
Sterols from Thai Marine Sponge Petrosia (Strongylophora) sp. and Their Cytotoxicity

Abstract
:1. Introduction
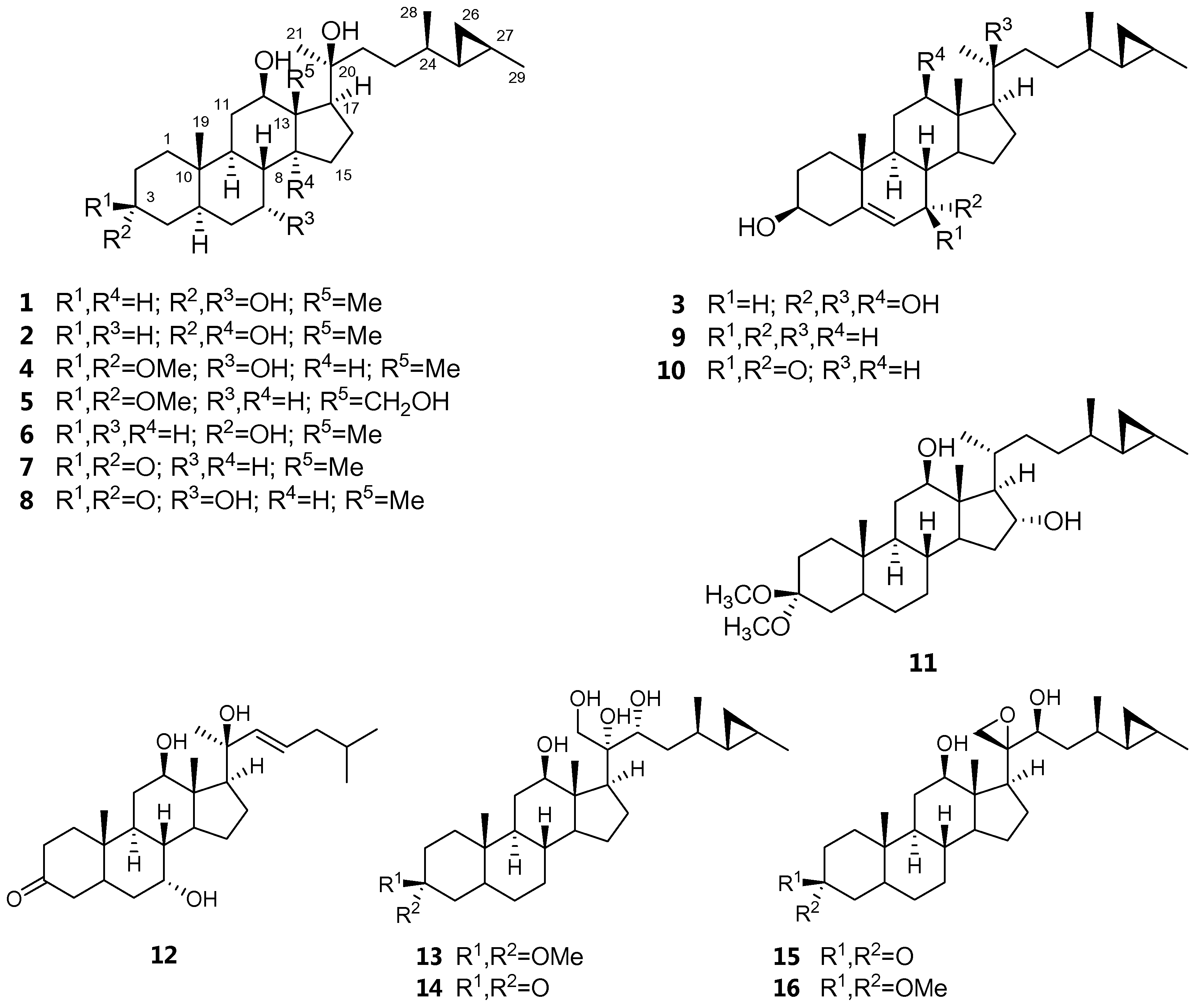
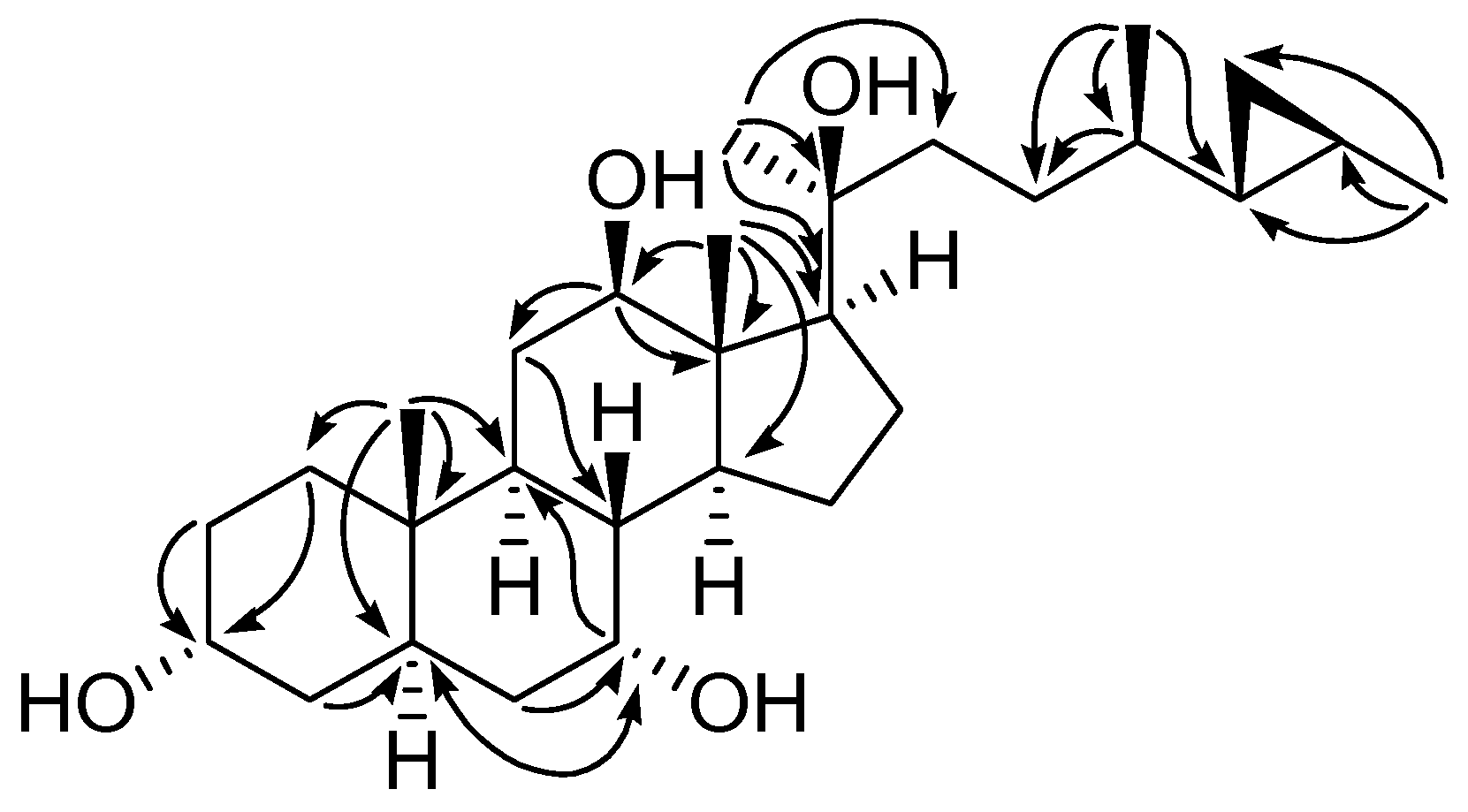
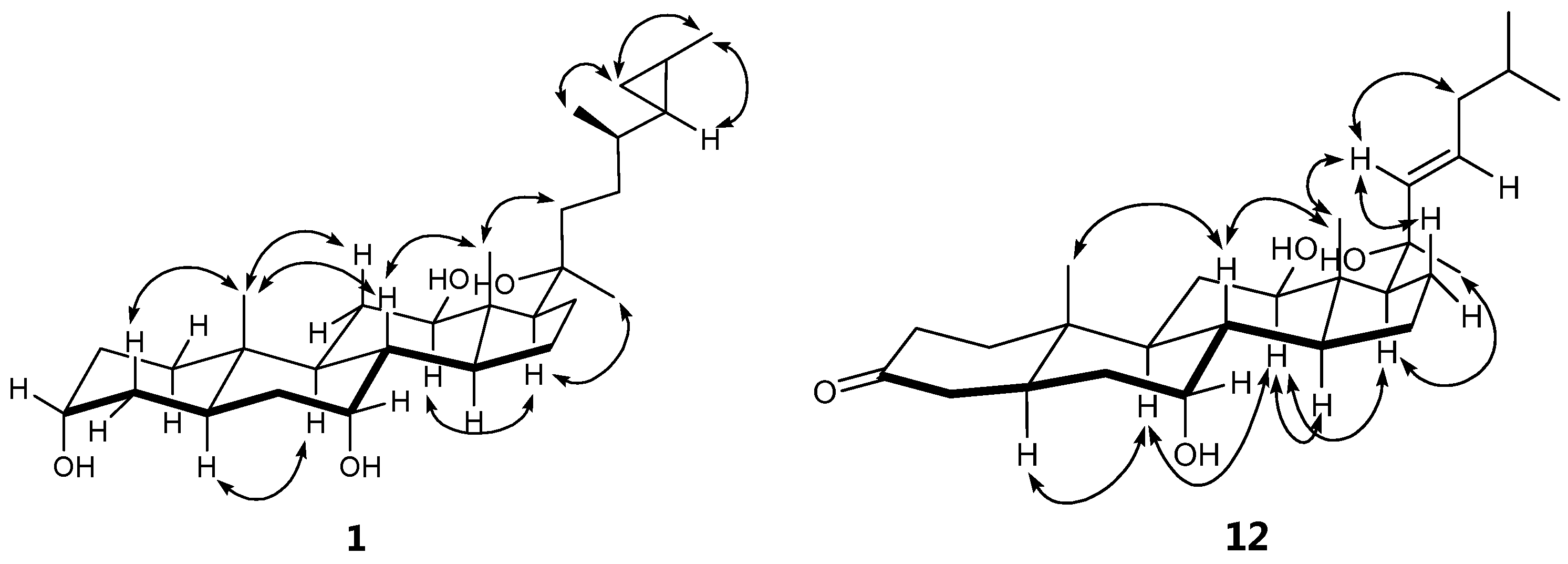
2. Results
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Animal Material
3.3. Extraction and Isolation
3.4. Cytotoxicity Assays
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mahidol, C.; Prawat, H.; Sangpetsiripan, S.; Ruchirawat, S. Bioactive scalaranes from the Thai sponge Hyrtios gumminae. J. Nat. Prod. 2009, 72, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Prawat, H.; Mahidol, C.; Kaweetripob, W.; Prachyawarakorn, V.; Tuntiwachwuttikul, P.; Ruchirawat, S. Sesquiterpene isocyanides, isothiocyanates, thiocyanates, and formamides from the Thai sponge Halichondria sp. Tetrahedron 2016, 72, 4222–4229. [Google Scholar] [CrossRef]
- Prawat, H.; Mahidol, C.; Kaweetripob, W.; Wittayalai, W.; Ruchirawat, S. Iodo–sesquiterpene hydroquinone and brominated indole alkaloids from the Thai sponge Smenospongia sp. Tetrahedron 2012, 68, 6881–6886. [Google Scholar] [CrossRef]
- Umeyama, A.; Ito, S.; Yoshigaki, A.; Arihara, S. Two new 26,27-cyclosterols from the marine sponge Strongylophora corticata. J. Nat. Prod. 2000, 63, 1540–1542. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.H.; Cross, S.S.; Gunasekera, M.; Koehn, F.E. Weinbersterol disulfates A and B, antiviral steroid sulfates from the sponge Petrosia weinbergi. Tetrahedron 1991, 47, 1185–1190. [Google Scholar] [CrossRef]
- Mattia, C.A.; Mazzarella, L.; Puliti, R.; Sica, D.; Zollo, F. X-ray crystal structure determination of petrosterol p-bromobenzoate. A revision. Tetrahedron Lett. 1978, 19, 3953–3954. [Google Scholar] [CrossRef]
- Gabriel, A.F.; Li, Z.; Kusuda, R.; Tanaka, C.; Miyamoto, T. Six new polyacetylenic alcohols from the marine sponges Petrosia sp. and Halichondria sp. Chem. Pharm. Bull. 2015, 63, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Tsuda, Y.; Hamada, M.; Omori, M.; Mori, G.; Iguchi, K.; Naoki, H.; Fujita, T.; Van Soest, R.W.M. Acetylenic strongylodiols from a Petrosia (Strongylophora) Okinawan marine sponge. J. Nat. Prod. 2005, 68, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Lim, Y.J.; Im, K.S.; Jung, J.H.; Shim, C.J.; Lee, C.O.; Hong, J.; Lee, H. Cytotoxic polyacetylenes from the marine sponge Petrosia sp. J. Nat. Prod. 1999, 62, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Hitora, Y.; Takada, K.; Okada, S.; Ise, Y.; Matsunaga, S. (−)-Duryne and its homologues, cytotoxic acetylenes from a marine Sponge Petrosia sp. J. Nat. Prod. 2011, 74, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, K.; Yagyu, T.; Yoshioka, Y.; Fujiwara, T.; Kanamoto, A.; Okamoto, T.; Ojika, M. Petrosiols A–E, neurotrophic diyne tetraols isolated from the Okinawan sponge Petrosia strongylata. Tetrahedron 2013, 69, 101–106. [Google Scholar] [CrossRef]
- Hitora, Y.; Takada, K.; Okada, S.; Matsunaga, S. Miyakosynes A–F, cytotoxic methyl branched acetylenes from a marine sponge Petrosia sp. Tetrahedron 2011, 67, 4530–4534. [Google Scholar] [CrossRef]
- Lee, J.-S.; Abdjul, D.B.; Yamazaki, H.; Takahashi, O.; Kirikoshi, R.; Ukai, K.; Namikoshi, M. Strongylophorines, new protein tyrosine phosphatase 1B inhibitors, from the marine sponge Strongylophora strongilata collected at Iriomote Island. Bioorg. Med. Chem. Lett. 2015, 25, 3900–3902. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Mitome, H.; Miyaoka, H.; Shintani, A.; Yamada, Y.; Van Soest, R.W.M. New strongylophorines from the Okinawan marine sponge Petrosia (Strongylophora) corticata. J. Nat. Prod. 2003, 66, 1600–1605. [Google Scholar] [CrossRef] [PubMed]
- Noda, A.; Sakai, E.; Kato, H.; Losung, F.; Mangindaan, R.E.P.; de Voogd, N.J.; Yokosawa, H.; Tsukamoto, S. Strongylophorines, meroditerpenoids from the marine sponge Petrosia corticata, function as proteasome inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 2650–2653. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, P.; Srinivasa Reddy, N.; Venkateswarlu, Y. A new 1,2-dihydroisoquinoline from the sponge Petrosia similis. J. Nat. Prod. 1999, 62, 780–781. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Takeuchi, T.; Kawabata, T.; Kato, H.; Yamakuma, M.; Matsuo, K.; El-Desoky, A.H.; Losung, F.; Mangindaan, R.E.P.; de Voogd, N.J.; et al. Halenaquinone inhibits RANKL-induced osteoclastogenesis. Bioorg. Med. Chem. Lett. 2014, 24, 5315–5317. [Google Scholar] [CrossRef] [PubMed]
- Nukoolkarn, V.S.; Saen-oon, S.; Rungrotmongkol, T.; Hannongbua, S.; Ingkaninan, K.; Suwanborirux, K. Petrosamine, a potent anticholinesterase pyridoacridine alkaloid from a Thai marine sponge Petrosia n. sp. Bioorg. Med. Chem. 2008, 16, 6560–6567. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Chen, Y.-J.; Higuchi, K.; Aoki, S.; Kitagawa, I. Marine natural products. XXXVII. Aragusterolketals A and C, two novel cytotoxic steroids from a marine sponge of Xestospongia sp. Chem. Pharm. Bull. 1996, 44, 1840–1842. [Google Scholar] [CrossRef]
- Iguchi, K.; Fujita, M.; Nagaoka, H.; Mitome, H.; Yamada, Y. Aragusterol A: A potent antitumor marine steroid from the Okinawan sponge of the genus, Xestospongia. Tetrahedron Lett. 1993, 34, 6277–6280. [Google Scholar] [CrossRef]
- Kobayashi, J.; Ishida, K.; Naitoh, K.; Shigemori, H. Xestokerols A, B, and C, new C29 steroids with a cyclopropane ring from the Okinawan marine sponge Xestospongia sp. J. Nat. Prod. 1993, 56, 1350–1355. [Google Scholar] [CrossRef]
- Nguyen, X.C.; Longeon, A.; Pham, V.C.; Urvois, F.; Bressy, C.; Trinh, T.T.V.; Nguyen, H.N.; Phan, V.K.; Chau, V.M.; Briand, J.-F.; et al. Antifouling 26,27-cyclosterols from the Vietnamese marine sponge Xestospongia testudinaria. J. Nat. Prod. 1993, 76, 1313–1318. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.H.; Minh, C.V.; Kiem, P.V.; Huong, H.T.; Ha, T.T.; Dat, N.T.; Nhiem, N.X.; Cuong, N.X.; Hyun, J.-H.; Kang, H.-K.; et al. A new C29-sterol with a cyclopropane ring at C-25 and 26 from the Vietnamese marine sponge Lanthella sp. Arch. Pharm. Res. 2009, 32, 1695–1698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Timmermann, B.N. Withanolide structural revisions by 13C NMR spectroscopic analysis inclusive of the γ-gauche effect. J. Nat. Prod. 2016, 79, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Mandeau, A.; Debitus, C.; Ariès, M.F.; David, B. Isolation and absolute configuration of new bioactive marine steroids from Euryspongia n. sp. Steroids 2005, 70, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.; Griffiths, J.B. (Eds.) Mammalian Cell Culture: Essential Techniques; John Wiley and Sons: New York, NY, USA, 1997.
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936–942. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Position | δH, mult. (J in Hz) | |||
|---|---|---|---|---|
| 1 a | 2 b | 3 a | 4 b | |
| 1 | 1.50, m | 1.39, m | 1.06, m | 1.23, ddd (13.6, 13.6, 3.6) |
| 1.53, m | 1.80, m | 1.75, m | ||
| 2 | 1.28, m | 1.29, m | 1.81 c, m | 1.46, m |
| 1.82, m | 1.64, m | 1.90, m | 1.89, m | |
| 3 | 4.27, s | 4.05, s | 3.76, dddd (10.6, 10.6, 5.6, 5.6) | |
| 4 | 1.61, m | 1.40 c, m | 2.63, t (12.1) | 1.37, m |
| 1.50, m | 2.69, dd (13.0, 4.4) | 1.69, m | ||
| 5 | 1.92, m | 1.56, m | 2.30, t (13.3) | |
| 6 | 1.65, m | 1.40 c, m | 5.91, d (4.9) | 1.52 c, m |
| 1.64, m | 1.62, m | |||
| 7 | 4.13, s | 1.26, m | 4.15, s | 4.06, s |
| 1.56, m | ||||
| 8 | 1.59, m | 1.79, m | 1.61, m | 1.51 c, m |
| 9 | 2.92, m | 1.40 c, m | 1.85, m | 1.75, m |
| 11 | 1.63, m | 1.27, m | 1.80 c, m | 1.57, m |
| 2.10, m | 1.77, m | 2.07, m | 2.00, m | |
| 12 | 3.76, dd (10.8, 2.6) | 3.97, dd (11.0, 4.9) | 3.84, dd (10.9, 4.1) | 3.72, dd (10.9, 4.1) |
| 14 | 1.93, m | 2.05 d, m | 1.85, m | |
| 15 | 1.43, m | 1.47, m | 1.48, m | 1.40, m |
| 2.12, m | 1.62, m | 2.25, m | 2.10, m | |
| 16 | 1.67, m | 1.53, m | 1.70, m | 1.63, m |
| 1.78, m | 1.82, m | 1.82 c, m | 1.76, m | |
| 17 | 1.99, m | 2.62, t (9.9) | 2.05 d, m | 1.95, m |
| 18 | 1.23, s | 0.92, s | 1.22, s | 1.17, s |
| 19 | 0.91, s | 0.82, s | 1.07, s | 0.83, s |
| 20 | ||||
| 21 | 1.35, s | 1.37, s | 1.32, s | |
| 22 | 1.78, m | 1.60, m | 1.80 c, m | 1.52, m |
| 2.12, m | 1.81, m | 2.15, t (12.9) | 2.11, m | |
| 23 | 1.78, m | 1.29, m | 1.49, m | 1.48, m |
| 1.97, m | 1.54, m | 2.00, m | 1.97, m | |
| 24 | 0.70, m | 0.65, m | 0.72, m | 0.69, m |
| 25 | 0.20 c, m | 0.16 d, m | 0.21 e, m | 0.20 d, m |
| 26 | 0.10, m | 0.10, m | 0.11, m | 0.10, m |
| 0.20 c, m | 0.16 d, m | 0.21 e, m | 0.20 d, m | |
| 27 | 0.54, m | 0.49, m | 0.54, m | 0.53, m |
| 28 | 1.02, d (6.6) | 0.92, d (6.0) | 1.03, d (6.2) | 1.01, d (6.7) |
| 29 | 1.06, d (5.7) | 1.03, d (6.0) | 1.06, d (6.0) | |
| 3-OMe | 3.07, s | |||
| 3-OMe | 3.16, s | |||
| Position | δC | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 a | 2 b | 3 a | 4 b | 5 a | 11 b | 12 a | 13 b | |
| 1 | 32.9 | 32.3 | 37.6 | 35.4 | 34.9 | 35.4 | 38.7 | 35.0 |
| 2 | 29.9 | 29.0 | 32.4 | 28.9 | 28.25 | 28.3 | 38.4 | 28.3 c |
| 3 | 65.8 | 66.4 | 71.1 | 100.7 | 100.3 | 100.3 | 210.3 | 100.3 |
| 4 | 36.75 | 35.8 | 43.3 | 35.8 | 35.3 | 34.9 | 44.66 | 35.4 |
| 5 | 45.6 | 38.9 | 145.1 | 35.23 | 42.3 | 42.4 | 39.7 | 42.4 |
| 6 | 38.0 | 28.33 | 125.5 | 37.6 | 28.34 | 28.4 | 38.0 | 28.4 c |
| 7 | 67.0 | 26.5 | 64.6 | 66.7 | 31.6 | 31.5 | 66.3 | 31.4 |
| 8 | 39.1 | 36.8 | 37.1 | 39.0 | 34.6 | 33.7 | 38.8 | 33.9 |
| 9 | 32.1 | 46.4 | 42.6 | 45.2 | 53.1 | 52.8 | 44.71 | 52.7 |
| 10 | 36.79 | 36.3 | 37.9 | 36.3 | 35.7 | 35.7 | 36.1 | 35.7 |
| 11 | 29.7 | 28.27 | 30.0 | 30.0 | 31.2 | 31.1 | 30.3 | 29.1 |
| 12 | 78.4 | 71.8 | 78.1 | 78.2 | 80.6 | 79.8 | 78.0 | 78.0 |
| 13 | 49.1 | 52.7 | 48.8 | 49.0 | 50.5 | 49.3 | 49.1 | 49.3 |
| 14 | 50.0 | 87.2 | 49.1 | 49.8 | 54.9 | 51.3 | 49.5 | 54.5 |
| 15 | 23.5 | 33.1 | 24.0 | 23.5 | 23.4 | 36.1 c | 23.6 | 22.8 |
| 16 | 25.6 | 24.0 | 25.8 | 25.6 | 23.9 | 74.2 | 25.1 | 23.5 |
| 17 | 66.0 | 57.6 | 65.8 | 65.9 | 61.2 | 67.9 | 64.1 | 54.2 |
| 18 | 10.2 | 12.4 | 10.2 | 10.2 | 60.8 | 9.1 | 10.0 | 9.7 |
| 19 | 10.6 | 10.9 | 18.5 | 10.9 | 11.58 | 11.49 | 10.4 | 11.5 |
| 20 | 74.4 | 75.5 | 74.5 | 74.4 | 74.6 | 32.3 | 73.9 | 76.3 |
| 21 | 28.6 | 28.9 | 28.6 | 28.6 | 29.0 | 22.5 | 31.6 | 66.4 |
| 22 | 35.2 | 34.4 | 35.2 | 35.15 | 37.0 | 32.7 | 137.6 | 75.1 |
| 23 | 32.5 | 31.9 | 32.5 | 32.5 | 32.0 | 36.2 c | 125.8 | 37.7 |
| 24 | 39.5 | 39.0 | 39.5 | 39.5 | 38.8 | 38.7 | 42.4 | 34.7 |
| 25 | 27.7 | 27.1 | 27.7 | 27.7 | 27.1 | 27.3 | 28.9 | 27.9 |
| 26 | 11.9 | 11.5 | 11.9 | 11.9 | 11.64 | 11.53 | 22.5 c | 12.5 |
| 27 | 13.2 | 12.8 | 13.2 | 13.2 | 12.7 | 12.7 | 22.8 c | 12.3 |
| 28 | 20.6 | 20.1 | 20.6 | 20.7 | 19.8 | 19.9 | 18.7 | |
| 29 | 19.3 | 19.1 | 19.3 | 19.1 | 19.1 | 19.1 | ||
| 3-OMe | 47.3 | 47.45 | 47.4 | 47.38 d | ||||
| 3-OMe | 47.4 | 47.51 | 47.5 | 47.43 d | ||||
| Position | δH, mult. (J in Hz) | |||
|---|---|---|---|---|
| 5 a | 11 b | 12 a | 13 b | |
| 1 | 1.11, ddd (13.6, 13.6, 3.7) | 1.30, m | 1.30, ddd (13.4, 13.4, 4.8) | 1.05, ddd (13.6, 13.6, 3.7) |
| 1.62, m | 1.55, m | 1.88, m | 1.60, m | |
| 2 | 1.48, m | 1.40, m | 2.28, m | 1.43, ddd (14.0, 14.0, 4.0) |
| 1.89, ddd (12.9, 12.9, 3.8) | 1.89, dq (14.1, 3.2) | 2.36, m | 1.88, br dd (2.9, 14.0) | |
| 4 | 1.30, m | 1.12, m | 2.12, m | 1.28, m |
| 2.32, m | ||||
| 5 | 1.31, m | 1.30, m | 2.43, m | 1.29, m |
| 6 | 1.28, m | 1.20, m | 1.56, ddd (13.7, 13.7, 2.0) | 1.25, m |
| 1.28, m | 1.65, m | |||
| 7 | 0.81, m | 0.88, m | 4.07, brs | 0.88, m |
| 1.65, m | 1.64, m | 1.67, m | ||
| 8 | 1.32, m | 1.31, m | 1.51, ddd (11.2, 11.2, 2.2) | 1.30, m |
| 9 | 0.85, m | 0.88, m | 1.76, m | 0.84, m |
| 11 | 1.57, m | 1.25, m | 1.65, m | 1.26, m |
| 1.93, ddd (13.6, 4.0, 4.0) | 1.67, m | 2.04, m | 1.73, m | |
| 12 | 3.50, dd (11.0, 4.0) | 3.53, dd (10.9, 4.5) | 3.75, dd (10.9, 4.3) | 3.38, dd (11.0, 4.4) |
| 14 | 1.02, m | 1.30, m | 1.87, m | 1.76, m |
| 15 | 1.01, m | 1.42, m | 1.40, m | 1.50, m |
| 1.66, m | 1.67, m | 2.10, m | 1.75, m | |
| 16 | 1.75, m | 4.33, t (7.4) | 1.76, m | 1.20, m |
| 1.69, m | ||||
| 17 | 1.75, m | 1.40, m | 2.03, m | 0.96, m |
| 18 | 3.65, d (12.0) | 0.71, s | 1.07, s | 0.80, s |
| 4.02, d (12.0) | ||||
| 19 | 0.84, s | 0.79, s | 1.00, s | 0.80, s |
| 20 | 1.85, m | |||
| 21 | 1.44, s | 1.09, d (6.9) | 1.45, s | 3.83, d (11.5) |
| 3.98, d (11.5) | ||||
| 22 | 1.61, m | 0.88, m | 6.00, d (15.5) | 3.82, d (10.6) |
| 1.90, m | 1.26, m | |||
| 23 | 1.28, m | 1.44, m | 5.94, ddd (15.0, 7.5, 7.5) | 1.35, m |
| 1.47, m | 1.66, m | 1.67, m | ||
| 24 | 0.70, m | 0.65, m | 1.99, m | 0.93, m |
| 2.03, m | ||||
| 25 | 0.19, m | 0.16 c, m | 1.66, m | 0.25, m |
| 26 | 0.11, m | 0.13 c, m | 0.92, d (6.6) | 0.16, m |
| 0.18, m | 0.25, m | |||
| 27 | 0.51, m | 0.46, m | 0.91, d (6.6) | 0.53, m |
| 28 | 0.93, d (6.7) | 0.91, d (6.7) | 0.95, d (6.0) | |
| 29 | 1.02, d (6.0) | 1.00, d (6.0) | 1.03, d (6.0) | |
| 3-OMe | 3.14, s | 3.14, s | 3.14, s | |
| 3-OMe | 3.19, s | 3.19, s | 3.19, s | |
| Compounds | Cell Lines (IC50, µM); Values Are Expressed as Mean ± S.D. (n = 3). | ||||||
|---|---|---|---|---|---|---|---|
| MOLT-3 | HepG-2 | A549 | HuCCA-1 | HeLa | MDA-MB-231 | MRC-5 | |
| 1 | 17.86 ± 0.26 | 12.71 ± 1.67 | 20.04 ± 1.52 | 21.32 ± 1.84 | ND | ND | 40.13 ± 1.23 |
| 2 | 32.36 ± 1.08 | 44.78 ± 3.79 | 38.59 ± 0.24 | 37.92 ± 3.01 | ND | ND | 60.61 ± 12.75 |
| 3 | 8 (27.17) a | 44.61 ± 9.76 | 9 (54.35) a | 47.48 ± 6.96 | ND | ND | 24.70 (54.35) a |
| 4 | 0 (12.35) a | 20.32 (24.70) a | 2 (12.35) a | 0 (12.35) a | ND | ND | 5.45 (24.70) a |
| 6 | 20.07 ± 0.52 | 54.89 ± 2.04 | 34.73 ± 17.71 | 37.06 ± 0.36 | ND | ND | 63.36 ± 8.30 |
| 7 | 16.33 ± 18.02 | 26.46 ± 5.32 | 23.58 ± 2.00 | 25.07 ± 2.27 | 11.23 ± 0.05 | 27.03 ± 7.79 | ND |
| 8 | 43 (108.70) a | 18.23 ± 1.86 | 32.62 ± 3.64 | 103.35 ± 2.29 | ND | ND | 90.11 ± 6.88 |
| 10 | 2 (29.34) a | 49.75 ± 3.96 | 52.15 ± 3.35 | 44 (58.69) a | ND | ND | 44.77 ± 1.36 |
| 12 | 36.57 ± 1.11 | 56.50 ± 2.15 | 54.26 ± 3.84 | 66.11 ± 0.90 | ND | ND | 76.94 ± 10.65 |
| 13 | 14.90 ± 0.25 | 12.53 ± 0.84 | 17.91 ± 2.26 | 20.79 ± 2.32 | ND | ND | 37.68 ± 10.65 |
| 14 | 24.45 ± 3.11 | 41.41 ± 3.24 | 23.76 ± 1.87 | 34.41 ± 1.83 | ND | ND | 65.90 ± 4.54 |
| 15 | 12.84 ± 0.98 | 7.10 ± 4.76 | 37.93 ± 0.07 | 37.58 ± 1.40 | 6.11 ± 0.02 | 18.01 ± 5.74 | ND |
| 16 | 14.58 ± 0.36 | 11.31 ± 9.29 | 29.11 ± 8.25 | 34.60 ± 3.85 | ND | ND | 49.60 ± 6.67 |
| Doxorubicin b | ND | 0.55 ± 0.12 | 0.92 ± 0.06 | 2.24 ± 0.15 | 0.17 ± 0.10 | 1.78 ± 0.54 | 24.85 (50.00) a |
| Etoposide b | 0.07 ± 0.005 | 31.50 ± 15.56 | ND | ND | ND | ND | ND |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pailee, P.; Mahidol, C.; Ruchirawat, S.; Prachyawarakorn, V. Sterols from Thai Marine Sponge Petrosia (Strongylophora) sp. and Their Cytotoxicity. Mar. Drugs 2017, 15, 54. https://doi.org/10.3390/md15030054
Pailee P, Mahidol C, Ruchirawat S, Prachyawarakorn V. Sterols from Thai Marine Sponge Petrosia (Strongylophora) sp. and Their Cytotoxicity. Marine Drugs. 2017; 15(3):54. https://doi.org/10.3390/md15030054
Chicago/Turabian StylePailee, Phanruethai, Chulabhorn Mahidol, Somsak Ruchirawat, and Vilailak Prachyawarakorn. 2017. "Sterols from Thai Marine Sponge Petrosia (Strongylophora) sp. and Their Cytotoxicity" Marine Drugs 15, no. 3: 54. https://doi.org/10.3390/md15030054
APA StylePailee, P., Mahidol, C., Ruchirawat, S., & Prachyawarakorn, V. (2017). Sterols from Thai Marine Sponge Petrosia (Strongylophora) sp. and Their Cytotoxicity. Marine Drugs, 15(3), 54. https://doi.org/10.3390/md15030054