4.2. Synthesis of the Right-Hand Dipeptide Fragment
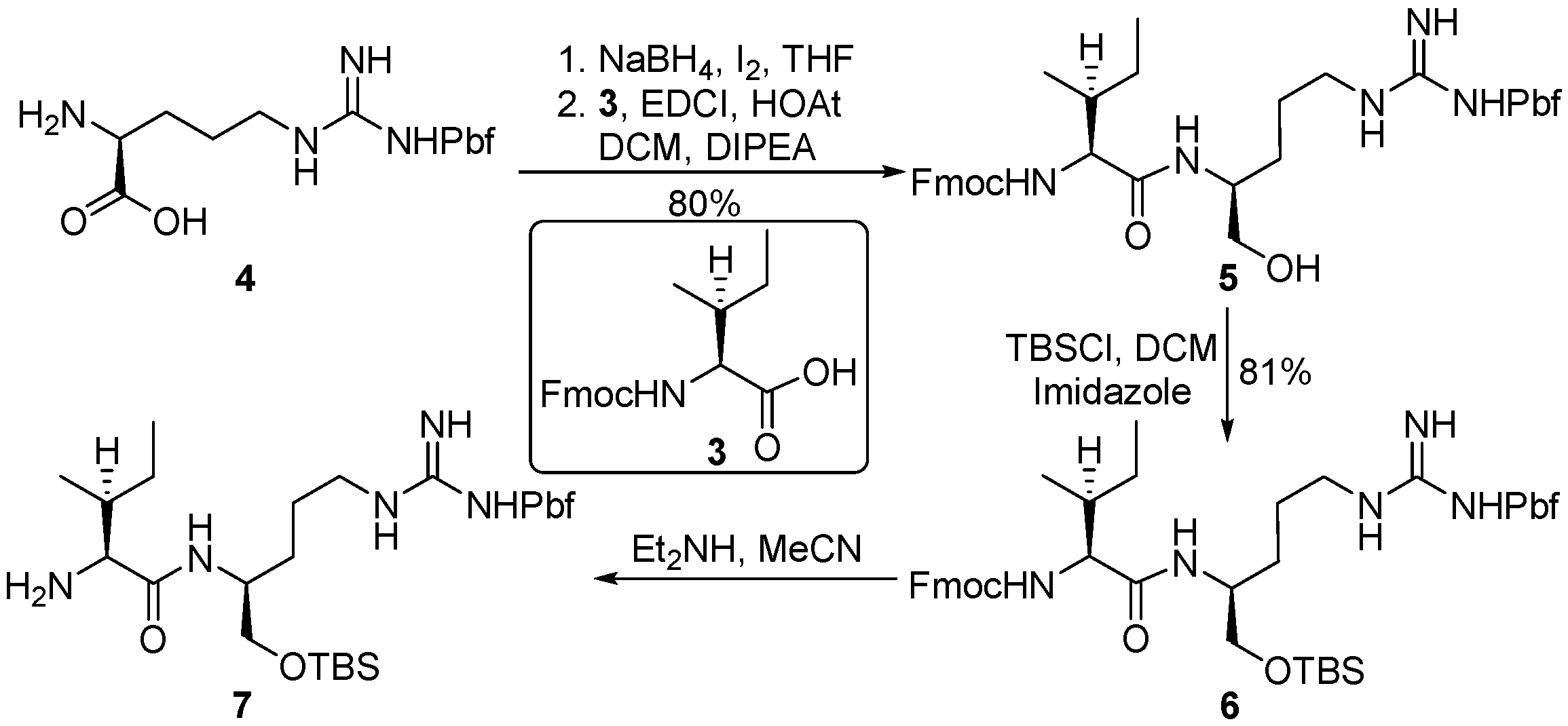
NaBH4 (2.13 g, 56.27 mmol) was suspended in THF (100 mL) at 0 °C, l-Pbf-Arg 4 (10.00 g, 23.45 mmol) was added in one portion. A solution of I2 (5.95 g, 23.45 mmol) in THF (20 mL) was dropwise added to the above amino acid solution within 0.5 h. After gas evolution ceased, the reaction mixture was heated to reflux for 18 h. The reaction temperature was cooled to room temperature, and methanol was carefully added to quench the reaction until the reaction solution became clear. Stir was continued for 0.5 h, the solution was concentrated in vacuo. The residue was dissolved by aqueous solution of KOH (50 mL, 20% in water) and stirred for 4 h, then the solution was extracted by CH2Cl2 (100 mL × 3). The combined organic layers were washed with water (50 mL), dried over anhydrous sodium sulfate and concentrated in vacuo to afford the amino alcohol (6.28 g, 65%) as crude oil.
To a solution of amino alcohol (0.58 g, 1.42 mmol) in CH2Cl2 (20 mL), Fmoc-Ile-OH 3 (0.50 g, 1.42 mmol), HOAt (0.29 g, 2.12 mmol), and EDCI (0.27 g, 1.42 mmol) were sequentially added at 0 °C, after DIPEA (0.47 mL, 2.83 mmol) was added via a syringe, the reaction mixture was brought to room temperature and stirred for 16 h. Volatiles were removed in vacuo. The residue was dissolved in ethyl acetate (150 mL) and washed with saturated aqueous solution of NH4Cl (30 mL), NaHCO3 (30 mL), and brine (30 mL). The organic phase was dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was purified by flash chromatography to give dipeptide 5 (0.84 g, 80%) as yellow oil.
Compound 5 (0.53 g, 0.70 mmol) was dissolved in DCM (5 mL) at 0 °C, after imidazole (0.24 g, 3.5 mmol) and TBSCl (0.21 g, 1.40 mmol) were added, the reaction mixture was brought to room temperature and stirred for 16 h. The reaction mixture was diluted by DCM (50 mL), the organic solution was washed with saturated aqueous solution of NH4Cl (30 mL), brine (30 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was purified by flash chromatography to afford compound 6 (0.49g, 81%). [α] = −28.7 (c 1.0, CH2Cl2); 1H-NMR (500 MHz, CDCl3) δ 7.72–7.70 (m, 2H), 7.53–7.51 (m, 2H), 7.37–7.32 (m, 2H), 7.25–7.22 (m, 2H), 6.45 (br, 1H), 6.13 (br, 2H), 5.63 (br, 1H), 4.41–4.37 (dd, J = 7.5, 10.5 Hz, 1H), 4.26–4.22 (m, 1H), 4.14–4.10 (m, 1H), 4.03–3.90 (m, 1H), 3.52 (s, 2H), 3.18 (br, 1H), 3.10 (br, 1H), 2.89 (s, 2H), 2.54 (s, 3H), 2.48 (s, 3H), 2.04 (s, 3H), 1.88–1.78 (m, 2H), 1.57–1.44 (m, 4H), 1.41 (s, 6H), 1.16–1.12 (m, 1H), 0.90–0.88 (m, 6H), 0.85 (s, 9H), 0.01 (s, 6H). 13C-NMR (125 MHz, CDCl3) δ 171.79, 158.74, 156.68, 156.22, 143.94, 143.69, 141.38, 138.45, 132.40, 127.82, 127.21, 125.11, 124.58, 120.07, 117.46, 88.34, 67.26, 65.02, 60.03, 47.22, 43.35, 41.22, 37.50, 29.15, 28.65, 25.94, 25.75, 25.33, 25.10, 19.30, 18.34, 17.95, 15.54, 12.50, 11.46, −5.42, −5.44 ppm; HRMS (ESI) calculated for C17H20O3 [M + Na]+ 884.4530, found 884.4434.
Compound 6 (55 mg, 0.064 mmol) was dissolved in CH3CN (2 mL) and cooled to 0 °C, after diethylamine (0.07 mL, 0.64 mmol) was added, the reaction mixture was brought to room temperature and monitored by TLC. Upon the consumption of all starting materials, the reaction mixture was concentrated in vacuo. The residue was dissolved in DCM (2 mL) and concentrated in vacuo, these procedures were repeated twice. The residue was dried under high vacuum for 1 h to give the crude amine 7, which was used directly without further purification.
4.3. Synthesis of the Hhpba Fragment
4.3.1. Synthesis of s-Hhpba (S-2)
Compound
15 [
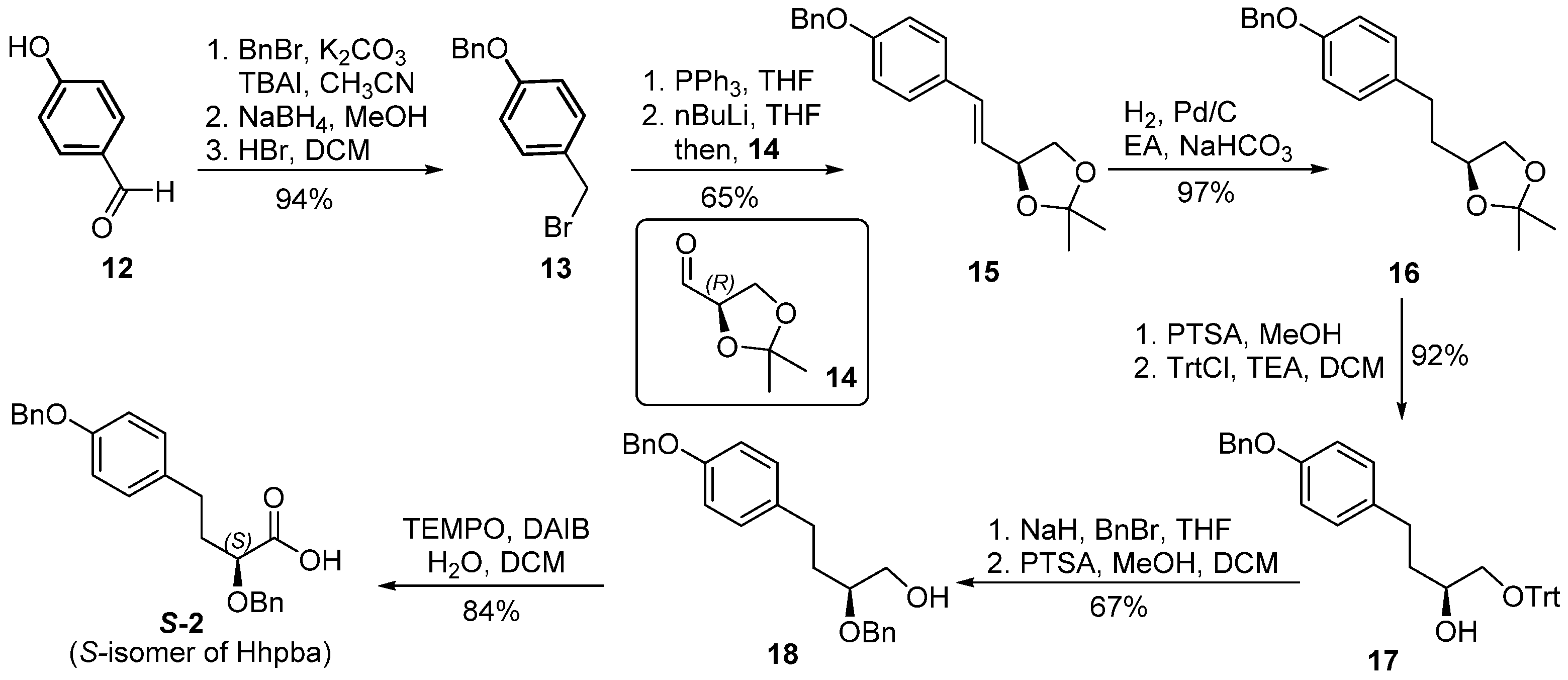
39] (3.55 g, 11.42 mmol) was dissovled in ethyl acetate (50 mL), Pd-C (355 mg, 10% on charcoal) and sodium bicarbonate (10.00 g, 114.24 mmol) were added. The reaction vessel was sealed and the atmosphere was changed to hydrogen. The reaction mixture was stirred at room temperature under hydrogen (balloon) atmosphere and monitored by thin layer chromatography. Upon completion of the starting material, the reaction mixture was filtered through a pad of silica gel and eluted with ethyl acetate (20 mL × 2). The combined organic filtrate was washed with brine (30 mL), dried over anhydrous sodium sulfate and concentrated in vacuo to afford compound
16 (3.46 g, 97%) as an oil. An analytical sample was obtained by chromatography. [α]
= 1.5 (
c 1.1, CHCl
3);
1H-NMR (500 MHz, CDCl
3) δ 7.48–7.35 (m, 5H), 7.16 (d,
J = 8.0 Hz, 2H), 6.95 (d,
J = 8.0 Hz, 2H), 5.07 (s, 2H), 4.19–4.08 (m, 1H), 4.04 (dd,
J = 7.9, 5.8 Hz, 1H), 3.56 (t,
J = 7.5 Hz, 1H), 2.79–2.60 (m, 2H), 2.04–1.73 (m, 2H), 1.49 (s, 3H), 1.41 (s, 3H).
13C-NMR (125 MHz, CDCl
3) δ 157.16, 137.21, 133.92, 129.34, 128.61, 127.95, 127.51, 114.84, 108.74, 75.41, 70.05, 69.40, 35.59, 31.19, 27.08, 25.83 ppm; HRMS (ESI) calculated for C
20H
24O
3 [M + Na]
+ 335.1725, found 335.1744.
Compound 16 (0.50 g, 1.60 mmol) was dissolved in methanol (10 mL) and cooled to 0 °C, after PTSA (30 mg, 0.16 mmol) was added, the reaction mixture was stirred at room temperature for 16 h. The reaction solution was concentrated in vacuo, the residue was dissolved in ethyl acetate (50 mL) and washed with saturated aqueous solution of sodium bicarbonate (50 mL) and brine (50 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was purified by flash chromatography to afford the desired diol compound S1 (0.43 g, 99%) as clear oil. [α] = −14.7 (c 1.0, MeOH); 1H-NMR (500 MHz, CDCl3) δ 7.47–7.29 (m, 5H), 7.12 (d, J = 8.5 Hz, 2H), 6.91 (d, J = 8.6 Hz, 2H), 5.04 (s, 2H), 3.78–3.57 (m, 2H), 3.46 (dd, J = 11.1, 7.6 Hz, 1H), 2.80–2.55 (m, 2H), 2.28 (s, 1H), 2.08 (s, 1H), 1.72 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 157.16, 137.20, 134.01, 129.33, 129.31, 128.57, 128.55, 127.89, 127.47, 127.44, 114.92, 114.88, 77.27, 77.01, 76.76, 71.52, 70.12, 66.82, 34.87, 30.90 ppm; HRMS (ESI) calculated for C17H20O3 [M + Na]+ 295.1412, found 295.1456.
Diol compound S1 (136 mg, 0.50 mmol) was dissolved in DCM (5 mL) and cooled to 0 °C, after triethyl amine (0.35 mL, 2.5 mmol) and triphenylmethyl chloride (0.15 g, 0.53 mmol) were added, the reaction mixture was stirred at room temperature for 4 h. The reaction was diluted with DCM (50 mL) and extracted with brine (50 mL). Layers were separated, the organic phase was dried over anhydrous sodium sulfate, concentrated in vacuo, and purified by flash chromatography to afford compound 17 (0.24 g, 93%) as clear oil. [α] = 2.7 (c 1.0, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.52–7.21 (m, 20H), 7.09 (d, J = 8.5 Hz, 2H), 6.91 (d, J = 8.5 Hz, 2H), 5.06 (s, 2H), 3.81 (s, 1H), 3.21 (dd, J = 9.4, 3.3 Hz, 1H), 3.09 (dd, J = 9.4, 7.3 Hz, 1H), 2.74–2.52 (m, 2H), 2.36 (s, 1H), 1.83–1.64 (m, 2H). 13C-NMR (125 MHz, CDCl3) δ 157.08, 143.90, 137.30, 134.31, 129.37, 128.72, 128.59, 127.90, 127.48, 127.15, 114.83, 86.73, 70.23, 70.12, 67.71, 35.20, 30.83 ppm; HRMS (ESI) calculated for C36H34O3 [M + Na]+ 537.2508, found 537.2553.
To a solution of compound 17 (0.43 g, 0.84 mmol) dissolved in THF (3 mL), NaH (0.1g, 4.17 mmol) was added at 0 °C. The reaction mixture was stirred at 0 °C for 0.5 h, then benzyl bromide (0.15 mL, 1.25 mmol) was added. Upon completion of the addition, the reaction mixture was brought to room temperature and stirred for 16 h. The reaction was quenched by saturated aqueous solution of NH4Cl (5 mL). Volatiles were removed in vacuo. The residue was extracted with ethyl acetate (50 mL × 2). The combined organic phases were washed with brine (50 mL), dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was purified by flash chromatography to give the desired fully protected compound S2 (0.48 g, 95%) as clear oil. [α] = −2.3 (c 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.56–7.17 (m, 25H), 7.09 (d, J = 8.5 Hz, 2H), 6.91 (d, J = 8.5 Hz, 2H), 5.04 (s, 2H), 4.71 (d, J = 11.6 Hz, 1H), 4.50 (d, J = 11.6 Hz, 1H), 3.54 (dt, J = 9.2, 4.7 Hz, 1H), 3.21 (qd, J = 9.9, 4.8 Hz, 2H), 2.67–2.48 (m, 2H), 1.93–1.80 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ 156.95, 144.13, 138.88, 137.24, 134.58, 129.32, 128.75, 128.56, 128.35, 127.88, 127.77, 127.47, 126.94, 114.71, 86.61, 77.73, 72.04, 70.06, 65.79, 34.02, 30.71 ppm; HRMS (ESI) calculated for C43H40O3 [M + Na]+ 627.2977, found 627.2941.
The above intermediate S2 (0.50 g, 0.83 mmol) was dissolved in methanol–DCM (4 mL, 1:1) and cooled to 0 °C. After PTSA (16 mg, 0.08 mmol) was added, the reaction mixture was stirred at room temperature for 4 h. Then the reaction was concentrated in vacuo. The residue was purified by flash chromatography to produce compound 18 (0.21 g, 71%). [α] = 2.0 (c 1.1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.49–7.29 (m, 10H), 7.09 (d, J = 8.5 Hz, 2H), 6.91 (d, J = 8.5 Hz, 2H), 5.06 (s, 2H), 4.66–4.53 (m, 2H), 3.75 (dd, J = 11.2, 3.1 Hz, 1H), 3.64–3.48 (m, 2H), 2.66 (ddd, J = 8.8, 6.6, 4.7 Hz, 2H), 2.03–1.76 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ 157.08, 138.38, 137.18, 134.17, 129.27, 128.57, 128.53, 127.92, 127.86, 127.83, 127.48, 114.85, 78.96, 71.58, 70.08, 64.13, 32.81, 30.70 ppm; HRMS (ESI) calculated for C24H26O3 [M + Na]+ 385.1882, found 385.1920.
Compound 18 (0.21 g, 0.59 mmol) was dissolved in DCM–H2O (3 mL, 2:1) at 0 °C. DAIB (16 mg, 0.08 mmol) and TEMPO (5 mg, 0.29 mmol) were sequentially added, the reaction mixture was stirred in dark for 16 h at room temperature. The reaction was diluted by DCM (50 mL) and washed with brine (20 mL). The organic layer was separated, dried over sodium sulfate, and concentrated in vacuo. The residue was purified by flash chromatography to provide acid S-2 (0.19 g, 84%) as clear oil. [α] = −15.5 (c 1.0, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.47–7.30 (m, 10H), 7.09 (d, J = 8.5 Hz, 2H), 6.91 (d, J = 8.5 Hz, 2H), 5.04 (s, 2H), 4.74 (d, J = 11.5 Hz, 1H), 4.45 (d, J = 11.4 Hz, 1H), 3.98 (t, J = 6.2 Hz, 1H), 2.72 (ddt, J = 43.4, 13.9, 7.7 Hz, 2H), 2.14–2.07 (m, 2H). 13C-NMR (125 MHz, CDCl3) δ 177.06, 157.24, 137.23, 133.32, 129.46, 128.56, 128.53, 128.16, 128.07, 127.89, 127.46, 114.93, 72.58, 70.12, 34.49, 30.42 ppm; HRMS (ESI) calculated for C24H24O4 [M + H]+ 377.1675, found 377.1695.
4.3.2. Synthesis of r-Hhpba (21)
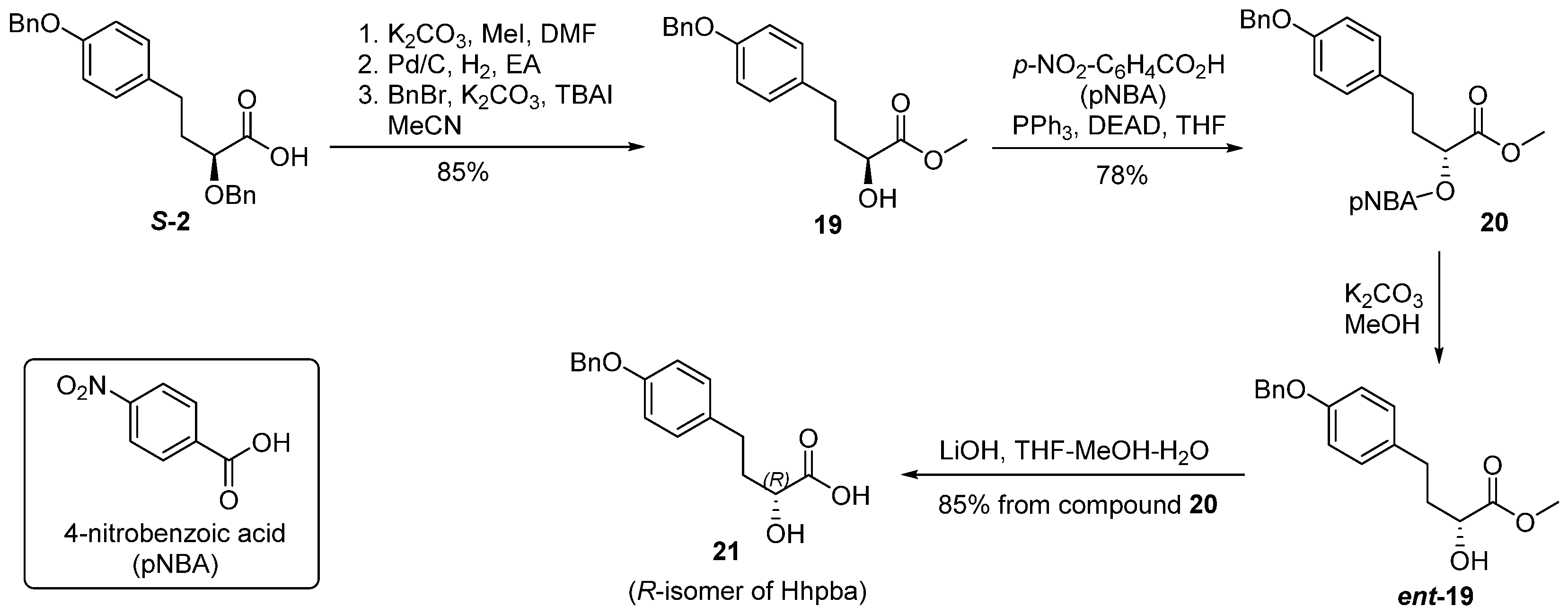
Compound S-2 (40 mg, 0.11 mmol) was dissolved in DMF (2 mL) at 0 °C, Cs2CO3 (30 mg, 0.21 mmol) and CH3I (33 μL, 0.53 mmol) were added at 0 °C. The reaction was brought to room temperature and stirred for 16 h. The reactant was diluted by ethyl acetate (50 mL). The organic solution was washed with brine (30 mL × 3), dried over anhydrous sodium sulfate, and concentrated in vacuo. The residue was purified by flash chromatography to give the corresponding methyl ester S3 (40 mg, 96%) as an oil. [α] = −19.2 (c 1.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.47–7.31 (m, 10H), 7.09 (d, J = 8.5 Hz, 2H), 6.91 (d, J = 8.5 Hz, 2H), 5.05 (s, 2H), 4.74 (d, J = 11.5 Hz, 1H), 4.42 (d, J = 11.5 Hz, 1H), 3.95 (dd, J = 8.2, 4.7 Hz, 1H), 3.74 (s, 3H), 2.81–2.63 (m, 2H), 2.12–2.02 (m, 2H). 13C-NMR (125 MHz, CDCl3) 173.28, 157.22, 137.59, 137.27, 133.44, 129.48, 128.57, 128.45, 128.15, 128.13, 127.90, 127.46, 114.91, 72.44, 70.12, 70.09, 51.84, 34.75, 30.52 ppm; HRMS (ESI) calculated for C25H26O4 [M + H]+ 391.1831, found 391.1889.
Methyl ester S3 (60 mg, 0.154 mmol) was dissolved in ethyl acetate (2 mL), after Pd/C (6 mg, 10% Pd on charcoal) was added under a protective flow of nitrogen, the reaction vessel was sealed and purged by hydrogen. The reaction mixture was stirred under hydrogen atmosphere (balloon) for 1 h. The reaction was monitored by TLC. Upon the complete consumption of starting material, the reaction solution was filtrated through a pad of celite and rinsed by ethyl acetate (5 mL). The combined filtrate was concentrated in vacuo to produce the desired compound S4 as colorless oil. An analytical sample was obtained via chromatography. [α] = 13.9 (c 1.0, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.09 (d, J = 8.5 Hz, 2H), 6.91 (d, J = 8.5 Hz, 2H), 5.80 (s, 1H), 4.21 (dd, J = 8.2, 4.1 Hz, 1H), 3.76 (s, 3H), 3.09 (d, J = 5.5 Hz, 1H), 2.75–2.58 (m, 2H), 2.25–1.83 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ 175.66, 153.88, 132.74, 129.56, 115.21, 69.60, 52.56, 35.93, 29.95 ppm; HRMS (ESI) calculated for C11H14O4 [M + Na]+ 233.0892, found 233.0823.
Intermediate S4, obtained from last step of reaction without further purification, was dissolved in acetonitrile (2 mL) at 0 °C. K2CO3 (30 mg, 0.21 mmol) was added to the solution, 10 min later, benzyl bromide (85 μL, 0.72 mmol) and TBAI (5 mg, 0.014 mmol) were sequentially added. The reaction mixture was stirred at room temperature for 16 h, and quenched by addition of saturated aqueous solution of NH4Cl (10 mL). Volatiles were removed in vacuo. The residue was extracted by ethyl acetate (30 mL × 2). The combined organic layers were washed with brine (20 mL), dried over anhydrous sodium sulfate, and concentrated in vacuo. The residue was purified by flash chromatography to afford compound 19 (40 mg, 88%). [α] = 10.9 (c 1.0, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.54–7.30 (m, 5H), 7.09 (d, J = 8.5 Hz, 2H), 6.91 (d, J = 8.5 Hz, 2H), 5.06 (s, 2H), 4.20 (dd, J = 8.0, 4.1 Hz, 1H), 3.76 (s, 3H), 2.91–2.60 (m, 3H), 2.22–1.80 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ 175.61, 157.06, 137.06, 133.26, 129.43, 128.47, 127.82, 127.37, 114.73, 69.95, 69.49, 52.46, 35.97, 29.99 ppm; HRMS (ESI) calculated for C18H20O4 [M + Na]+ 323.1362, found 323.1302.
Triphenylphosphine (40 mg, 0.133 mmol) was added to a solution of compound 19 (10 mg, 0.033 mmol) and 4-nitrobenzoic acid (11 mg, 0.066 mmol) dissolved in THF (2 mL) at 0 °C, after DEAD (10 μL, 0.066 mmol) was added, the reaction was stirred at room temperature for 16 h and then quenched by addition of saturated aqueous solution of NH4Cl (10 mL). Volatiles were removed in vacuo. The residue was extracted by ethyl acetate (30 mL × 2). The combined organic layers were washed with brine (20 mL), dried over anhydrous sodium sulfate, and concentrated in vacuo. The residue was purified by flash chromatography to produce ester 20 (12 mg, 78%) as an oil. 1H-NMR (300 MHz, CDCl3) δ 8.39–8.12 (m, 4H), 7.53–7.30 (m, 5H), 7.09 (d, J = 8.5 Hz, 2H), 6.91 (d, J = 8.5 Hz, 2H), 5.29 (t, J = 6.3 Hz, 1H), 5.05 (s, 2H), 3.78 (s, 3H), 2.80 (t, J = 7.6 Hz, 2H), 2.41–2.24 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ 169.98, 164.04, 157.32, 150.69, 136.89, 134.61, 132.25, 130.92, 129.30, 128.51, 127.90, 127.39, 123.50, 114.90, 72.80, 69.97, 52.47, 32.66, 30.54 ppm; HRMS (ESI) calculated for C25H23NO7 [M + Na]+ 472.1475, found 472.1515.
K2CO3 (6 mg, 0.044 mmol) was added to a solution of compound 20 (33 mg, 0.073 mmol) dissolved in methanol (2 mL) at 0 °C, the reaction mixture was then brought to room temperature and stirred for 10 h. Volatiles were removed in vacuo, the residue was dissolved in ethyl acetate (50 mL). The organic solution was washed with brine (30 mL), dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was purified by flash chromatography to afford the secondary alcohol compound ent-19 (20 mg, 85%). [α] = −10.2 (c 1.0, CHCl3).
LiOH (7 mg, 0.17 mmol) was added to a solution of the above secondary alcohol (10 mg, 33 μmol) dissolved in THF-MeOH-H2O (3 mL, v/v/v = 1/1/1) at 0 °C. The reaction mixture was stirred at room temperature for 3 h and concentrated in vacuo. The residue was diluted with water (5 mL) and extracted with Et2O (20 mL), the organic phase was discarded, the aqueous phase was adjusted to pH 3 with dilute HCl and extracted by ethyl acetate (30 mL × 3). The combined organic layers were washed with brine (30 mL), dried over anhydrous sodium sulfate, and concentrated in vacuo to give acid 21 (8 mg, 91%), which was ready for next step of transformation.
4.4. Synthesis of Nostosin B 1a and 1b
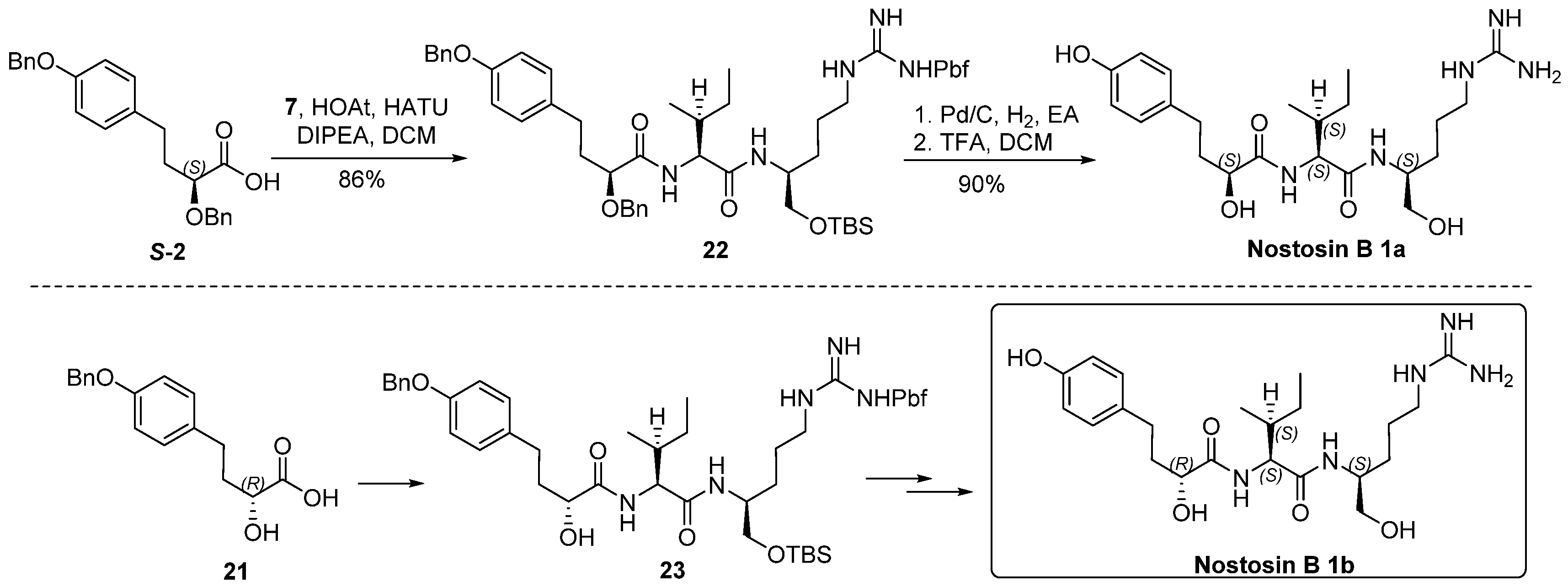
S-2 (20 mg, 0.053 mmol) and 7 (51 mg, 0.08 mmol) were dissolved in DCM (2 mL) at 0 °C, HOAT (14 mg, 0.11 mmol) and HATU (40 mg, 0.11 mmol) were added, followed by addition of DIPEA (44 μL, 0.26 mmol). The reaction mixture was warmed to room temperature and stirred for 16 h. Volatiles were removed in vacuo. The residue was dissolved in ethyl acetate (100 mL), washed with saturated aqueous solution of NH4Cl (30 mL), NaHCO3 (30 mL) and brine (30 mL), dried over Na2SO4, and concentrated in vacuo. The residue was purified by flash chromatography (EA/Hexanes) to provide tripeptide 22 (45 mg, 85%) as a viscous oil. [α] = −25.5 (c 1.0, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.51–7.27 (m, 10H), 7.20 (d, J = 8.5 Hz, 1H), 7.09 (d, J = 8.5 Hz, 2H), 6.91 (d, J = 8.5 Hz, 2H), 6.41 (d, J = 8.7 Hz, 1H), 6.09 (br, 2H), 5.03 (s, 2H), 4.60 (d, J = 11.1 Hz, 1H), 4.45 (d, J = 11.1 Hz, 1H), 4.21 (t, J = 7.8 Hz, 1H), 3.95 (brs, 1H), 3.86 (dd, J = 7.2, 4.2 Hz, 1H), 3.69–3.46 (m, 2H), 3.28–3.05 (m, 2H), 2.92 (s, 2H), 2.68–2.60 (m, 2H), 2.57 (s, 3H), 2.51 (s, 3H), 2.07 (s, 3H), 2.05–1.92 (m, 2H), 1.86–1.72 (m, 1H), 1.57–1.43 (m, 4H), 1.44 (s, 6H), 1.18–1.06 (m, 1H), 0.92 (d, J = 6.7 Hz, 3H), 0.88 (brs, 12H), 0.04 (s, 3H); 0.03 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 173.29, 171.24, 158.58, 157.25, 156.12, 138.34, 137.22, 136.96, 133.46, 132.31, 129.34, 128.68, 128.53, 128.28, 128.17, 127.86, 127.47, 127.43, 124.43, 117.31, 114.95, 86.21, 79.42, 72.80, 70.11, 65.10, 57.87, 50.36, 43.29, 41.09, 36.98, 34.83, 30.22, 29.24, 28.57, 25.86, 25.17, 25.11, 19.20, 18.28, 17.85, 15.53, 12.40, 11.08, −5.49, −5.52 ppm; HRMS (ESI) calculated for C55H79N5O8SSi [M + H]+ 998.5419, found 998.5486.
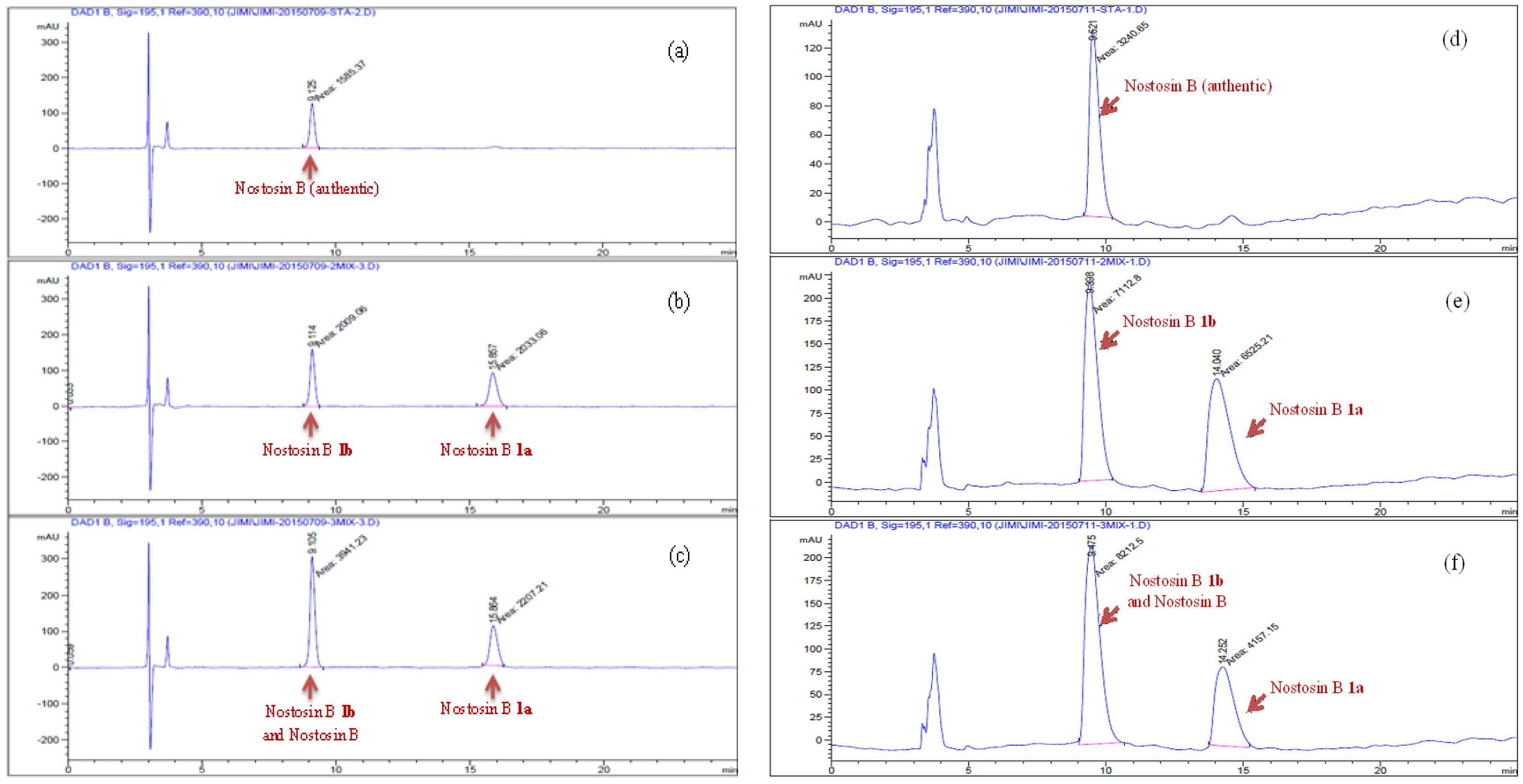
Tripeptide 22 (72 mg, 0.072 mmol) was dissolved in ethyl acetate (2 mL), Pd/C (8 mg, 10%) was added. Hydrogen was bubbled for 1 h at room temperature. The conversion of reaction was monitored by TLC. The mixture was concentrated and passed through a silica column. The filtrate was concentrated in vacuo, the residue was dried under high vacuum for 2 h and then it (56 mg, 0.068 mmol) was dissolved in DCM (2 mL), TFA (0.5 mL) was added at 0 °C. The reaction was stirred at room temperature for 2 h, volatiles were removed in vacuo. The residue was purified by HPLC and lyophilized to give nostosin B 1a (17 mg, 74%) as a powder. (Welch Materials. Inc. XB-C18, 5 μm, 4.6 × 250 mm, 1.0 mL/min, 18% MeCN:82% H2O within 30 min, DAD detector was adjusted to 195 nm wave length.). [α] = 1.6 (c 1.0, MeOH); 1H-NMR (400 MHz, DMSO-d6) δ 9.14 (brs, 1H), 7.86 (d, J = 8.4 Hz, 1H), 7.50–7.46 (m, 2H), 6.94 (d, J = 8.4 Hz, 2H), 6.64 (d, J = 8.4 Hz, 2H), 5.74 (brs, 1H), 4.65 (brs, 1H), 4.18 (dd, J = 9.2, 6.9 Hz, 1H), 3.84 (dd, J = 7.9, 3.8 Hz, 1H), 3.69 (td, J = 8.6, 3.8 Hz, 1H), 3.12–2.99 (m, 2H), 2.52–2.49 (m, 2H), 1.86–1.82 (m, 1H), 1.74–1.63 (m, 1H), 1.59–1.54 (m, 1H), 1.48–1.32 (m, 3H), 1.33–1.22 (m, 2H), 1.08–0.95 (m, 2H), 0.84–0.78 (m, 6H). 13C-NMR (100 MHz, DMSO-d6) δ 173.69, 171.06, 157.07, 155.78, 132.21, 129.56, 115.53, 70.86, 63.61, 56.41, 50.81, 41.17, 37.95, 37.39, 30.32, 28.24, 25.67, 24.67, 15.80, 11.46 ppm; HRMS (ESI) calculated for C22H37N5O5 [M + H]+ 452.2795, found 452.2856.
Compound 21 (8 mg, 0.03 mmol) and 7 (28 mg, 0.045 mmol) were dissolved in DCM (2 mL) at 0 °C, after HATU (23 mg, 0.06 mmol), HOAt (8 mg, 0.06 mmol) were added followed by addition of DIPEA (25 μL, 0.15 mmol), the reaction mixture was stirred at room temperature for 16 h. The reaction was concentrated in vacuo, the residue was dissolved in DCM (75 mL), the organic solution was washed sequentially by saturated aqueous solution of NH4Cl (30 mL), NaHCO3 (30 mL) and brine (30 mL). The organic layer was separated, dried over anhydrous sodium sulfate, and concentrated in vacuo. The residue was purified by flash chromatography to produce compound 23 (23 mg, 86%). [α] = −13.8 (c 1.0, CHCl3); 1H-NMR (500 MHz, CHCl3) δ 7.45–7.28 (m, 6H), 7.09 (d, J = 8.0 Hz, 2H), 6.91–6.82 (d, J = 8.0 Hz, 2H), 6.50 (d, J = 9.3 Hz, 1H), 6.36–6.21 (m, 2H), 6.00 (brs, 1H), 5.00 (s, 2H), 4.19 (t, J = 8.4 Hz, 1H), 4.10 (dd, J = 8.4, 3.4 Hz, 1H), 3.96 (brs, 1H), 3.54 (dd, J = 8.4, 4.2 Hz, 2H), 3.25–3.00 (m, 2H), 2.92 (s, 2H), 2.76–2.61 (m, 2H), 2.55 (s, 3H), 2.49 (s, 3H), 2.07 (s, 3H), 1.93–1.81 (m, 3H), 1.62–1.37 (m, 11H), 1.17–1.12 (m, 1H), 0.92–0.86 (m, 15H), 0.03 (s, 6H). 13C-NMR (125 MHz, CDCl3) δ 174.58, 171.80, 158.77, 157.17, 156.23, 138.40, 137.21, 133.50, 132.87, 132.33, 129.52, 128.53, 127.87, 127.47, 124.60, 117.48, 114.89, 86.36, 71.33, 70.05, 65.17, 58.09, 43.23, 40.90, 36.66, 36.40, 30.43, 29.69, 28.82, 28.58, 25.87, 25.29, 25.09, 19.23, 18.30, 17.88, 15.42, 12.45, 11.00, −5.47, −5.51 ppm; HRMS (ESI) calculated for C48H73N5O8SSi [M + H]+ 908.4949, found 908.5102.
Compound 23 (16 mg, 0.0176 mmol) was dissolved in ethyl acetate (2 mL), after Pd/C (5 mg, 10% Pd on charcoal) was added under a protective nitrogen flow, the reaction vessel was sealed and flashed by hydrogen. The reaction mixture was stirred under hydrogen atmosphere (balloon) at room temperature for 1 h, then it was filtered through a pad of celite to remove the catalyst, the filter cake was rinsed by ethyl acetate (5 mL × 2). The combined filtrate was concentrated in vacuo. The residue was dried under high vacuum for 2 h, and it (13 mg, 0.016 mmol) was subsequently dissolved in DCM (2 mL) at 0 °C, after TFA (0.5 mL) was added, the reaction mixture was stirred at room temperature for 2 h. Volatiles were removed in vacuo, the residue was purified by HPLC and lyophilized to give nostosin B 1b (6 mg, 75%) as a powder. (Agilent 1200; Column: Welch materials (Shanghai) Inc., Shanghai, China, XB-C18, 5 μm, 4.6 × 250 mm; flow rate: 1.0 mL/min; eluent: 18% CH3CN with 82% H2O; detector: DAD at 195 nm−1) [α] = −2.4 (c 1.1, MeOH); 1H-NMR (500 MHz, DMSO-d6) δ 9.13 (s, 1H), 7.84 (d, J = 8.5 Hz, 1H), 7.59 (d, J = 9.0 Hz, 1H), 7.54 (t, J = 6.1 Hz, 1H), 6.94 (d, J = 8.3 Hz, 2H), 6.65 (d, J = 8.3 Hz, 2H), 5.69 (d, J = 5.6 Hz, 1H), 4.70 (t, J = 5.3 Hz, 1H), 4.16–4.12 (m, 1H), 3.88 (ddd, J = 8.9, 5.7, 3.8 Hz, 1H), 3.69 (dq, J = 9.2, 4.1 Hz, 1H), 3,37-3.30 (m, 1H), 3.23–3.19 (m, 1H), 3.03 (dt, J = 12.1, 6.7 Hz, 2H), 2.52–2.46 (m, 2H), 1.87–1.78 (m, 1H), 1.72–1.54 (m, 3H), 1.52–1.36 (m, 3H), 1.33–1.24 (m, 1H), 1.08–1.00 (m, 1H), 0.85–0.75 (m, 6H). 13C-NMR (125 MHz, DMSO-d6) δ 173.41, 170.69, 156.65, 155.27, 131.75, 129.07, 115.03, 70.10, 63.17, 56.42, 50.23, 40.68, 37.02, 36.52, 29.86, 27.79, 25.10, 24.36, 15.28, 10.91 ppm; HRMS (ESI) calculated for C22H37N5O5 [M + H]+ 452.2795, found 452.2885.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}