I.; Nakajima, S.; Kawai, K.-I. Studies on Fungal Products. IX. Dethiosecoemestrin, a New Metabolite Related to Emestrin, from Emericella striata. Chem. Pharm. Bull. 1986, 34, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Ooike, M.; Nozawa, K.; Kawai, K.-I. An epitetrathiodioxopiperazine related to emestrin from Emericella foveolata. Phytochemistry 1997, 46, 123–126. [Google Scholar] [CrossRef]
- Kawahara, N.; Nakajima, S.; Yamazaki, M.; Kawai, K. Structure of a Novel Epidithiodioxopiperazine, Emethallicin A, a Potent Inhibitor of Histamine Release, from Emericella heterothallica. Chem. Pharm. Bull. 1989, 37, 2592–2595. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, N.; Nozawa, K.; Yamazaki, M.; Nakajima, S.; Kawai, K.-I. Structures of Novel Epipolythiodioxopiperazines, Emethallicins B, C, and D, Potent Inhibitors of Histamine Release, from Emericella heterothallica. Chem. Pharm. Bull. 1990, 38, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R.M.; Kishi, Y. Experimental Support for the Primary Stereoelectronic Effect Governing Baeyer–Villiger Oxidation and Criegee Rearrangement. J. Am. Chem. Soc. 1998, 120, 9392–9393. [Google Scholar] [CrossRef]
- Peng, J.; Clive, D.L.J. Synthesis of Dihydrooxepin Models Related to the Antitumor Antibiotic MPC1001. Org. Lett. 2007, 9, 2939–2941. [Google Scholar] [CrossRef] [PubMed]
- Gross, U.; Nieger, M.; Bräse, S. A Unified Strategy Targeting the Thiodiketopiperazine Mycotoxins Exserohilone, Gliotoxin, the Epicoccins, the Epicorazines, Rostratin A and Aranotin. Chem. Eur. J. 2010, 16, 11624–11631. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Lu, M.; Totokotsopoulos, S.; Heretsch, P.; Giguère, D.; Sun, Y.-P.; Sarlah, D.; Nguyen, T.H.; Wolf, I.C.; Smee, D.F.; et al. Synthesis and Biological Evaluation of Epidithio-, Epitetrathio-, and bis-(Methylthio)diketopiperazines: Synthetic Methodology, Enantioselective Total Synthesis of Epicoccin G, 8,8′-epi-ent-Rostratin B, Gliotoxin, Gliotoxin G, Emethallicin E, and Haematocin and Discovery of New Antiviral and Antimalarial Agents. J. Am. Chem. Soc. 2012, 134, 17320–17332. [Google Scholar] [CrossRef] [PubMed]
- Schuber, P.T.; Williams, R.M. Synthetic studies toward MPC1001: Preparation of a β-hydroxyl-tyrosine derivative. Tetrahedron Lett. 2012, 53, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, H.F.; Carreira, E.M. An Efficient Synthesis Strategy to the Core Structure of 6-5-6-5-6-Membered Epipolythiodiketopiperazines. Org. Lett. 2014, 16, 2854–2857. [Google Scholar] [CrossRef] [PubMed]
- Belov, D.S.; Ratmanova, N.K.; Andreev, I.A.; Kurkin, A.V. Synthesis of Bicyclic Proline Derivatives by the Aza-Cope-Mannich Reaction: Formal Synthesis of (±)-Acetylaranotin. Chem. Eur. J. 2015, 21, 4141–4147. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Liang, Y.-M. Copper-Catalyzed [2 + 2 + 3] Annulation of 1,6-Enynes with α-Bromo-1,3-Dicarbonyl Compounds: Synthesis of Dihydrooxepines. J. Org. Chem. 2017, 82, 7000–7007. [Google Scholar] [CrossRef] [PubMed]
- Codelli, J.A.; Puchlopek, A.L.A.; Reisman, S.E. Enantioselective Total Synthesis of (−)-Acetylaranotin, a Dihydrooxepine Epidithiodiketopiperazine. J. Am. Chem. Soc. 2012, 134, 1930–1933. [Google Scholar] [CrossRef] [PubMed]
- Onodera, H.; Hasegawa, A.; Tsumagari, N.; Nakai, R.; Ogawa, T.; Kanda, Y. MPC1001 and Its Analogues: New Antitumor Agents from the Fungus Cladorrhinum Species. Org. Lett. 2004, 6, 4101–4104. [Google Scholar] [CrossRef] [PubMed]
- Tsumaragi, N.; Nakai, R.; Onodera, H.; Hasegawa, A.; Rahayu, E.S.; Ando, K.; Yamashita, Y. MPC1001, a New Antitumor Antibiotic Produced by Cladorrhinum sp. J. Antibiot. 2004, 57, 532–534. [Google Scholar] [CrossRef]
- Wang, H.; Regan, C.J.; Codelli, J.A.; Romanato, P.; Puchlopek-Dermenci, A.L.A.; Reisman, S.E. Enantioselective Synthesis of (−)-Acetylapoaranotin. Org. Lett. 2017, 19, 1698–1701. [Google Scholar] [CrossRef] [PubMed]











































© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbero, H.; Díez-Poza, C.; Barbero, A. The Oxepane Motif in Marine Drugs. Mar. Drugs 2017, 15, 361. https://doi.org/10.3390/md15110361
Barbero H, Díez-Poza C, Barbero A. The Oxepane Motif in Marine Drugs. Marine Drugs. 2017; 15(11):361. https://doi.org/10.3390/md15110361
Chicago/Turabian StyleBarbero, Héctor, Carlos Díez-Poza, and Asunción Barbero. 2017. "The Oxepane Motif in Marine Drugs" Marine Drugs 15, no. 11: 361. https://doi.org/10.3390/md15110361
APA StyleBarbero, H., Díez-Poza, C., & Barbero, A. (2017). The Oxepane Motif in Marine Drugs. Marine Drugs, 15(11), 361. https://doi.org/10.3390/md15110361