
Transcriptome Analysis of Core Dinoflagellates Reveals a Universal Bias towards “GC” Rich Codons


Abstract
:1. Introduction
2. Results
2.1. GC Content
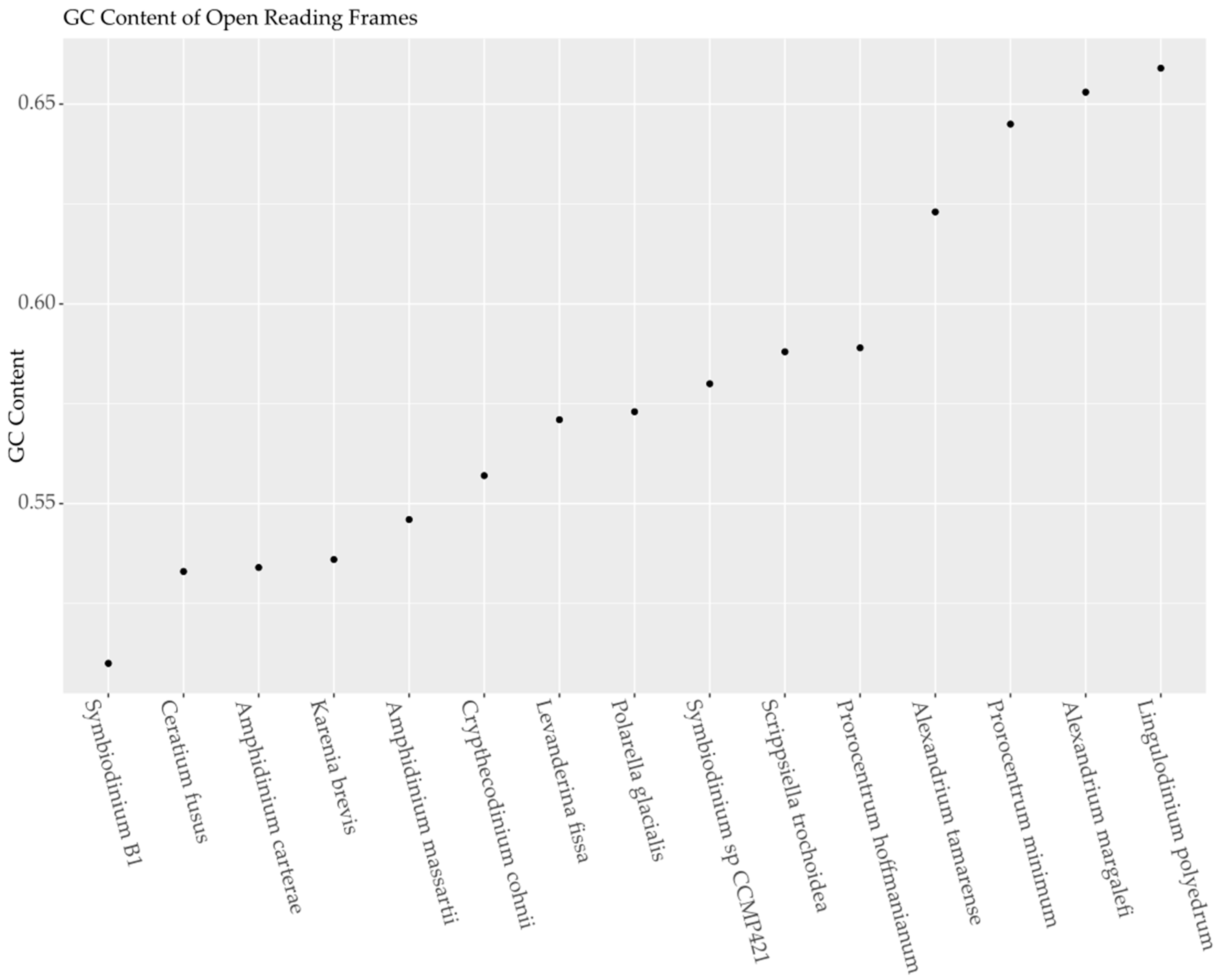
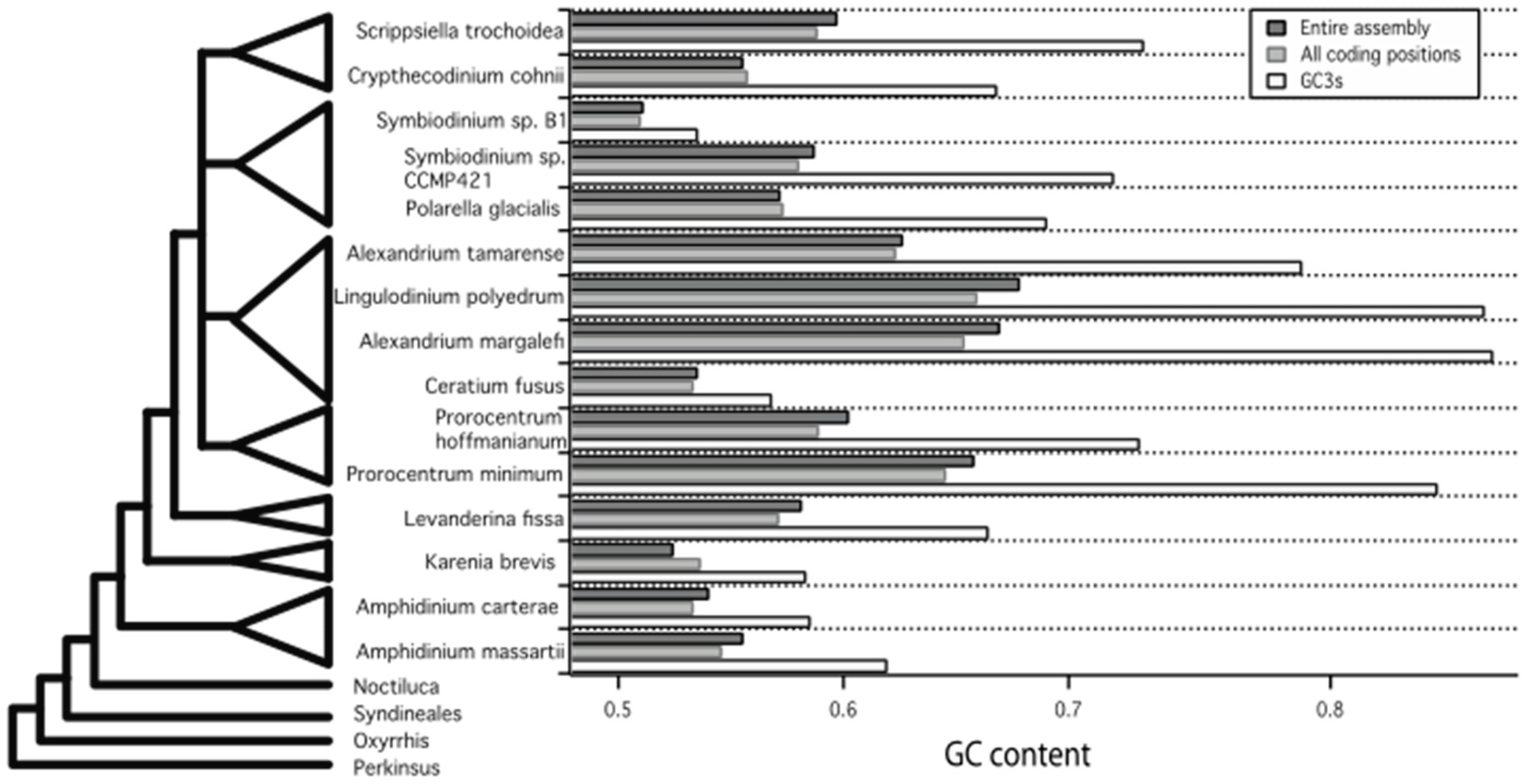
2.1.1. Total GC Content
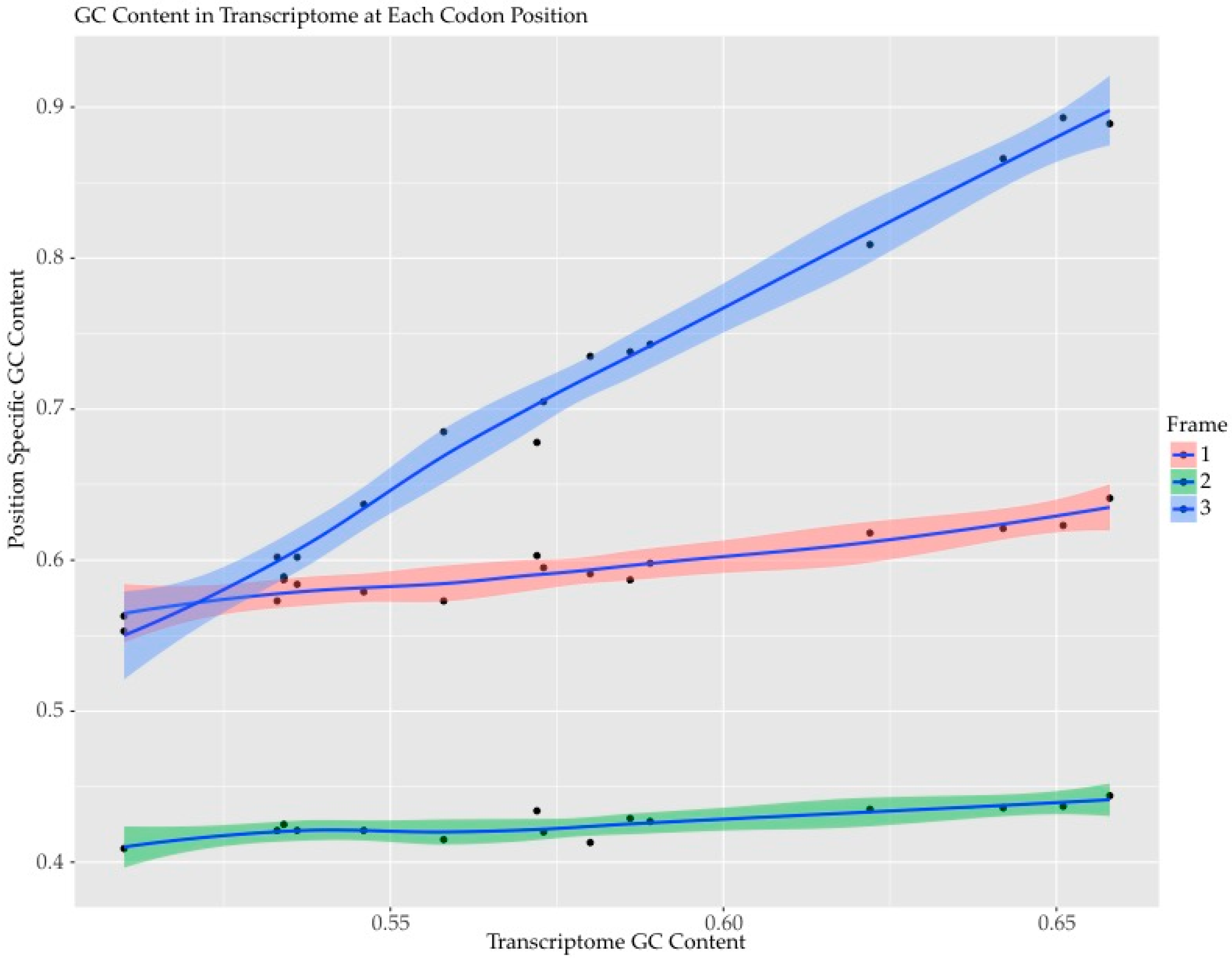
2.1.2. GC Content by Codon Position
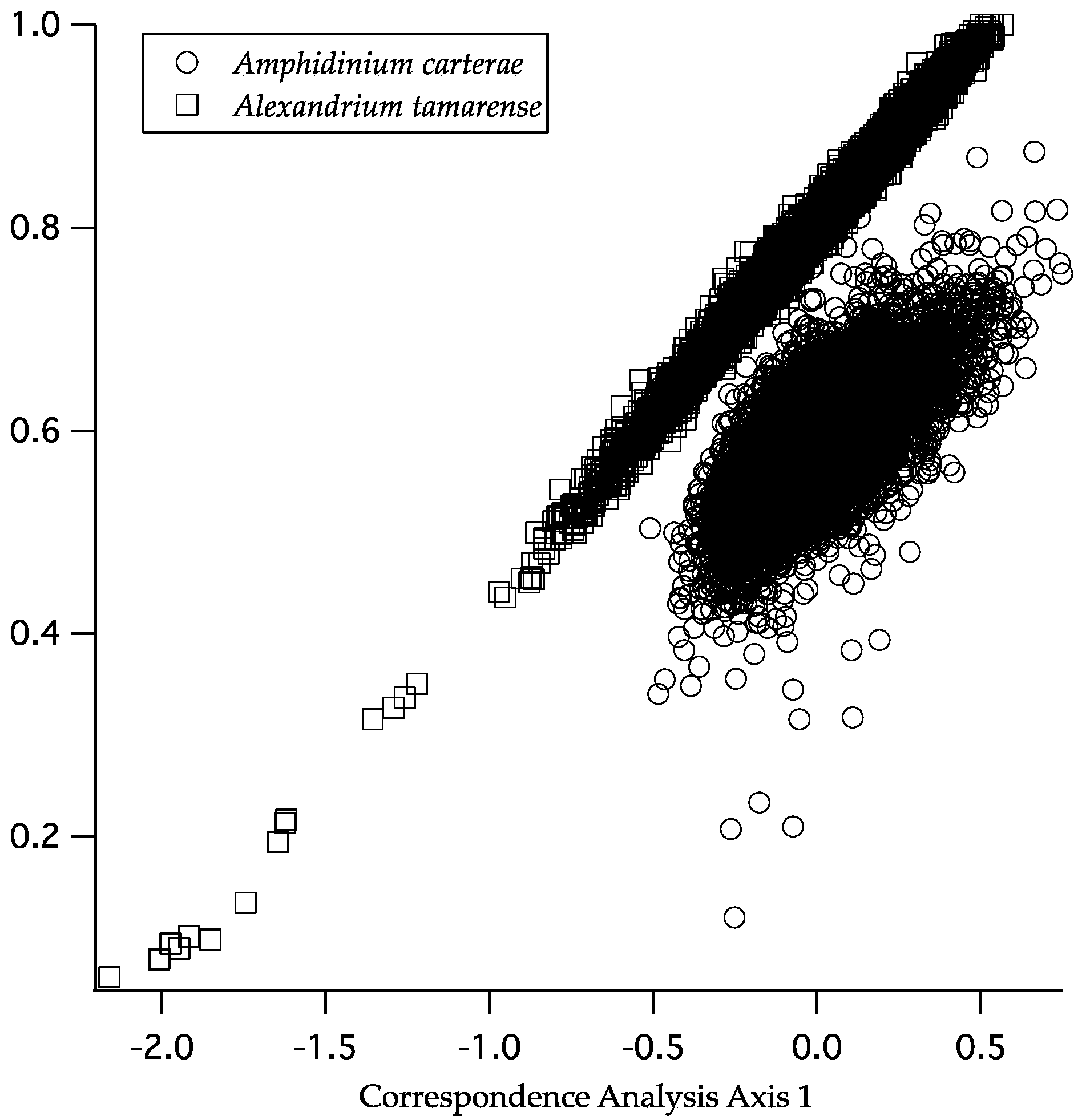
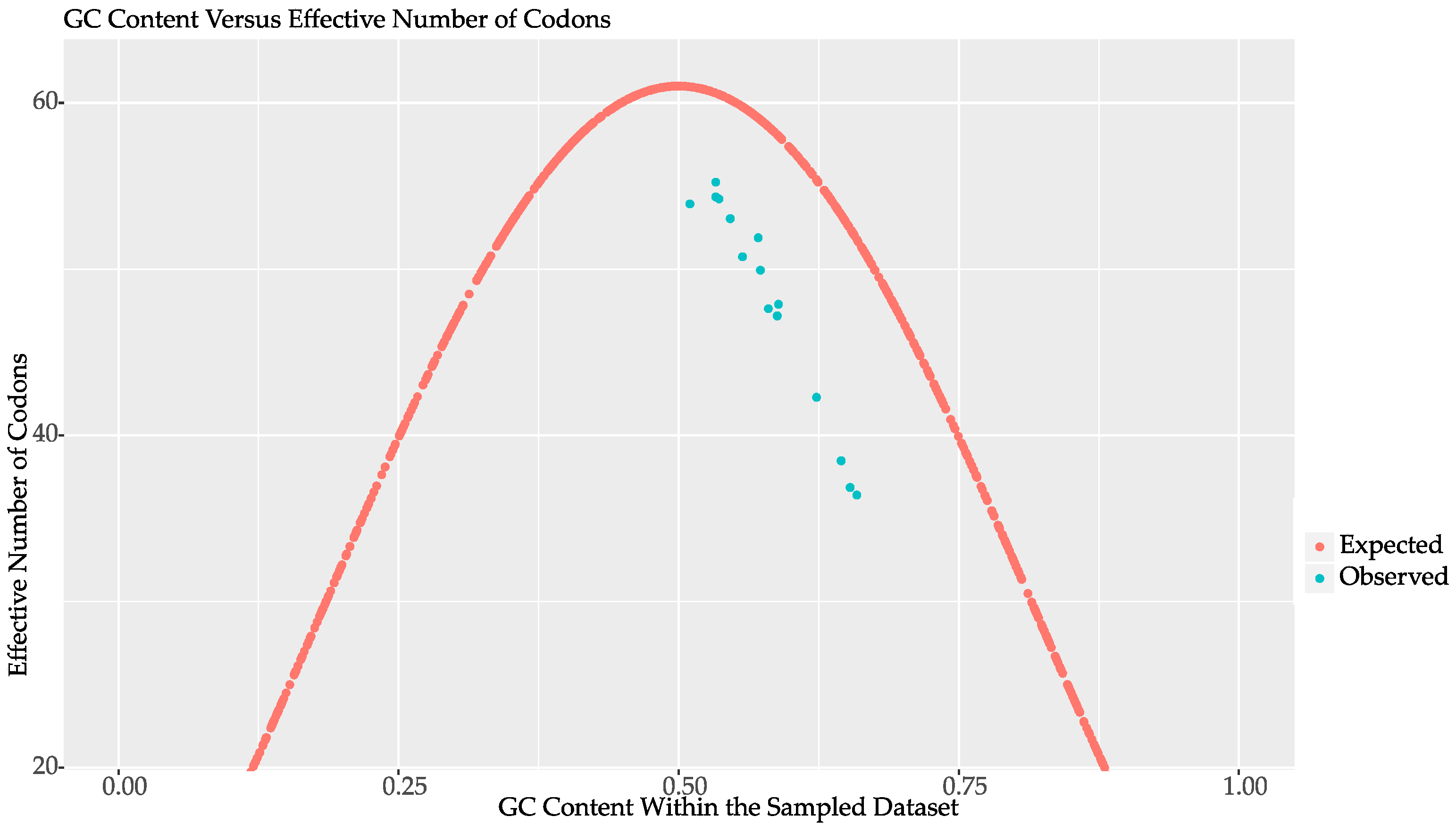
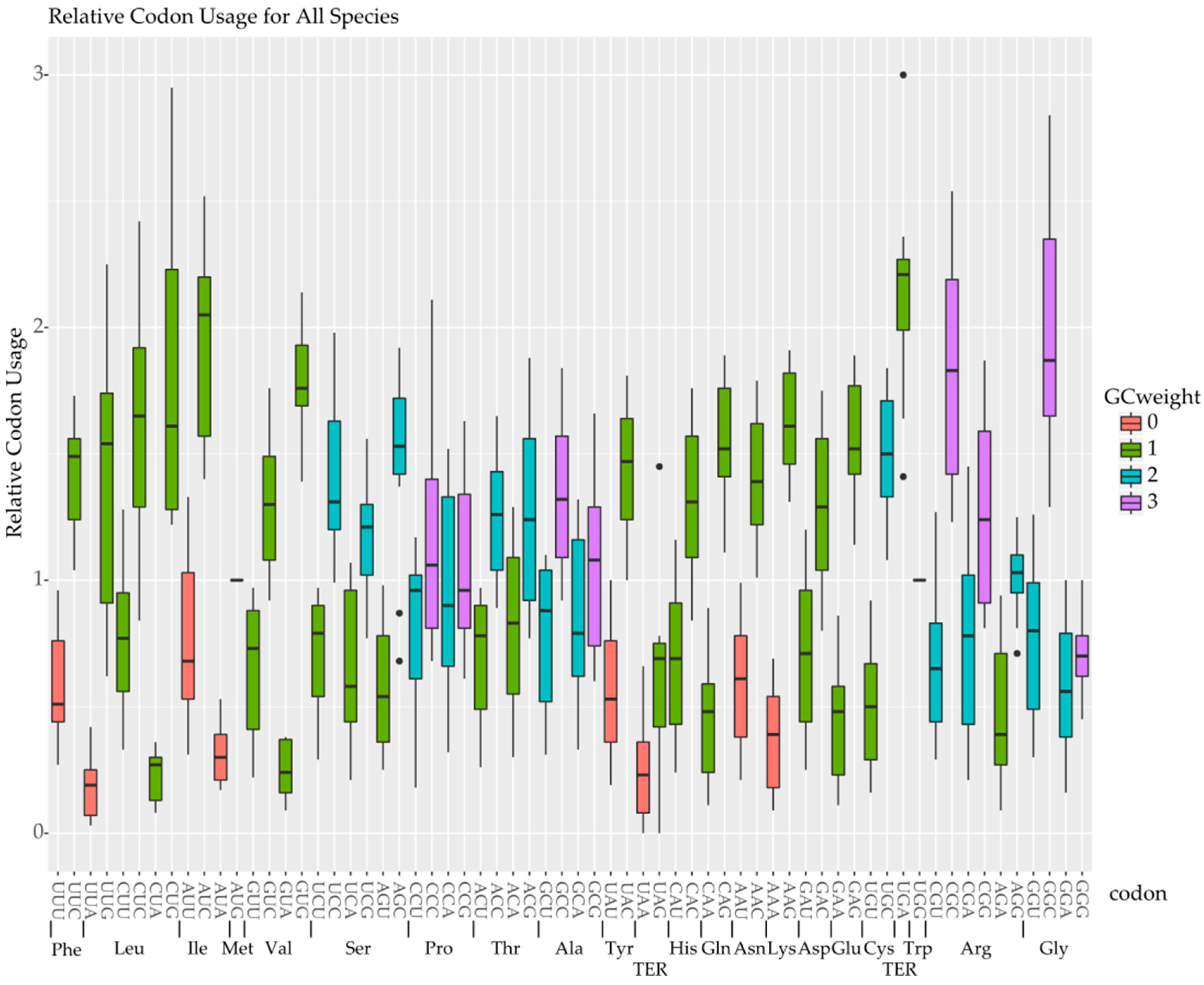
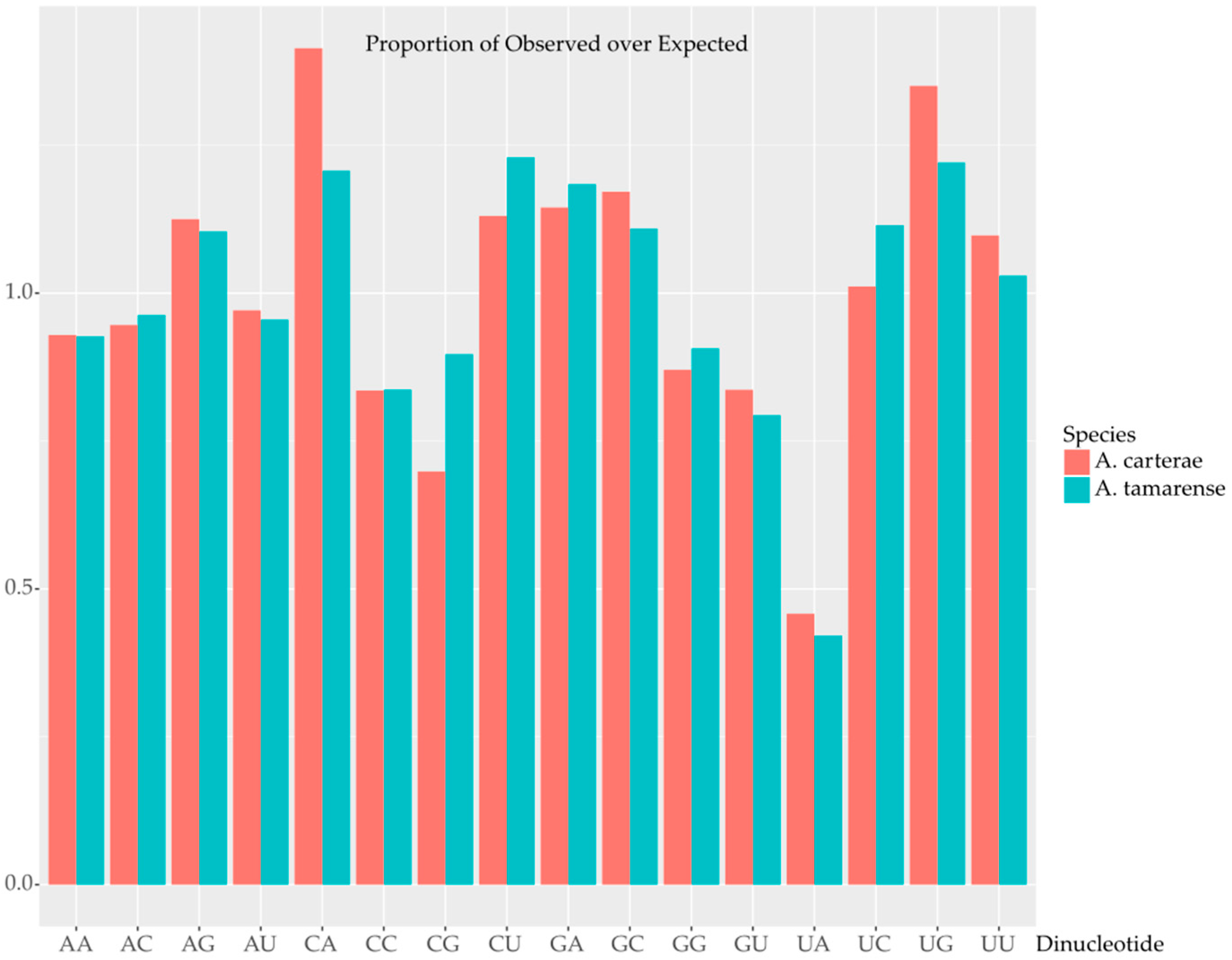
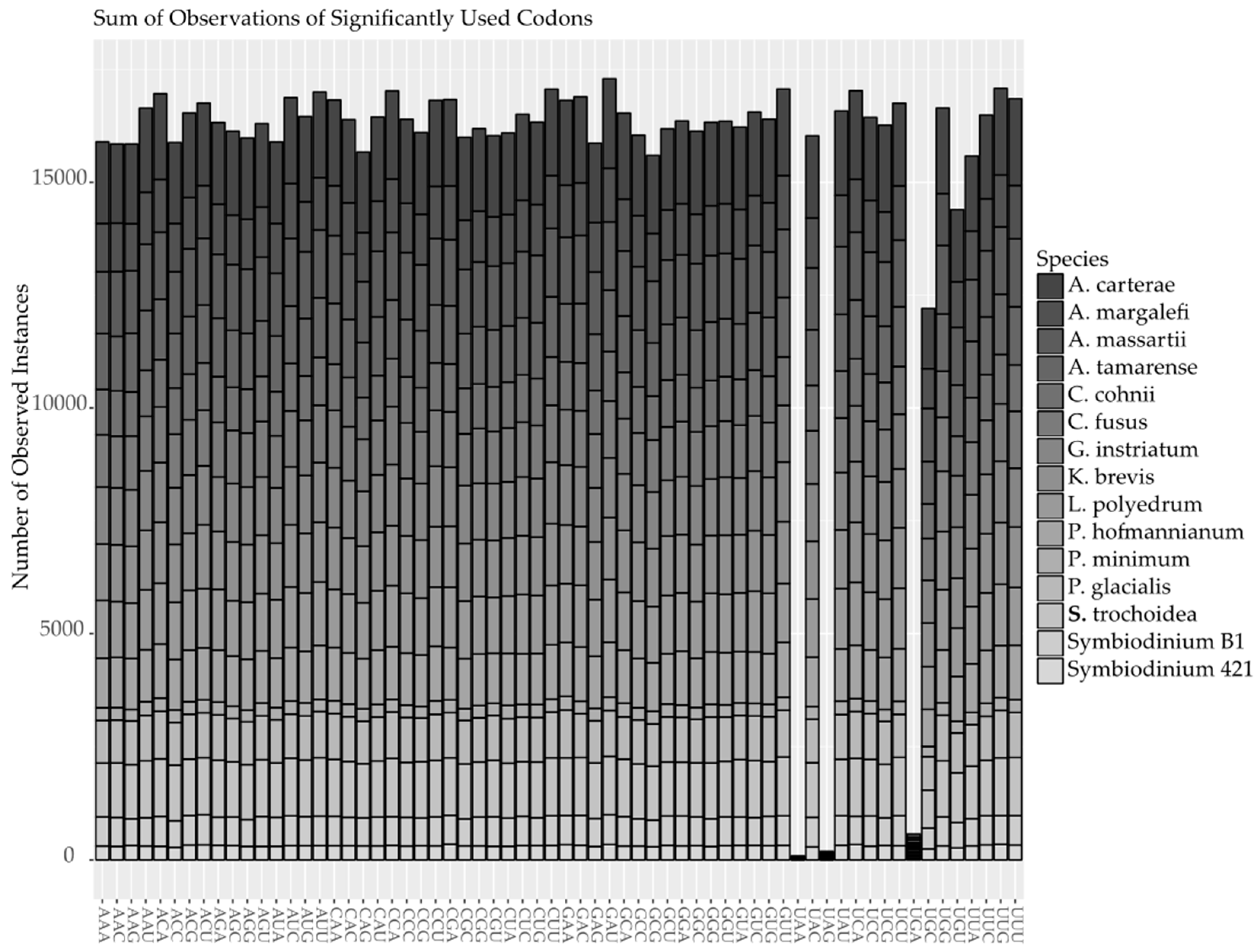
2.2. Codon Bias and Effective Codon Number
3. Discussion
4. Materials and Methods
4.1. ORF Extraction
4.2. Relative Synonomous Codon Usage Analyses
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Anderson, D.M. Red tides. Sci. Am. 1994, 271, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Z. Neurotoxins from marine dinoflagellates: A brief review. Mar. Drugs 2008, 6, 349–371. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Oshima, Y.; Yasumoto, T. Structures of two paralytic shellfish toxins, gonyautoxins V and VI, isolated from a tropical dinoflagellate, Pyrodinium bahamense var. compressa. Agric. Biol. Chem. 1982, 46, 1861–1864. [Google Scholar]
- Baden, D.G. Brevetoxins: Unique polyether dinoflagellate toxins. FASEB J. 1989, 3, 1807–1817. [Google Scholar] [PubMed]
- Seki, T.; Satake, M.; Mackenzie, L.; Kaspar, H.F.; Yasumoto, T. Gymnodimine, a new marine toxin of unprecedented structure isolated from New Zealand oysters and the dinoflagellate, Gymnodinium sp. Tetrahedron Lett. 1995, 36, 7093–7096. [Google Scholar] [CrossRef]
- Morse, D.; Milos, P.M.; Roux, E.; Hastings, J.W. Circadian regulation of bioluminescence in Gonyaulax involves translational control. Proc. Natl. Acad. Sci. USA 1989, 86, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Morey, J.S.; Monroe, E.A.; Kinney, A.L.; Beal, M.; Johnson, J.G.; Hitchcock, G.L.; Van Dolah, F.M. Transcriptomic response of the red tide dinoflagellate, Karenia brevis, to nitrogen and phosphorus depletion and addition. BMC Genom. 2011, 12, 346. [Google Scholar] [CrossRef] [PubMed]
- Lidie, K.B.; Ryan, J.C.; Barbier, M.; Van Dolah, F.M. Gene expression in Florida red tide dinoflagellate Karenia brevis: Analysis of an expressed sequence tag library and development of DNA microarray. Mar. Biotechnol. 2005, 7, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Proudfoot, N. Connecting transcription to messenger RNA processing. Trends Biochem. Sci. 2000, 25, 290–293. [Google Scholar] [CrossRef]
- Shirley, B.W.; Meagher, R.B. A potential role for RNA turnover in the light regualtion of plant gene expression: Ribulose-1, 5-bisphosphate carboxylase small subunit in soybean. Nucleic Acids Res. 1990, 18, 3377–3385. [Google Scholar] [CrossRef] [PubMed]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Baumgarten, S.; Bayer, T.; Aranda, M.; Liew, Y.J.; Carr, A.; Micklem, G.; Voolstra, C.R. Integrating microRNA and mRNA expression profiling in Symbiodinium microadriaticum, a dinoflagellate symbiont of reef-building corals. BMC Genom. 2013, 14, 704. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Qiu, L.; Hou, Z.; Zhang, Q.; Wu, J.; Gao, Q.; Song, L. Computational Identification of MicroRNAs from the Expressed Sequence Tags of Toxic Dinoflagellate Alexandrium tamarense. Evol. Bioinform. Online 2013, 9, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Morey, J.S.; Van Dolah, F.M. Global analysis of mRNA half-lives and de novo transcription in a dinoflagellate, Karenia brevis. PLoS ONE 2013, 8, e66347. [Google Scholar] [CrossRef] [PubMed]
- Novoa, E.M.; de Pouplana, L.R. Speeding with control: Codon usage, tRNAs, and ribosomes. Trends Genet. 2012, 28, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.D.; Li, Y.Q.; Chen, J.S.; Dong, H.J.; Guan, W.J.; Zhou, H. Whole genome analysis of non-optimal codon usage in secretory signal sequences of Streptomyces coelicolor. Biosystems 2006, 85, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ma, P.; Shah, P.; Rokas, A.; Liu, Y.; Johnson, C.H. Non-optimal codon usage is a mechanism to achieve circadian clock conditionality. Nature 2013, 495, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Guo, J.; Cha, J.; Chae, M.; Chen, S.; Barral, J.M.; Sachs, M.S.; Liu, Y. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature 2013, 495, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Hershberg, R.; Petrov, D.A. General rules for optimal codon choice. PLoS Genet. 2009, 5, e1000556. [Google Scholar] [CrossRef] [PubMed]
- Peden, J.F. Analysis of Codon Usage. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 2000. [Google Scholar]
- Bensoussan, C.; Rival, N.; Hanquet, G.; Colobert, F.; Reymond, S.; Cossy, J. Isolation, structural determination and synthetic approaches toward amphidinol 3. Nat. Prod. Rep. 2014, 31, 468–488. [Google Scholar] [CrossRef] [PubMed]
- Houdai, T.; Matsuoka, S.; Matsumori, N.; Murata, M. Membrane-permeabilizing activities of amphidinol 3, polyene-polyhydroxy antifungal from a marine dinoflagellate. Biochim. Biophys. Acta Biomembr. 2004, 1667, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Irwin, B.; Heck, J.D.; Hatfield, G.W. Codon Pair Utilization Biases Influence Translational Elongation Step Times. J. Biol. Chem. 1995, 270, 22801–22806. [Google Scholar] [CrossRef] [PubMed]
- Kudla, G.; Murray, A.W.; Tollervey, D.; Plotkin, J.B. Coding-sequence determinants of gene expression in Escherichia coli. Science 2009, 324, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Houdai, T.; Matsuoka, S.; Murata, M.; Satake, M.; Ota, S.; Oshima, Y.; Rhodes, L.L. Acetate labeling patterns of dinoflagellate polyketides, amphidinols 2, 3 and 4. Tetrahedron 2001, 57, 5551–5555. [Google Scholar] [CrossRef]
- Kellmann, R.; Stüken, A.; Orr, R.J.; Svendsen, H.M.; Jakobsen, K.S. Biosynthesis and molecular genetics of polyketides in marine dinoflagellates. Mar. Drugs 2010, 8, 1011–1148. [Google Scholar] [CrossRef] [PubMed]
- Van Wagoner, R.M.; Satake, M.; Wright, J.L. Polyketide biosynthesis in dinoflagellates: What makes it different? Nat. Prod. Rep. 2014, 31, 1101–1137. [Google Scholar] [CrossRef] [PubMed]
- Bachvaroff, T.R.; Place, A.R.; Coats, D.W. Expressed sequence tags from Amoebophrya sp. infecting Karlodinium veneficum: Comparing host and parasite sequences. J. Eukaryot. Microbiol. 2009, 56, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.W.; Place, A.R. Proliferation of 5-hydroxymethyl uracil in the genomes of dinoflagellates is synapomorphic to dinokaryon containing species. In Harmful Algae, Proceedings of the 15th International Conference on Harmful Algae, Changwon, Korea, 29 October–2 November 2012; Kim, H.G., Reguera, B., Hallengraeff, G.M., Lee, C.K., Han, M.S., Choi, J.K., Eds.; Maple Design Agency: Busan, Korea, 2014; pp. 153–156. [Google Scholar]
- Jones, G.D.; Williams, E.P.; Place, A.R.; Jagus, R.; Bachvaroff, T.R. The alveolate translation initiation factor 4E family reveals a custom toolkit for translational control in core dinoflagellates. BMC Evol. Biol. 2015, 15, 14. [Google Scholar] [CrossRef] [PubMed]
- Presnyak, V.; Alhusaini, N.; Chen, Y.H.; Martin, S.; Morris, N.; Kline, N.; Olson, S.; Weinberg, D.; Baker, K.E.; Graveley, B.R.; et al. Codon optimality is a major determinant of mRNA stability. Cell 2015, 160, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Janouškovec, J.; Gavelis, G.S.; Burki, F.; Dinh, D.; Bachvaroff, T.R.; Gornik, S.G.; Bright, K.J.; Imanian, B.; Strom, S.L.; Delwiche, C.F.; et al. Major transitions in dinoflagellate evolution unveiled by phylotranscriptomics. Proc. Natl. Acad. Sci. USA 2017, 114, E171–E180. [Google Scholar] [CrossRef] [PubMed]
- Gornik, S.G.; Ford, K.L.; Mulhern, T.D.; Bacic, A.; McFadden, G.I.; Waller, R.F. Loss of nucleosomal DNA condensation coincides with appearance of a novel nuclear protein in dinoflagellates. Curr. Biol. 2012, 22, 2303–2312. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Henikoff, S. Chromatin: Packaging without nucleosomes. Curr. Biol. 2012, 22, R1040–R1043. [Google Scholar] [CrossRef] [PubMed]
- Spector, D.L.; Triemer, R.E. Chromosome structure and mitosis in the dinoflagellates: An ultrastructural approach to an evolutionary problem. Biosystems 1981, 14, 289–298. [Google Scholar] [CrossRef]
- Chatton, E. Les Péridiniens Parasites: Morphologie, Reproduction, Ethologie. Arch. Zool. Exp. Gen. 1920, 59, l–475. [Google Scholar]
- Bütschli, O. Protozoa, Bronns Klassen und Ordnungen im Tierreich; CF Winter: Leipzig, Germany, 1880. [Google Scholar]
- Shoguchi, E.; Shinzato, C.; Kawashima, T.; Gyoja, F.; Mungpakdee, S.; Koyanagi, R.; Takeuchi, T.; Hisata, K.; Tanaka, M.; Fujiwara, M.; et al. Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr. Biol. 2013, 23, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Cheng, S.; Song, B.; Zhong, X.; Lin, X.; Li, W.; Li, L.; Zhang, Y.; Zhang, H.; Ji, Z.; et al. The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 2015, 350, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Aranda, M.; Li, Y.; Liew, Y.J.; Baumgarten, S.; Simakov, O.; Wilson, M.C.; Piel, J.; Ashoor, H.; Bougouffa, S.; Bajic, V.B.; et al. Genomes of coral dinoflagellate symbionts highlight evolutionary adaptations conducive to a symbiotic lifestyle. Sci. Rep. 2016, 6, 39734. [Google Scholar] [CrossRef] [PubMed]
- Bachvaroff, T.R.; Gornik, S.G.; Concepcion, G.T.; Waller, R.F.; Mendez, G.S.; Lippmeier, J.C.; Delwiche, C.F. Dinoflagellate phylogeny revisited: Using ribosomal proteins to resolve deep branching dinoflagellate clades. Mol. Phylogenet. Evol. 2014, 70, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species Name | Genes | Codons | Stops | CBI | Fop | Nc | GC3s | Transcriptome GC Content |
|---|---|---|---|---|---|---|---|---|
| Symbiodinium sp. B1 DRX021274-5 | 4270 | 1,445,562 | 19 | 0.007 | 0.420 | 53.911 | 0.535 | 0.510 |
| Ceratium fusus SRX554108-9 | 8133 | 2,611,156 | 47 | 0.047 | 0.443 | 55.218 | 0.568 | 0.533 |
| Karenia brevis (combined strains) SRX551359, SRX551258-61 | 8748 | 2,985,699 | 51 | 0.048 | 0.444 | 54.211 | 0.583 | 0.536 |
| Amphidinium carterae CCMP1314 SRX722011 | 12,578 | 1,866,927 | 88 | 0.025 | 0.431 | 54.339 | 0.585 | 0.533 |
| Amphidinium massartii SRX551299 | 9848 | 3,234,642 | 74 | 0.055 | 0.448 | 53.029 | 0.619 | 0.546 |
| Levanderina fissa SRX730950-55 | 8601 | 2,852,315 | 93 | 0.107 | 0.477 | 51.881 | 0.664 | 0.571 |
| Crypthecodinium cohnii SRX551367-8, SRX551296-97 | 6884 | 2,285,099 | 26 | 0.095 | 0.472 | 50.736 | 0.668 | 0.557 |
| Polarella glacialis SRX554324 | 6600 | 2,028,399 | 41 | 0.112 | 0.481 | 49.930 | 0.690 | 0.573 |
| Symbiodinium sp. CCMP421 SRX551158 | 3962 | 1,244,352 | 18 | 0.124 | 0.489 | 47.615 | 0.720 | 0.580 |
| Prorocentrum hoffmanianum SRX722009 | 7764 | 2,511,354 | 39 | 0.112 | 0.480 | 47.879 | 0.731 | 0.589 |
| Scrippsiella trochoidea CCMP3099 SRX551166-8 | 8303 | 2,785,323 | 33 | 0.107 | 0.478 | 47.182 | 0.733 | 0.588 |
| Alexandrium tamarense SRX554029-30 | 8583 | 2,785,359 | 35 | 0.160 | 0.508 | 42.275 | 0.803 | 0.623 |
| Prorocentrum minimum CCMP2233 SRX551159-61 | 1862 | 599,994 | 7 | 0.204 | 0.534 | 38.466 | 0.863 | 0.645 |
| Lingulodinium polyedrum SRX554063-5 | 8701 | 2,854,932 | 53 | 0.210 | 0.536 | 36.406 | 0.884 | 0.659 |
| Alexandrium margalefi SRX551350 | 7597 | 2,148,204 | 81 | 0.194 | 0.527 | 36.853 | 0.888 | 0.653 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, E.; Place, A.; Bachvaroff, T. Transcriptome Analysis of Core Dinoflagellates Reveals a Universal Bias towards “GC” Rich Codons. Mar. Drugs 2017, 15, 125. https://doi.org/10.3390/md15050125
Williams E, Place A, Bachvaroff T. Transcriptome Analysis of Core Dinoflagellates Reveals a Universal Bias towards “GC” Rich Codons. Marine Drugs. 2017; 15(5):125. https://doi.org/10.3390/md15050125
Chicago/Turabian StyleWilliams, Ernest, Allen Place, and Tsvetan Bachvaroff. 2017. "Transcriptome Analysis of Core Dinoflagellates Reveals a Universal Bias towards “GC” Rich Codons" Marine Drugs 15, no. 5: 125. https://doi.org/10.3390/md15050125
APA StyleWilliams, E., Place, A., & Bachvaroff, T. (2017). Transcriptome Analysis of Core Dinoflagellates Reveals a Universal Bias towards “GC” Rich Codons. Marine Drugs, 15(5), 125. https://doi.org/10.3390/md15050125